
Research Progress on Neuroprotection of Insulin-like Growth Factor-1 towards Glutamate-Induced Neurotoxicity


Abstract
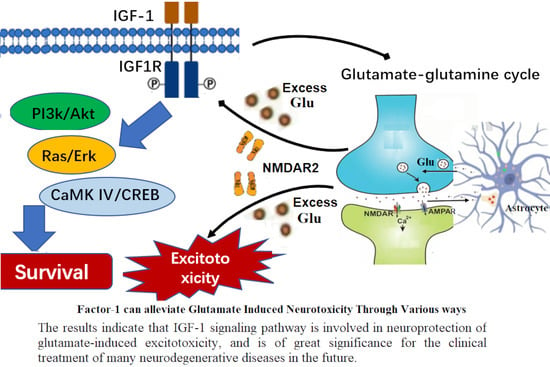
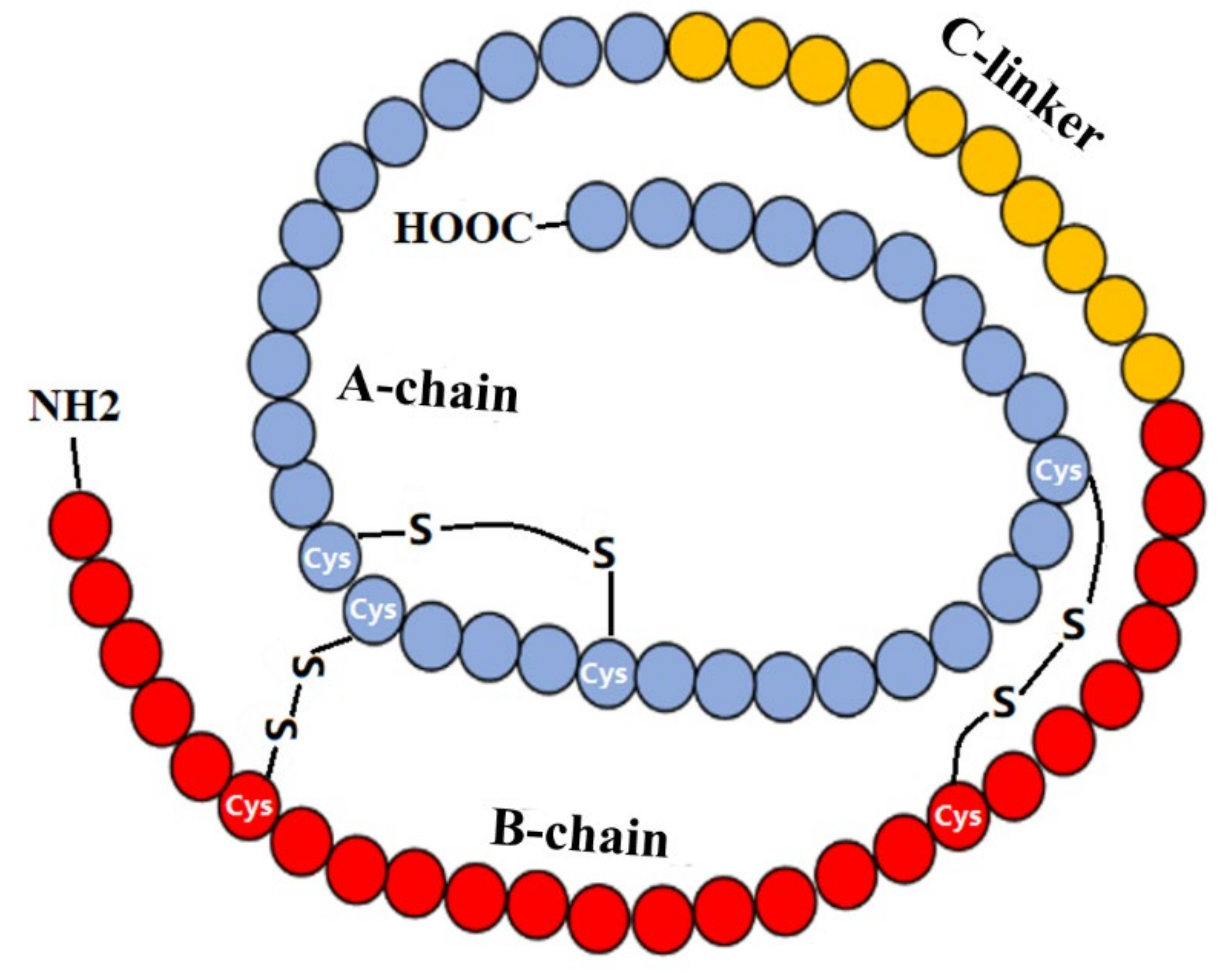
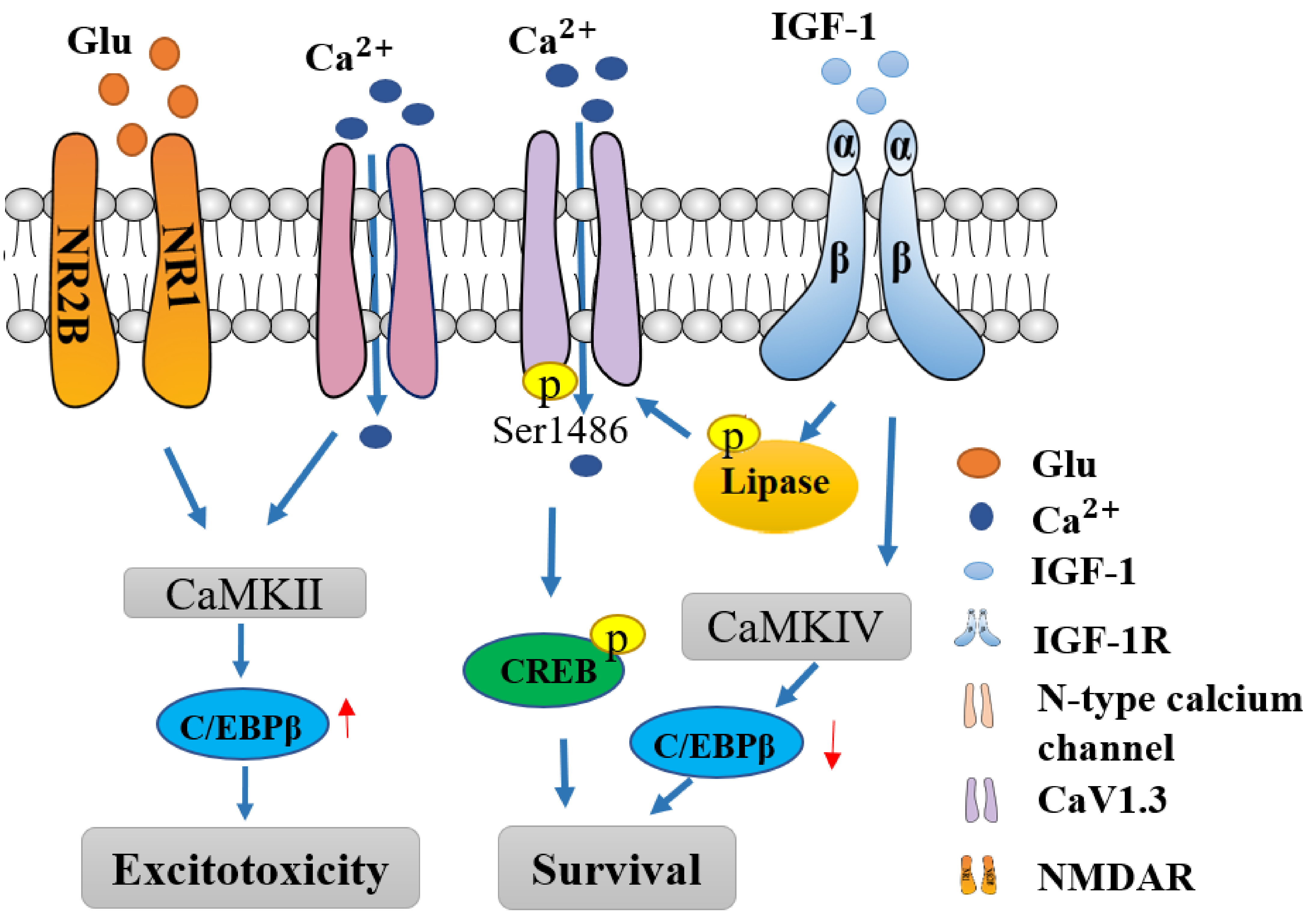
:
1. Introduction
2. IGF-1 and IGF-1R
3. Glutamate Excitotoxicity
3.1. NMDAR Mediated Excitotoxicity
3.2. AMPAR Mediated Excitotoxicity
3.3. Calcium Channels Mediated Excitotoxicity
4. The Reciprocal Cross-Talk between IGF-1R and Glutamate Receptors
5. IGF-1 Modulation of Glutamate-Induced Synaptic Plasticity
6. IGF-1 Modulates Calcium Pathway
7. IGF-I Confers Neuroprotection towards Neurological Diseases with Glutamate Excitotoxicity
8. Summary
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lewitt, M.S.; Boyd, G.W. The Role of Insulin-Like Growth Factors and Insulin-Like Growth Factor-Binding Proteins in the Nervous System. Biochem. Insights 2019, 12, 1178626419842176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagami, T.; Yamamoto, Y.; Koma, H. Pathophysiological Roles of Intracellular Proteases in Neuronal Development and Neurological Diseases. Mol. Neurobiol. 2019, 56, 3090–3112. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur J. Pharmacol. 2013, 698, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Olloquequi, J.; Cornejo-Cordova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.A.; Valcarcel-Ares, M.N.; Ashpole, N.M. Preclinical and clinical evidence of IGF-1 as a prognostic marker and acute intervention with ischemic stroke. J. Cereb. Blood. Flow. Metab. 2021, 41, 2475–2491. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Tong, W. IGF-1: An endogenous link between traumatic brain injury and Alzheimer disease? J. Neurosurg. Sci. 2017, 61, 416–421. [Google Scholar] [CrossRef]
