
Bone Marrow-Derived Cells in Endometrial Cancer Pathogenesis: Insights from Breast Cancer

Abstract
:1. Introduction
2. Endometrial Cancer
2.1. Types of Endometrial Cancer
2.2. Role of Estrogen in EC Pathogenesis
2.3. Role of Obesity in EC Pathogenesis
2.4. Role of p53 and PTEN Mutations in EC
3. Cancer Stem Cells
4. Pathways Involved in eCSC Maintenance
5. Endometrial Cancer Microenvironment
5.1. Cancer-Associated Fibroblasts in EC
5.2. Endothelial Cells in EC
5.3. Immune Cells in EC
6. Bone Marrow Niche in Hormone-Driven Cancers—Using Breast Cancer (BC) as a Model
6.1. Concept of BC Dormancy
6.2. Role of BM Niche in BC Dormancy
6.2.1. Perivascular Niche
6.2.2. Endosteal Niche
7. Perspective: BM-Derived Cells in EC Progression—Insights from BC
7.1. Role of CXCL12-CXCR4 Axis in BMDC Recruitment/Parallels with BC
7.2. Role of Specific BMDC Population: BM-MSC
7.3. Role of BM-MSC Exosomes in Promoting Dormancy
7.4. Role of Sex Hormones
7.5. Pathways Regulating Endometrial CSC Self Renewal/Maintenance: Parallels with BC
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- National Cancer Institute. Cancer Stat Facts: Uterine Cancer 2021. Available online: https://seer.cancer.gov/statfacts/html/corp.html (accessed on 15 January 2022).
- Henley, S.J.; Miller, J.W.; Dowling, N.F.; Benard, V.B.; Richardson, L.C. Uterine Cancer Incidence and Mortality—United States, 1999–2016. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1333–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.H.; Broaddus, R.R. Endometrial Cancer. N. Engl. J. Med. 2020, 383, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Giannone, G.; Attademo, L.; Scotto, G.; Genta, S.; Ghisoni, E.; Tuninetti, V.; Aglietta, M.; Pignata, S.; Valabrega, G. Endometrial Cancer Stem Cells: Role, Characterization and Therapeutic Implications. Cancers 2019, 11, 1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, K.K.; Thiel, K.W.; Goodheart, M.J.; De Geest, K.; Jia, Y.; Yang, S. Endometrial cancer. Obs. Gynecol. Clin. North. Am. 2012, 39, 255–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, M.J.; Summers, G.K.; Crotzer, D. Endometrial Cancer; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Gatius, S.; Cuevas, D.; Fernandez, C.; Roman-Canal, B.; Adamoli, V.; Piulats, J.M.; Eritja, N.; Martin-Satue, M.; Moreno-Bueno, G.; Matias-Guiu, X. Tumor Heterogeneity in Endometrial Carcinoma: Practical Consequences. Pathobiology 2018, 85, 35–40. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.A.; Ramkissoon, S.H.; Bryan, M.; Pliner, L.F.; Dontu, G.; Patel, P.S.; Amiri, S.; Pine, S.R.; Rameshwar, P. Delineation of breast cancer cell hierarchy identifies the subset responsible for dormancy. Sci. Rep. 2012, 2, 906. [Google Scholar] [CrossRef]
- Tempest, N.; Maclean, A.; Hapangama, D.K. Endometrial Stem Cell Markers: Current Concepts and Unresolved Questions. Int. J. Mol. Sci. 2018, 19, 3240. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, S.S.; Zhang, X.D.; Hondermarck, H.; Tanwar, P.S. The Emerging Role of the Microenvironment in Endometrial Cancer. Cancers 2018, 10, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Sanchis, C.; Cervello, I.; Khurana, S.; Faus, A.; Verfaillie, C.; Simon, C. Contribution of different bone marrow-derived cell types in endometrial regeneration using an irradiated murine model. Fertil Steril 2015, 103, 1596–1605.e1591. [Google Scholar] [CrossRef] [PubMed]
- Morelli, S.S.; Rameshwar, P.; Goldsmith, L.T. Experimental evidence for bone marrow as a source of nonhematopoietic endometrial stromal and epithelial compartment cells in a murine model. Biol. Reprod. 2013, 89, 7. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Taylor, H.S. Contribution of bone marrow-derived stem cells to endometrium and endometriosis. Stem. Cells 2007, 25, 2082–2086. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Yi, K.W. Bone marrow-derived stem cells contribute to regeneration of the endometrium. Clin. Exp. Reprod. Med. 2018, 45, 149–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruppender, N.S.; Morrissey, C.; Lange, P.H.; Vessella, R.L. Dormancy in solid tumors: Implications for prostate cancer. Cancer Metastasis Rev. 2013, 32, 501–509. [Google Scholar] [CrossRef]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Setiawan, V.W.; Yang, H.P.; Pike, M.C.; McCann, S.E.; Yu, H.; Xiang, Y.-B.; Wolk, A.; Wentzensen, N.; Weiss, N.S.; Webb, P.M.; et al. Type I and II endometrial cancers: Have they different risk factors? J. Clin. Oncol. 2013, 31, 2607–2618. [Google Scholar] [CrossRef]
