
A Journey through Time on the Discovery of Cell Cycle Regulation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Principles
2. The First Cell Cycle Control Discovery: The Cell Cycle Engine
3. Functional Issues at the Molecular Level
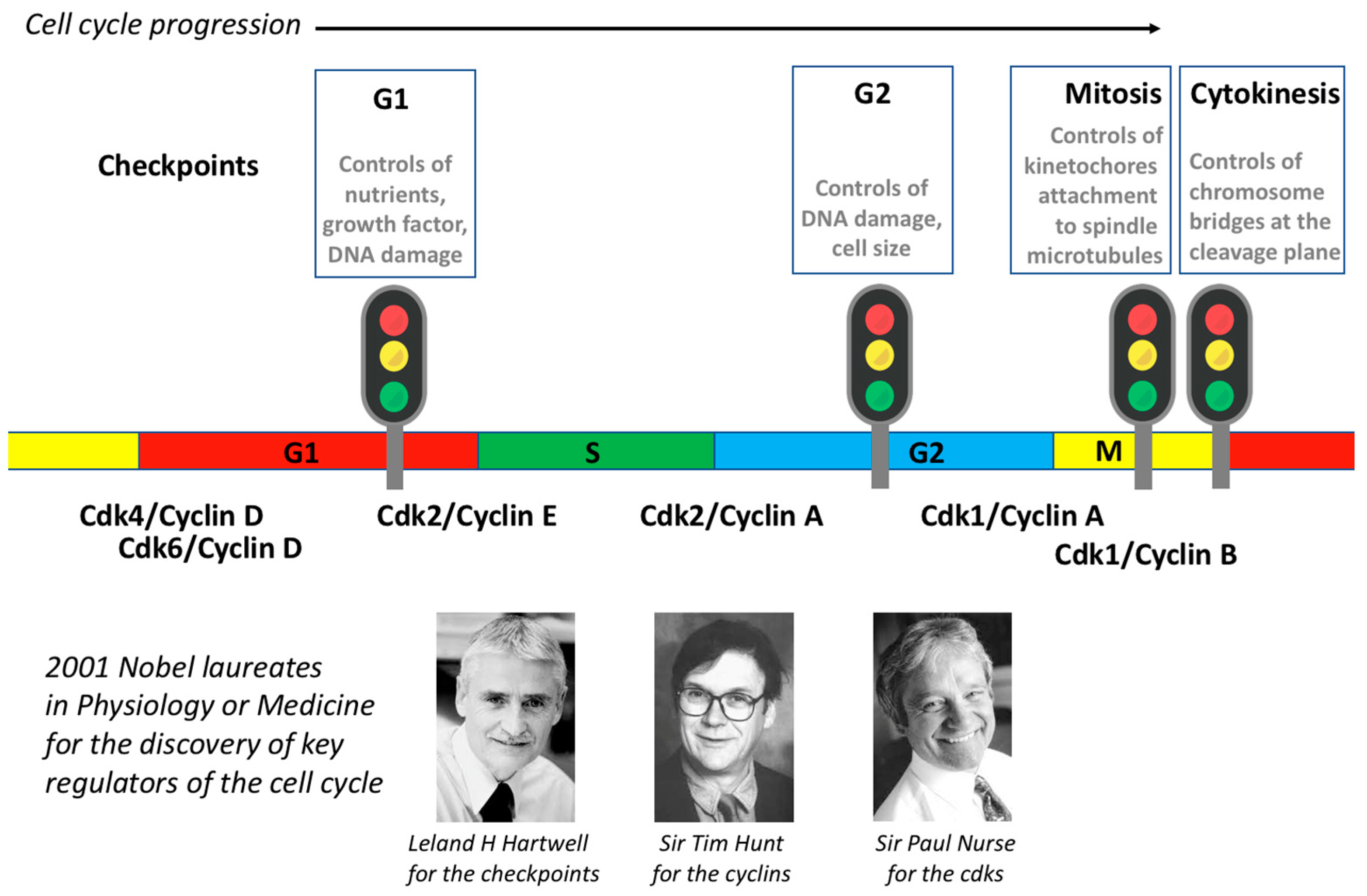
4. Checkpoints
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Hooke, R. Micrographia, or Some Physiological Descriptions of Minute Bodies Made by Magnifying Glasses, with Observations and Inquiries Thereupon; Library of Congress: London, UK, 1665. [Google Scholar]
- Virchow, R. Die Cellularpathologie in Ihrer Begründung auf Physiologische und Pathologische Gewebelehre; Verlag von August Hirschwald: Berlin, Germany, 1859. [Google Scholar]
- Flemming, W. Zellsubstanz, Kern und Zelltheilung; Von Verlag, F.C.W., Ed.; Vogel: Leipzig, Germany, 1882. [Google Scholar]
- Von Waldeyer-Hartz, W. Über Karyokinese und ihre Beziehungen zu den Befruchtungsvorgängen. Arch. Mikrosk. Anat. Entwickl. 1888, 32, 1–122. [Google Scholar]
- Pelc, S.R. Autoradiograph technique. Nature 1947, 160, 749. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.; Pelc, S.R. Synthesis of nucleoprotein in bean root cells. Nature 1951, 167, 599–600. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.; Pelc, S.R. Synthesis of Deoxyribonucleic Acid in Normal and Irradiated Cells and Its Relation to Chromosome Breakage. Heredity 1953, 6, 261–273. [Google Scholar]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H. Genetical implications of the structure of deoxyribonucleic acid. Nature 1953, 171, 964–967. [Google Scholar] [CrossRef]
- Lajtha, L.G.; Olivier, R.; Ellis, F. Incorporation of 32P and adenine 14C into DNA by human bone marrow cells In Vitro. Br. J. Cancer 1954, 8, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Lajtha, L.G. On the concept of the cell cycle. J. Cell. Comp. Physiol. 1963, 62 (Suppl. S1), 143–145. [Google Scholar]
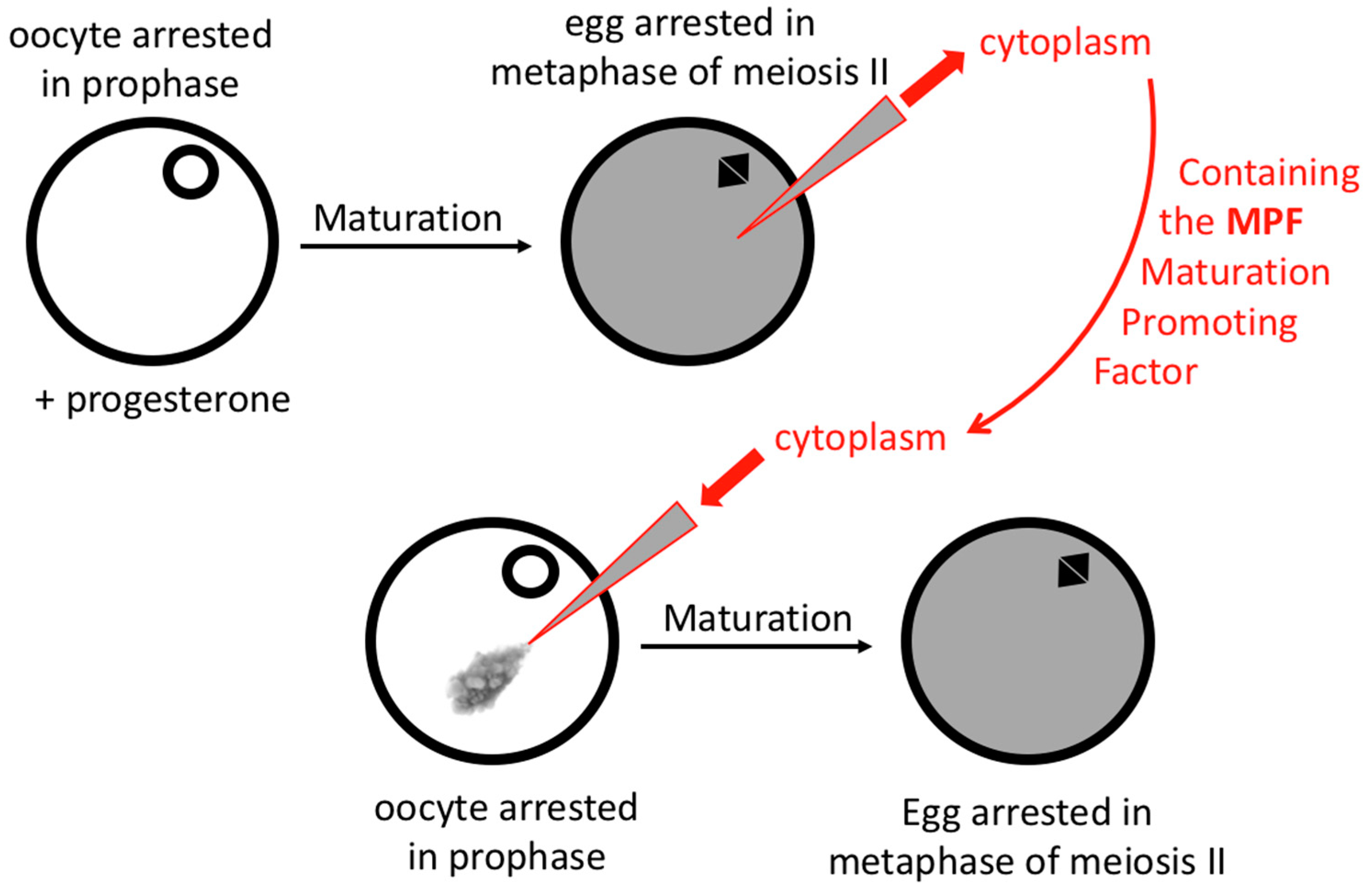
- Masui, Y. Relative roles of the pituitary, follicle cells, and progesterone in the induction of oocyte maturation in Rana pipiens. J. Exp. Zool. 1967, 166, 365–375. [Google Scholar] [CrossRef]
- Masui, Y.; Markert, C.L. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J. Exp. Zool. 1971, 177, 129–145. [Google Scholar] [CrossRef]