- Carro, E.; Trejo, J.L.; Nunez, A.; Torres-Aleman, I. Brain repair and neuroprotection by serum insulin-like growth factor I. Mol. Neurobiol. 2003, 27, 153–162. [Google Scholar] [CrossRef]
- Salmon, W.D., Jr.; Daughaday, W.H. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J. Lab. Clin. Med. 1957, 49, 825–836. [Google Scholar]
- Froesch, E.R.; Buergi, H.; Ramseier, E.B.; Bally, P.; Labhart, A. Antibody-Suppressible and Nonsuppressible Insulin-Like Activities in Human Serum and Their Physiologic Significance. An Insulin Assay with Adipose Tissue of Increased Precision and Specificity. J. Clin. Investig. 1963, 42, 1816–1834. [Google Scholar] [CrossRef] [Green Version]
- Daughaday, W.H.; Hall, K.; Raben, M.S.; Salmon, W.D., Jr.; van den Brande, J.L.; van Wyk, J.J. Somatomedin: Proposed designation for sulphation factor. Nature 1972, 235, 107. [Google Scholar] [CrossRef]
- Rinderknecht, E.; Humbel, R.E. Polypeptides with nonsuppressible insulin-like and cell-growth promoting activities in human serum: Isolation, chemical characterization, and some biological properties of forms I and II. Proc. Natl. Acad. Sci. USA 1976, 73, 2365–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laron, Z. Somatomedin-1 (recombinant insulin-like growth factor-1): Clinical pharmacology and potential treatment of endocrine and metabolic disorders. BioDrugs 1999, 11, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Ranke, M.B. Insulin-like growth factor binding-protein-3 (IGFBP-3). Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, F.; Takaku, F.; Nagao, M.; Sugimura, T. Cysteine-rich regions conserved in amino-terminal halves of raf gene family products and protein kinase C. Jpn. J. Cancer Res. 1986, 77, 1183–1187. [Google Scholar]