- Malik, T.Y.; Chishti, U.; Aziz, A.B.; Sheikh, I. Comparison of Risk Factors and survival of Type 1 and Type II Endometrial Cancers. Pak. J. Med. Sci. 2016, 32, 886–890. [Google Scholar] [CrossRef]
- Murali, R.; Soslow, R.A.; Weigelt, B. Classification of endometrial carcinoma: More than two types. Lancet Oncol. 2014, 15, e268–e278. [Google Scholar] [CrossRef]
- Moore, K.N.; Fader, A.N. Uterine papillary serous carcinoma. Clin. Obs. Gynecol. 2011, 54, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Wilczyński, M.; Danielska, J.; Wilczyński, J. An update of the classical Bokhman’s dualistic model of endometrial cancer. Prz Menopauzalny 2016, 15, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Talhouk, A.; McAlpine, J.N. New classification of endometrial cancers: The development and potential applications of genomic-based classification in research and clinical care. Gynecol. Oncol. Res. Pract. 2016, 3, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, B.G.; Carr, B.R. The Normal Menstrual Cycle and the Control of Ovulation. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Gompel, A. Progesterone and endometrial cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2020, 69, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Constantine, G.D.; Kessler, G.; Graham, S.; Goldstein, S.R. Increased Incidence of Endometrial Cancer Following the Women’s Health Initiative: An Assessment of Risk Factors. J. Womens Health 2019, 28, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Onstad, M.A.; Schmandt, R.E.; Lu, K.H. Addressing the Role of Obesity in Endometrial Cancer Risk, Prevention, and Treatment. J. Clin. Oncol. 2016, 34, 4225–4230. [Google Scholar] [CrossRef] [PubMed]
- Trojano, G.; Olivieri, C.; Tinelli, R.; Damiani, G.R.; Pellegrino, A.; Cicinelli, E. Conservative treatment in early stage endometrial cancer: A review. Acta Biomed. 2019, 90, 405–410. [Google Scholar] [CrossRef]
- Nakamura, M.; Obata, T.; Daikoku, T.; Fujiwara, H. The Association and Significance of p53 in Gynecologic Cancers: The Potential of Targeted Therapy. Int. J. Mol. Sci. 2019, 20, 5482. [Google Scholar] [CrossRef] [Green Version]
- Schultheis, A.M.; Martelotto, L.G.; De Filippo, M.R.; Piscuglio, S.; Ng, C.K.; Hussein, Y.R.; Reis-Filho, J.S.; Soslow, R.A.; Weigelt, B. TP53 Mutational Spectrum in Endometrioid and Serous Endometrial Cancers. Int. J. Gynecol. Pathol. 2016, 35, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Wild, P.J.; Ikenberg, K.; Fuchs, T.J.; Rechsteiner, M.; Georgiev, S.; Fankhauser, N.; Noske, A.; Roessle, M.; Caduff, R.; Dellas, A.; et al. p53 suppresses type II endometrial carcinomas in mice and governs endometrial tumour aggressiveness in humans. EMBO Mol. Med. 2012, 4, 808–824. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, A.; Minaguchi, T.; Fujieda, K.; Hosokawa, Y.; Nishida, K.; Shikama, A.; Tasaka, N.; Sakurai, M.; Ochi, H.; Satoh, T. Abnormal accumulation of p53 predicts radioresistance leading to poor survival in patients with endometrial carcinoma. Oncol. Lett. 2019, 18, 5952–5958. [Google Scholar] [CrossRef] [PubMed]