- Rao, P.N.; Johnson, R.T. Mammalian cell fusion: Studies on the regulation of DNA synthesis and mitosis. Nature 1970, 225, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.T.; Rao, P.N. Mammalian cell fusion: Induction of premature chromosome condensation in interphase nuclei. Nature 1970, 226, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Lohka, M.J.; Masui, Y. Formation In Vitro of sperm pronuclei and mitotic chromosomes induced by amphibian ooplasmic components. Science 1983, 220, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Lohka, M.J.; Maller, J.L. Induction of nuclear envelope breakdown, chromosome condensation, and spindle formation in cell-free extracts. J. Cell Biol. 1985, 101, 518–523. [Google Scholar] [CrossRef] [Green Version]
- Hartwell, L.H. Macromolecule synthesis in temperature-sensitive mutants of yeast. J. Bacteriol. 1967, 93, 1662–1670. [Google Scholar] [CrossRef] [Green Version]
- Hartwell, L.H. Three additional genes required for deoxyribonucleic acid synthesis in Saccharomyces cerevisiae. J. Bacteriol. 1973, 115, 966–974. [Google Scholar] [CrossRef] [Green Version]
- Hartwell, L.H.; Culotti, J.; Pringle, J.R.; Reid, B.J. Genetic control of the cell division cycle in yeast. Science 1974, 183, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Nurse, P.; Thuriaux, P.; Nasmyth, K. Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 1976, 146, 167–178. [Google Scholar] [CrossRef]
- Nasmyth, K.; Nurse, P. Cell division cycle mutants altered in DNA replication and mitosis in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 1981, 182, 119–124. [Google Scholar] [CrossRef]
- Thuriaux, P.; Nurse, P.; Carter, B. Mutants altered in the control co-ordinating cell division with cell growth in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 1978, 161, 215–220. [Google Scholar] [CrossRef]
- Nurse, P.; Thuriaux, P. Regulatory genes controlling mitosis in the fission yeast Schizosaccharomyces pombe. Genetics 1980, 96, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Beach, D.; Durkacz, B.; Nurse, P. Functionally homologous cell cycle control genes in budding and fission yeast. Nature 1982, 300, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Hindley, J.; Phear, G.A. Sequence of the cell division gene CDC2 from Schizosaccharomyces pombe; patterns of splicing and homology to protein kinases. Gene 1984, 31, 129–134. [Google Scholar] [CrossRef]
- Simanis, V.; Nurse, P. The cell cycle control gene cdc2+ of fission yeast encodes a protein kinase potentially regulated by phosphorylation. Cell 1986, 45, 261–268. [Google Scholar] [CrossRef]
- Lee, M.G.; Nurse, P. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 1987, 327, 31–35. [Google Scholar] [CrossRef]