- Obata, T.; Nakamura, M.; Mizumoto, Y.; Iizuka, T.; Ono, M.; Terakawa, J.; Daikoku, T.; Fujiwara, H. Dual expression of immunoreactive estrogen receptor beta and p53 is a potential predictor of regional lymph node metastasis and postoperative recurrence in endometrial endometrioid carcinoma. PLoS ONE 2017, 12, e0188641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavropoulos, A.; Varras, M.; Vasilakaki, T.; Varra, V.K.; Tsavari, A.; Varra, F.N.; Nonni, A.; Kavantzas, N.; Lazaris, A.C. Expression of p53 and PTEN in human primary endometrial carcinomas: Clinicopathological and immunohistochemical analysis and study of their concomitant expression. Oncol. Lett. 2019, 17, 4575–4589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallares, J.; Bussaglia, E.; Martinez-Guitarte, J.L.; Dolcet, X.; Llobet, D.; Rue, M.; Sanchez-Verde, L.; Palacios, J.; Prat, J.; Matias-Guiu, X. Immunohistochemical analysis of PTEN in endometrial carcinoma: A tissue microarray study with a comparison of four commercial antibodies in correlation with molecular abnormalities. Mod. Pathol. 2005, 18, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Bussaglia, E.; del Rio, E.; Matias-Guiu, X.; Prat, J. PTEN mutations in endometrial carcinomas: A molecular and clinicopathologic analysis of 38 cases. Hum. Pathol. 2000, 31, 312–317. [Google Scholar] [CrossRef]
- Mutter, G.L.; Lin, M.C.; Fitzgerald, J.T.; Kum, J.B.; Baak, J.P.; Lees, J.A.; Weng, L.P.; Eng, C. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J. Natl. Cancer Inst. 2000, 92, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, H.B.; Stefansson, I.; Kalvenes, M.B.; Das, S.; Akslen, L.A. Loss of PTEN expression is associated with metastatic disease in patients with endometrial carcinoma. Cancer 2002, 94, 2185–2191. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Gao, J.; Luo, F.; Rui, C.; Zheng, T.; Wang, D.; Wang, Y.; Roberts, T.M.; Liu, P.; Zhao, J.J.; et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene 2018, 37, 341–351. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [Green Version]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology 2014, 28, 1101–1107, 1110. [Google Scholar]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleffel, S.; Schatton, T. Tumor dormancy and cancer stem.m cells: Two sides of the same coin? Adv. Exp. Med. Biol. 2013, 734, 145–179. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell. Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef]
- Sell, S. Stem cell origin of cancer and differentiation therapy. Crit. Rev. Oncol. Hematol. 2004, 51, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.A.; Friel, A.M.; Kumar, B.; Zhang, L.; Rueda, B.R.; Gargett, C.E. Evidence for Cancer Stem Cells in Human Endometrial Carcinoma. Cancer Res. 2009, 69, 8241–8248. [Google Scholar] [CrossRef] [Green Version]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Butti, R.; Gunasekaran, V.P.; Kumar, T.V.S.; Banerjee, P.; Kundu, G.C. Breast cancer stem cells: Biology and therapeutic implications. Int. J. Biochem. Cell Biol. 2019, 107, 38–52. [Google Scholar] [CrossRef]
- Vinson, K.E.; George, D.C.; Fender, A.W.; Bertrand, F.E.; Sigounas, G. The Notch pathway in colorectal cancer. Int. J. Cancer 2016, 138, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Maliekal, T.T.; Bajaj, J.; Giri, V.; Subramanyam, D.; Krishna, S. The role of Notch signaling in human cervical cancer: Implications for solid tumors. Oncogene 2008, 27, 5110–5114. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tang, P.; Li, S.; Qin, X.; Yang, H.; Wu, C.; Liu, Y. Notch signaling pathway networks in cancer metastasis: A new target for cancer therapy. Med. Oncol. 2017, 34, 180. [Google Scholar] [CrossRef]
- Dong, B.; Li, S.; Zhu, S.; Yi, M.; Luo, S.; Wu, K. MiRNA-mediated EMT and CSCs in cancer chemoresistance. Exp. Hematol. Oncol. 2021, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Lang, B.; Meng, L.R. Blocking NOTCH pathway can enhance the effect of EGFR inhibitor through targeting CD133+ endometrial cancer cells. Cancer Biol. Ther. 2018, 19, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bley, N.; Hmedat, A.; Muller, S.; Rolnik, R.; Rausch, A.; Lederer, M.; Huttelmaier, S. Musashi-1-A Stemness RBP for Cancer Therapy? Biology 2021, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Greve, B.; Kelsch, R.; Müller-Uthoff, H.; Weiss, K.; Kharabi Masouleh, B.; Sibrowski, W.; Kiesel, L.; Buchweitz, O. The adult stem cell marker Musashi-1 modulates endometrial carcinoma cell cycle progression and apoptosis via Notch-1 and p21WAF1/CIP1. Int. J. Cancer 2011, 129, 2042–2049. [Google Scholar] [CrossRef]
- Taciak, B.; Pruszynska, I.; Kiraga, L.; Bialasek, M.; Krol, M. Wnt signaling pathway in development and cancer. J. Physiol Pharm. 2018, 69, 185–196. [Google Scholar] [CrossRef]
- Wend, P.; Holland, J.D.; Ziebold, U.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Semin Cell Dev. Biol. 2010, 21, 855–863. [Google Scholar] [CrossRef]
- Fatima, I.; Barman, S.; Rai, R.; Thiel, K.W.W.; Chandra, V. Targeting Wnt Signaling in Endometrial Cancer. Cancers 2021, 13, 2351. [Google Scholar] [CrossRef]
- Chen, H.; Wang, Y.; Xue, F. Expression and the clinical significance of Wnt10a and Wnt10b in endometrial cancer are associated with the Wnt/beta-catenin pathway. Oncol. Rep. 2013, 29, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, N.; Inagaki, T.; Kusunoki, S.; Okabe, H.; Yamada, I.; Matsumoto, A.; Terao, Y.; Takeda, S.; Kato, K. SPARC was overexpressed in human endometrial cancer stem-like cells and promoted migration activity. Gynecol. Oncol. 2014, 134, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Yan, L.; Zhao, X.; Li, C.; Fu, Y. microRNA-21 overexpression contributes to cell proliferation by targeting PTEN in endometrioid endometrial cancer. Oncol. Lett. 2012, 4, 1290–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Yue, Y.; Wang, R.; Zhang, Y.; Jin, Q.; Zhou, X. Overexpression of microRNA-194 suppresses the epithelial-mesenchymal transition in targeting stem cell transcription factor Sox3 in endometrial carcinoma stem cells. Tumour Biol. 2017, 39, 1010428317706217. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, W.; Huang, K.; Wang, Y.; Li, J.; Yang, X. MicroRNA-34a inhibits cells proliferation and invasion by downregulating Notch1 in endometrial cancer. Oncotarget 2017, 8, 111258–111270. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, T.; Huang, Y. MicroRNA-134 suppresses endometrial cancer stem cells by targeting POGLUT1 and Notch pathway proteins. FEBS Lett. 2015, 589, 207–214. [Google Scholar] [CrossRef]
- Ding, D.C.; Liu, H.W.; Chang, Y.H.; Chu, T.Y. Expression of CD133 in endometrial cancer cells and its implications. J. Cancer 2017, 8, 2142–2153. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Zhang, X.; Mizumoto, Y.; Maida, Y.; Bono, Y.; Takakura, M.; Kyo, S. Molecular characterization of CD133+ cancer stem-like cells in endometrial cancer. Int. J. Oncol. 2014, 44, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef]
- Park, J.Y.; Hong, D.; Park, J.Y. Association between Morphological Patterns of Myometrial Invasion and Cancer Stem Cell Markers in Endometrial Endometrioid Carcinoma. Pathol. Oncol. Res. 2019, 25, 123–130. [Google Scholar] [CrossRef]
- Elbasateeny, S.S.; Salem, A.A.; Abdelsalam, W.A.; Salem, R.A. Immunohistochemical expression of cancer stem cell related markers CD44 and CD133 in endometrial cancer. Pathol. Res. Pract. 2016, 212, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kyo, S.; Nakamura, M.; Mizumoto, Y.; Maida, Y.; Bono, Y.; Takakura, M.; Fujiwara, H. Imatinib sensitizes endometrial cancer cells to cisplatin by targeting CD117-positive growth-competent cells. Cancer Lett. 2014, 345, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [Green Version]
- Rahadiani, N.; Ikeda, J.-i.; Mamat, S.; Matsuzaki, S.; Ueda, Y.; Umehara, R.; Tian, T.; Wang, Y.; Enomoto, T.; Kimura, T.; et al. Expression of aldehyde dehydrogenase 1 (ALDH1) in endometrioid adenocarcinoma and its clinical implications. Cancer Sci. 2011, 102, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Kitson, S.J.; Rosser, M.; Fischer, D.P.; Marshall, K.M.; Clarke, R.B.; Crosbie, E.J. Targeting Endometrial Cancer Stem Cell Activity with Metformin Is Inhibited by Patient-Derived Adipocyte-Secreted Factors. Cancers 2019, 11, 653. [Google Scholar] [CrossRef] [Green Version]