- Hirt, H.; Páy, A.; Györgyey, J.; Bakó, L.; Németh, K.; Bögre, L.; Schweyen, R.J.; Heberle-Bors, E.; Dudits, D. Complementation of a yeast cell cycle mutant by an alfalfa cDNA encoding a protein kinase homologous to p34cdc2. Proc. Natl. Acad. Sci. USA 1991, 88, 1636–1640. [Google Scholar] [CrossRef] [Green Version]
- Lohka, M.J.; Hayes, M.K.; Maller, J.L. Purification of maturation promoting factor, an intracellular regulator of early mitotic events. Proc. Natl. Acad. Sci. USA 1988, 85, 3009–3013. [Google Scholar] [CrossRef] [Green Version]
- Dunphy, W.G.; Brizuela, L.; Beach, D.; Newport, J. The Xenopus cdc2 protein is a component of MPF, a cytoplasmic regulator of mitosis. Cell 1988, 54, 423–431. [Google Scholar] [CrossRef]
- Gautier, J.; Norbury, C.; Lohka, M.; Nurse, P.; Maller, J. Purified maturation-promoting factor contains the product of a Xenopus homolog of the fission yeast cell cycle control gene cdc2+. Cell 1988, 54, 433–439. [Google Scholar] [CrossRef]
- Evans, T.; Rosenthal, E.T.; Youngblom, J.; Distel, D.; Hunt, T. Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 1983, 33, 389–396. [Google Scholar] [CrossRef]
- Picard, A.; Peaucellier, G.; le Bouffant, F.; Le Peuch, C.; Dorée, M. Role of protein synthesis and proteases in production and inactivation of maturation-promoting activity during meiotic maturation of starfish oocytes. Dev. Biol. 1985, 109, 311–320. [Google Scholar] [CrossRef]
- Draetta, G.; Luca, F.; Westendorf, J.; Brizuela, L.; Ruderman, J.; Beach, D. Cdc2 protein kinase is complexed with both cyclin A and B: Evidence for proteolytic inactivation of MPF. Cell 1989, 56, 829–838. [Google Scholar] [CrossRef]
- Labbe, J.C.; Capony, J.P.; Caput, D.; Cavadore, J.C.; Derancourt, J.; Kaghad, M.; Lelias, J.M.; Picard, A.; Dorée, M. MPF from starfish oocytes at first meiotic metaphase is a heterodimer containing one molecule of cdc2 and one molecule of cyclin B. EMBO J. 1989, 8, 3053–3058. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Arion, D.; Golsteyn, R.; Pines, J.; Brizuela, L.; Hunt, T.; Beach, D. Cyclin is a component of the sea urchin egg M-phase specific histone H1 kinase. EMBO J. 1989, 8, 2275–2282. [Google Scholar] [CrossRef] [PubMed]
- Gautier, J.; Minshull, J.; Lohka, M.; Glotzer, M.; Hunt, T.; Maller, J.L. Cyclin is a component of maturation-promoting factor from Xenopus. Cell 1990, 60, 487–494. [Google Scholar] [CrossRef]
- Hunt, T. Cyclins and their partners: From a simple idea to complicated reality. Semin. Cell Biol. 1991, 2, 213–222. [Google Scholar]
- Paris, J.; Le Guellec, R.; Couturier, A.; Le Guellec, K.; Omilli, F.; Camonis, J.; MacNeill, S.; Philippe, M. Cloning by differential screening of a Xenopus cDNA coding for a protein highly homologous to cdc2. Proc. Natl. Acad. Sci. USA 1991, 88, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Murray, A.W.; Kirschner, M.W. Dominoes and clocks: The union of two views of the cell cycle. Science 1989, 246, 614–621. [Google Scholar] [CrossRef] [Green Version]
- Elledge, S.J.; Spottswood, M.R. A new human p34 protein kinase, CDK2, identified by complementation of a cdc28 mutation in Saccharomyces cerevisiae, is a homolog of Xenopus Eg1. EMBO J. 1991, 10, 2653–2659. [Google Scholar] [CrossRef]
- Ninomiya-Tsuji, J.; Nomoto, S.; Yasuda, H.; Reed, S.I.; Matsumoto, K. Cloning of a human cDNA encoding a CDC2-related kinase by complementation of a budding yeast cdc28 mutation. Proc. Natl. Acad. Sci. USA 1991, 88, 9006–9010. [Google Scholar] [CrossRef] [Green Version]
- Th’ng, J.P.; Wright, P.S.; Hamaguchi, J.; Lee, M.G.; Norbury, C.J.; Nurse, P.; Bradbury, E.M. The FT210 cell line is a mouse G2 phase mutant with a temperature-sensitive CDC2 gene product. Cell 1990, 63, 313–324. [Google Scholar] [CrossRef]
- Tsai, L.H.; Harlow, E.; Meyerson, M. Isolation of the human cdk2 gene that encodes the cyclin A- and adenovirus E1A-associated p33 kinase. Nature 1991, 353, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Koff, A.; Cross, F.; Fisher, A.; Schumacher, J.; Leguellec, K.; Philippe, M.; Roberts, J.M. Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell 1991, 66, 1217–1228. [Google Scholar] [CrossRef]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quandt., E.; Ribeiro, M.P.C.; Clotet, J. Atypical cyclins: The extended family portrait. Cell. Mol. Life Sci. 2020, 77, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef]
- Lau, H.W.; Ma, H.T.; Yeung, T.K.; Tam, M.Y.; Zheng, D.; Chu, S.K.; Poon, R.Y.C. Quantitative differences between cyclin-dependent kinases underlie the unique functions of CDK1 in human cells. Cell Rep. 2021, 37, 109808. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef] [Green Version]
- Khodjakov, A.; Rieder, C.L. The nature of cell-cycle checkpoints: Facts and fallacies. J. Biol. 2009, 8, 88. [Google Scholar] [CrossRef] [Green Version]
- Temin, H.M. Stimulation by serum of multiplication of stationary chicken cells. J. Cell. Physiol. 1971, 78, 161–170. [Google Scholar] [CrossRef]