- Kusunoki, S.; Kato, K.; Tabu, K.; Inagaki, T.; Okabe, H.; Kaneda, H.; Suga, S.; Terao, Y.; Taga, T.; Takeda, S. The inhibitory effect of salinomycin on the proliferation, migration and invasion of human endometrial cancer stem-like cells. Gynecol. Oncol. 2013, 129, 598–605. [Google Scholar] [CrossRef]
- Talkowski, K.; Kiełbasiński, K.; Peszek, W.; Grabarek, B.O.; Boroń, D.; Oplawski, M. Salinomycin Modulates the Expression of mRNAs and miRNAs Related to Stemness in Endometrial Cancer. Curr. Pharm. Biotechnol. 2021, 22, 317–326. [Google Scholar] [CrossRef]
- Zhou, X.; Zhou, Y.-P.; Huang, G.-R.; Gong, B.-L.; Yang, B.; Zhang, D.-X.; Hu, P.; Xu, S.-R. Expression of the stem cell marker, Nanog, in human endometrial adenocarcinoma. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2011, 30, 262–270. [Google Scholar] [CrossRef]
- Kong, F.F.; Li, D.; Yang, H.; Ma, J.; Pan, X.; Liu, H.X.; Huo, J.N.; Ma, X.X. Preliminary identification of endometrial cancer stem cells in vitro and in vivo. Biochem. Biophys Res. Commun. 2017, 490, 506–513. [Google Scholar] [CrossRef]
- Lu, H.; Ju, D.-D.; Yang, G.-D.; Zhu, L.-Y.; Yang, X.-M.; Li, J.; Song, W.-W.; Wang, J.-H.; Zhang, C.-C.; Zhang, Z.-G.; et al. Targeting cancer stem cell signature gene SMOC-2 Overcomes chemoresistance and inhibits cell proliferation of endometrial carcinoma. EBioMedicine 2019, 40, 276–289. [Google Scholar] [CrossRef] [Green Version]
- Larue, L.; Bellacosa, A. Epithelial–mesenchymal transition in development and cancer: Role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, A.I.; Trinidad, J.R.; Sandiford, O.; Etchegaray, J.P.; Rameshwar, P. Epigenetic dynamics in cancer stem cell dormancy. Cancer Metastasis Rev. 2020, 39, 721–738. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Donnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, K.S.; Tham, S.T.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-associated fibroblasts promote proliferation of endometrial cancer cells. PLoS ONE 2013, 8, e68923. [Google Scholar] [CrossRef]
- Subramaniam, K.S.; Omar, I.S.; Kwong, S.C.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-associated fibroblasts promote endometrial cancer growth via activation of interleukin-6/STAT-3/c-Myc pathway. Am. J. Cancer Res. 2016, 6, 200–213. [Google Scholar] [PubMed]
- Wang, X.; Zhang, W.; Sun, X.; Lin, Y.; Chen, W. Cancer-associated fibroblasts induce epithelial-mesenchymal transition through secreted cytokines in endometrial cancer cells. Oncol. Lett. 2018, 15, 5694–5702. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.; Tian, W.Y.; Wang, Y.M.; Zhang, Y.F.; Guo, F.; Zhao, J.; Gao, C.; Xue, F.X. Cancer-associated fibroblasts promote the progression of endometrial cancer via the SDF-1/CXCR4 axis. J. Hematol. Oncol. 2016, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Walentowicz-Sadlecka, M.; Sadlecki, P.; Bodnar, M.; Marszalek, A.; Walentowicz, P.; Sokup, A.; Wilinska-Jankowska, A.; Grabiec, M. Stromal derived factor-1 (SDF-1) and its receptors CXCR4 and CXCR7 in endometrial cancer patients. PLoS ONE 2014, 9, e84629. [Google Scholar] [CrossRef]
- Yu, Y.; Shi, X.; Shu, Z.; Xie, T.; Huang, K.; Wei, L.; Song, H.; Zhang, W.; Xue, X. Stromal cell-derived factor-1 (SDF-1)/CXCR4 axis enhances cellular invasion in ovarian carcinoma cells via integrin beta1 and beta3 expressions. Oncol. Res. 2013, 21, 217–225. [Google Scholar] [CrossRef]