- Pardee, A.B. A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. USA 1974, 71, 1286–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinert, T.; Hartwell, L. Control of G2 delay by the rad9 gene of Saccharomyces cerevisiae. J. Cell Sci. 1989, 12, 145–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiestl, R.H.; Reynolds, P.; Prakash, S.; Prakash, L. Cloning and sequence analysis of the Saccharomyces cerevisiae RAD9 gene and further evidence that its product is required for cell cycle arrest induced by DNA damage. Mol. Cell. Biol. 1989, 9, 1882–1896. [Google Scholar] [PubMed] [Green Version]
- Zirkle, R.E. Ultraviolet-microbeam irradiation of newt-cell cytoplasm: Spindle destruction, false anaphase, and delay of true anaphase. Radiat. Res. 1970, 41, 516–537. [Google Scholar] [CrossRef]
- Sluder, G.; Thompson, E.A.; Miller, F.J.; Hayes, J.; Rieder, C.L. The checkpoint control for anaphase onset does not monitor excess numbers of spindle poles or bipolar spindle symmetry. J. Cell Sci. 1997, 110 Pt 4, 421–429. [Google Scholar] [CrossRef]
- Mikhailov, A.; Cole, R.W.; Rieder, C.L. DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr. Biol. 2002, 12, 1797–1806. [Google Scholar] [CrossRef] [Green Version]
- Rieder, C.L.; Maiato, H. Stuck in division or passing through: What happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell. 2004, 7, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Norden, C.; Mendoza, M.; Dobbelaere, J.; Kotwaliwale, C.V.; Biggins, S.; Barral, Y. The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosome breakage. Cell 2006, 125, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.; Norden, C.; Durrer, K.; Rauter, H.; Uhlmann, F.; Barral, Y. A mechanism for chromosome segregation sensing by the NoCut checkpoint. Nat. Cell Biol. 2009, 11, 477–483. [Google Scholar] [CrossRef]
- Steigemann, P.; Wurzenberger, C.; Schmitz, M.H.; Held, M.; Guizetti, J.; Maar, S.; Gerlich, D.W. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009, 136, 473–484. [Google Scholar] [CrossRef] [Green Version]
- Petsalaki, E.; Zachos, G. The Abscission Checkpoint: A Guardian of Chromosomal Stability. Cells 2021, 10, 3350. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uzbekov, R.; Prigent, C. A Journey through Time on the Discovery of Cell Cycle Regulation. Cells 2022, 11, 704. https://doi.org/10.3390/cells11040704
Uzbekov R, Prigent C. A Journey through Time on the Discovery of Cell Cycle Regulation. Cells. 2022; 11(4):704. https://doi.org/10.3390/cells11040704
Chicago/Turabian StyleUzbekov, Rustem, and Claude Prigent. 2022. "A Journey through Time on the Discovery of Cell Cycle Regulation" Cells 11, no. 4: 704. https://doi.org/10.3390/cells11040704