- Corcoran, K.E.; Trzaska, K.A.; Fernandes, H.; Bryan, M.; Taborga, M.; Srinivas, V.; Packman, K.; Patel, P.S.; Rameshwar, P. Mesenchymal stem cells in early entry of breast cancer into bone marrow. PLoS ONE 2008, 3, e2563. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, E.D. Effect of estrogen on angiogenesis in co-cultures of human endometrial cells and microvascular endothelial cells. Hum. Reprod. 2003, 18, 2039–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamat, A.A.; Merritt, W.M.; Coffey, D.; Lin, Y.G.; Patel, P.R.; Broaddus, R.; Nugent, E.; Han, L.Y.; Landen, C.N., Jr.; Spannuth, W.A.; et al. Clinical and biological significance of vascular endothelial growth factor in endometrial cancer. Clin. Cancer Res. 2007, 13, 7487–7495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahecha, A.M.; Wang, H. The influence of vascular endothelial growth factor-A and matrix metalloproteinase-2 and -9 in angiogenesis, metastasis, and prognosis of endometrial cancer. Onco. Targets 2017, 10, 4617–4624. [Google Scholar] [CrossRef] [Green Version]
- Habeeb, O.; Goodglick, L.; Soslow, R.A.; Rao, R.G.; Gordon, L.K.; Schirripa, O.; Horvath, S.; Braun, J.; Seligson, D.B.; Wadehra, M. Epithelial membrane protein-2 expression is an early predictor of endometrial cancer development. Cancer 2010, 116, 4718–4726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, L.K.; Kiyohara, M.; Fu, M.; Braun, J.; Dhawan, P.; Chan, A.; Goodglick, L.; Wadehra, M. EMP2 regulates angiogenesis in endometrial cancer cells through induction of VEGF. Oncogene 2013, 32, 5369–5376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paprocka, M.; Kieda, C.; Kantor, A.; Bielawska-Pohl, A.; Dus, D.; Czekanski, A.; Heimrath, J. Increased Endothelial Progenitor Cell Number in Early Stage of Endometrial Cancer. Int. J. Gynecol. Cancer 2017, 27, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Jiang, T.; Zhao, W.B.; Wang, F.; Wang, G.L.; Cui, M.; Wen, Z.Q. Gene alterations in tumor-associated endothelial cells from endometrial cancer. Int. J. Mol. Med. 2008, 22, 619–632. [Google Scholar]
- Zhang, J.; Song, H.; Lu, Y.; Chen, H.; Jiang, S.; Li, L. Effects of estradiol on VEGF and bFGF by Akt in endometrial cancer cells are mediated through the NF-kappaB pathway. Oncol. Rep. 2016, 36, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Seo, K.H.; Lee, H.S.; Jung, B.; Ko, H.M.; Choi, J.H.; Park, S.J.; Choi, I.H.; Lee, H.K.; Im, S.Y. Estrogen enhances angiogenesis through a pathway involving platelet-activating factor-mediated nuclear factor-kappaB activation. Cancer Res. 2004, 64, 6482–6488. [Google Scholar] [CrossRef] [Green Version]
- Bruno, V.; Corrado, G.; Baci, D.; Chiofalo, B.; Carosi, M.A.; Ronchetti, L.; Piccione, E.; Albini, A.; Noonan, D.M.; Piaggio, G.; et al. Endometrial Cancer Immune Escape Mechanisms: Let Us Learn From the Fetal-Maternal Interface. Front. Oncol. 2020, 10, 156. [Google Scholar] [CrossRef]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Wang, Z.; Fu, L.; Xu, T. Macrophage Polarization in the Development and Progression of Ovarian Cancers: An Overview. Front. Oncol. 2019, 9, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; He, Y.; Peng, F.; Yang, J.; Yuan, C. Endometrial Cancer Cells Promote M2-Like Macrophage Polarization by Delivering Exosomal miRNA-21 under Hypoxia Condition. J. Immunol. Res. 2020, 2020, 9731049. [Google Scholar] [CrossRef] [PubMed]
- Degos, C.; Heinemann, M.; Barrou, J.; Boucherit, N.; Lambaudie, E.; Savina, A.; Gorvel, L.; Olive, D. Endometrial Tumor Microenvironment Alters Human NK Cell Recruitment, and Resident NK Cell Phenotype and Function. Front. Immunol. 2019, 10, 877. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.V.; Shen, Z.; Rodriguez-Garcia, M.; Usherwood, E.J.; Tafe, L.J.; Wira, C.R. Endometrial Cancer Suppresses CD8+ T Cell-Mediated Cytotoxicity in Postmenopausal Women. Front. Immunol. 2021, 12, 657326. [Google Scholar] [CrossRef]
- Walker, N.D.; Patel, J.; Munoz, J.L.; Hu, M.; Guiro, K.; Sinha, G.; Rameshwar, P. The bone marrow niche in support of breast cancer dormancy. Cancer Lett. 2016, 380, 263–271. [Google Scholar] [CrossRef]
- Risson, E.; Nobre, A.R.; Maguer-Satta, V.; Aguirre-Ghiso, J.A. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat. Cancer 2020, 1, 672–680. [Google Scholar] [CrossRef]
- Redig, A.J.; McAllister, S.S. Breast cancer as a systemic disease: A view of metastasis. J. Intern. Med. 2013, 274, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef] [Green Version]
- Sandiford, O.A.; Donnelly, R.J.; El-Far, M.H.; Burgmeyer, L.M.; Sinha, G.; Pamarthi, S.H.; Sherman, L.S.; Ferrer, A.I.; DeVore, D.E.; Patel, S.A.; et al. Mesenchymal Stem Cell-Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res. 2021, 81, 1567–1582. [Google Scholar] [CrossRef]
- Kusumbe, A.P. Vascular niches for disseminated tumour cells in bone. J. Bone Oncol. 2016, 5, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Kopp, H.G.; Avecilla, S.T.; Hooper, A.T.; Rafii, S. The bone marrow vascular niche: Home of HSC differentiation and mobilization. Physiology 2005, 20, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A.; et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Carlson, P.; Dasgupta, A.; Grzelak, C.A.; Kim, J.; Barrett, A.; Coleman, I.M.; Shor, R.E.; Goddard, E.T.; Dai, J.; Schweitzer, E.M.; et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 2019, 21, 238–250. [Google Scholar] [CrossRef]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamma, R.; Ribatti, D. Bone Niches, Hematopoietic Stem Cells, and Vessel Formation. Int. J. Mol. Sci. 2017, 18, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [Green Version]
- Sinha, G.; Ferrer, A.I.; Ayer, S.; El-Far, M.H.; Pamarthi, S.H.; Naaldijk, Y.; Barak, P.; Sandiford, O.A.; Bibber, B.M.; Yehia, G.; et al. Specific N-cadherin-dependent pathways drive human breast cancer dormancy in bone marrow. Life Sci. Alliance 2021, 4, 1–22. [Google Scholar] [CrossRef]
- Huijgens, A.N.; Mertens, H.J. Factors predicting recurrent endometrial cancer. Facts Views Vis. Obgyn. 2013, 5, 179–186. [Google Scholar]
- Guzha, B.; Adams, T.; Rogers, L.; Mbatani, N.; Wu, H.t.; Fakie, N.; Opie, J.; Denny, L. Endometrial cancer with bone marrow metastases: A management dilemma. S. Afr. J. Gynaecol. Oncol. 2017, 9, 22–24. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Mamillapalli, R.; Mutlu, L.; Du, H.; Taylor, H.S. Chemoattraction of bone marrow-derived stem cells towards human endometrial stromal cells is mediated by estradiol regulated CXCL12 and CXCR4 expression. Stem. Cell Res. 2015, 15, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, K.W.; Mamillapalli, R.; Sahin, C.; Song, J.; Tal, R.; Taylor, H.S. Bone marrow-derived cells or C-X-C motif chemokine 12 (CXCL12) treatment improve thin endometrium in a mouse model. Biol. Reprod. 2019, 100, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Khatun, M.; Sorjamaa, A.; Kangasniemi, M.; Sutinen, M.; Salo, T.; Liakka, A.; Lehenkari, P.; Tapanainen, J.S.; Vuolteenaho, O.; Chen, J.C.; et al. Niche matters: The comparison between bone marrow stem cells and endometrial stem cells and stromal fibroblasts reveal distinct migration and cytokine profiles in response to inflammatory stimulus. PLoS ONE 2017, 12, e0175986. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, T.L.; Rojas, A.; Zelenko, Z.; Aghajanova, L.; Erikson, D.W.; Barragan, F.; Meyer, M.; Tamaresis, J.S.; Hamilton, A.E.; Irwin, J.C.; et al. Perivascular human endometrial mesenchymal stem cells express pathways relevant to self-renewal, lineage specification, and functional phenotype. Biol. Reprod. 2012, 86, 58. [Google Scholar] [CrossRef]
- Taghdiri Nooshabadi, V.; Verdi, J.; Ebrahimi-Barough, S.; Mowla, J.; Atlasi, M.A.; Mazoochi, T.; Valipour, E.; Shafiei, S.; Ai, J.; Banafshe, H.R. Endometrial Mesenchymal Stem Cell-Derived Exosome Promote Endothelial Cell Angiogenesis in a Dose Dependent Manner: A New Perspective on Regenerative Medicine and Cell-Free Therapy. Arch. NeuroSci. 2019, 6, e94041. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liang, S.; Yang, F.; Sun, Y.; Niu, L.; Ren, Y.; Wang, H.; He, Y.; Du, J.; Yang, J.; et al. Biological characteristics of endometriotic mesenchymal stem cells isolated from ectopic lesions of patients with endometriosis. Stem. Cell Res. 2020, 11, 346. [Google Scholar] [CrossRef]
- Yao, Y.; Chen, R.; Wang, G.; Zhang, Y.; Liu, F. Exosomes derived from mesenchymal stem cells reverse EMT via TGF-beta1/Smad pathway and promote repair of damaged endometrium. Stem. Cell Res. 2019, 10, 225. [Google Scholar] [CrossRef]
- Xiao, B.; Zhu, Y.; Huang, J.; Wang, T.; Wang, F.; Sun, S. Exosomal transfer of bone marrow mesenchymal stem cell-derived miR-340 attenuates endometrial fibrosis. Biol. Open 2019, 8, bio039958. [Google Scholar] [CrossRef] [Green Version]
- Morelli, S.S.; Resnikoff, A.N.; Haberl, J.; Wojtczuk, A.S.; Rameshwar, P.; Goldsmith, L.T. Bone Marrow-Derived Cells in Murine Endometrium Express Estrogen Receptor Alpha. Endocr. Rev. 2015, 36. [Google Scholar]
- Morelli, S.S.; Resnikoff, A.N.; Haberl, J.; Wojtczuk, A.S.; Rameshwar, P.; Goldsmith, L.T. Bone Marrow-Derived Cells in Murine Endometrium Express Estrogen Receptor Beta. Reprod. Sci. 2015, 22, 83A. [Google Scholar]
- Morelli, S.S.; Resnikoff, A.N.; Wojtczuk, A.S.; Rameshwar, P.; Goldsmith, L.T. Bone Marrow-Derived Murine Endometrial Cells Express Progesterone Receptor. Reprod. Sci. 2016, 23, 336A. [Google Scholar]
- Schuring, A.N.; Braun, J.; Wullner, S.; Kiesel, L.; Gotte, M. mRNA-expression of ERalpha, ERbeta, and PR in clonal stem cell cultures obtained from human endometrial biopsies. Sci. World J. 2011, 11, 1762–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhailik, A.; Mazella, J.; Liang, S.; Tseng, L. Notch ligand-dependent gene expression in human endometrial stromal cells. Biochem. Biophys Res. Commun. 2009, 388, 479–482. [Google Scholar] [CrossRef]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting Notch signalling pathway of cancer stem cells. Stem. Cell Investig. 2018, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-SaInt Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Capulli, M.; Hristova, D.; Valbret, Z.; Carys, K.; Arjan, R.; Maurizi, A.; Masedu, F.; Cappariello, A.; Rucci, N.; Teti, A. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br. J. Cancer 2019, 121, 157–171. [Google Scholar] [CrossRef] [Green Version]


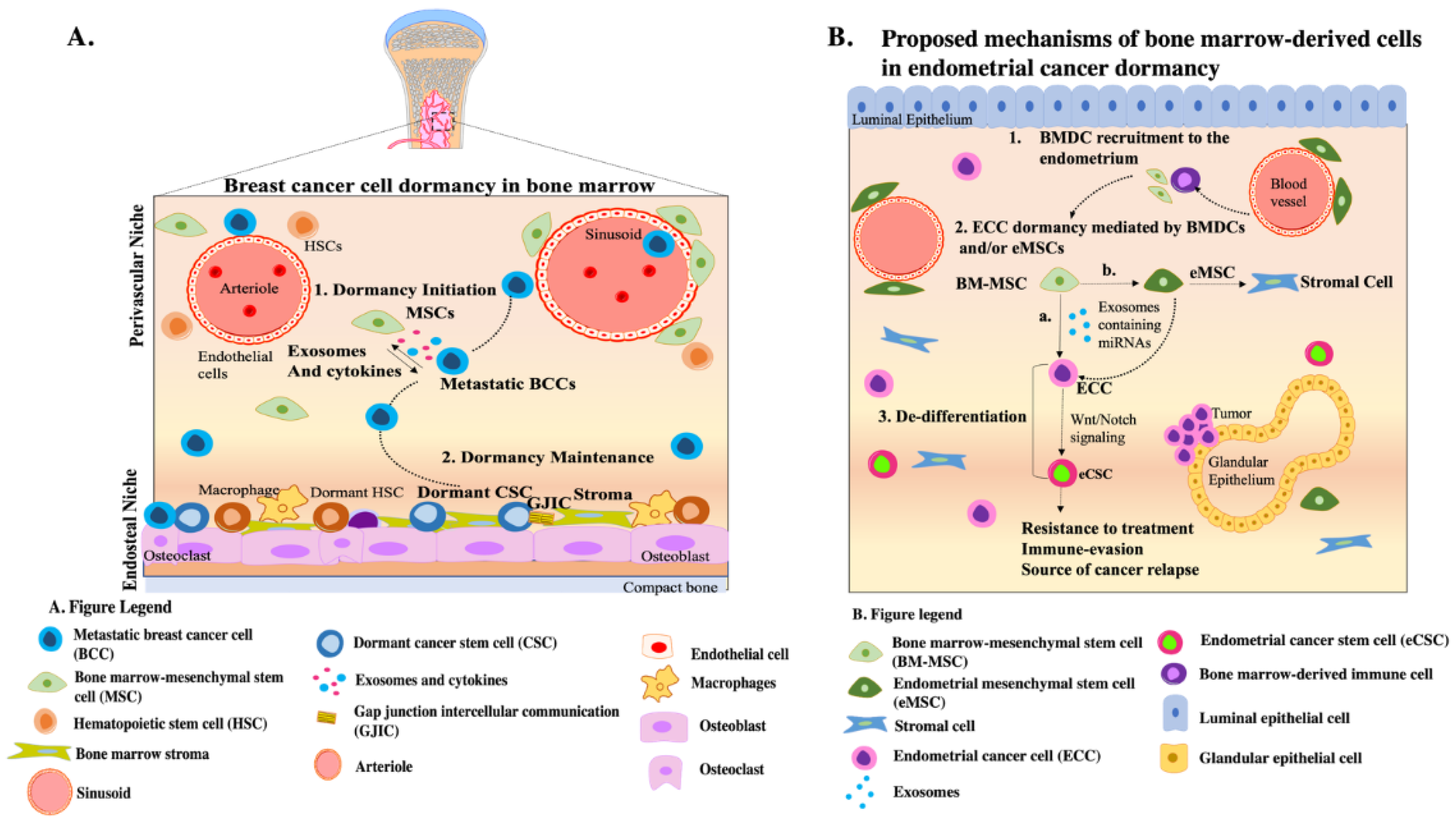{kind=link}
| Name | Type | Function(s) within EC | References |
|---|---|---|---|
| CD133 | pentaspan transmembrane glycoprotein | Modulation of stem cell genes, invasiveness, chemoresistance, tumorigenesis | [70,71] |
| CD44 | transmembrane glycoprotein | Crosstalk with microenvironment, progression, poor prognosis, co-expression with CD133 | [72,73,74] |
| CD117 | type III receptor tyrosine kinase | Proliferation, aggression, independent prognostic factor | [75] |
| ALDH | enzyme | Drug resistance, independent prognostic factor | [76,77,78] |
| Notch | signaling pathway | Cell proliferation, apoptosis | [60] |
| Musashi-1 | RNA-binding protein | Involved in Notch pathway; cell proliferation and apoptosis | [60] |
| Wnt/β-catenin | signaling pathway | Proliferation, migration, invasiveness, tumorigenicity | [79,80] |
| NANOG | homeobox transcription factor | Self-renewal | [50,81] |
| OCT-4 | transcription factor | Self-renewal | [50,82] |
| SOX-2 | transcription factor | Self-renewal | [50,82] |
| SMOC-2 | protein | Reduce expression of stemness-related transcription factors, activate Wnt pathway | [83] |
| miRNA-21 | miRNA | Cell proliferation | [84] |
| miRNA-194 | miRNA | Inhibits EMT | [66] |
| miRNA-34a | miRNA | Inhibits Notch pathway | [67] |
| miRNA-134 | miRNA | Inhibits Notch pathway | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrer, A.I.; Einstein, E.; Morelli, S.S. Bone Marrow-Derived Cells in Endometrial Cancer Pathogenesis: Insights from Breast Cancer. Cells 2022, 11, 714. https://doi.org/10.3390/cells11040714
Ferrer AI, Einstein E, Morelli SS. Bone Marrow-Derived Cells in Endometrial Cancer Pathogenesis: Insights from Breast Cancer. Cells. 2022; 11(4):714. https://doi.org/10.3390/cells11040714
Chicago/Turabian StyleFerrer, Alejandra I., Ella Einstein, and Sara S. Morelli. 2022. "Bone Marrow-Derived Cells in Endometrial Cancer Pathogenesis: Insights from Breast Cancer" Cells 11, no. 4: 714. https://doi.org/10.3390/cells11040714