- Vajdos, F.F.; Ultsch, M.; Schaffer, M.L.; Deshayes, K.D.; Liu, J.; Skelton, N.J.; de Vos, A.M. Crystal structure of human insulin-like growth factor-1: Detergent binding inhibits binding protein interactions. Biochemistry 2001, 40, 11022–11029. [Google Scholar] [CrossRef]
- Keating, G.M. Mecasermin. BioDrugs 2008, 22, 177–188. [Google Scholar] [CrossRef]
- Mofid, M.R.; Babaeipour, V.; Jafari, S.; Haddad, L.; Moghim, S.; Ghanavi, J. Efficient process development for high-level production, purification, formulation, and characterization of recombinant mecasermin in Escherichia coli. Biotechnol. Appl. Biochem. 2021, 68, 776–788. [Google Scholar] [CrossRef]
- Slaaby, R. Specific insulin/IGF1 hybrid receptor activation assay reveals IGF1 as a more potent ligand than insulin. Sci. Rep. 2015, 5, 7911. [Google Scholar] [CrossRef] [Green Version]
- Clemmons, D.R. Role of IGF Binding Proteins in Regulating Metabolism. Trends Endocrinol. Metab. 2016, 27, 375–391. [Google Scholar] [CrossRef]
- Pan, W.; Kastin, A.J. Interactions of IGF-1 with the blood-brain barrier in vivo and in situ. Neuroendocrinol. 2000, 72, 171–178. [Google Scholar] [CrossRef]
- Romero, C.J.; Pine-Twaddell, E.; Sima, D.I.; Miller, R.S.; He, L.; Wondisford, F.; Radovick, S. Insulin-like growth factor 1 mediates negative feedback to somatotroph GH expression via POU1F1/CREB binding protein interactions. Mol. Cell. Biol. 2012, 32, 4258–4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, A.H.; Vahdatpour, C.; Sanfeliu, A.; Tropea, D. The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neurosci. 2016, 325, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Ying, J.; Jin, L.; Li, C.; Gui, S.; Li, Z.; Wang, R.; Zuo, Z.; Zhang, Y. Correction for: The role of serum growth hormone and insulin-like growth factor-1 in adult humans brain morphology. Aging 2021, 13, 22623–22624. [Google Scholar] [CrossRef]
- Nishijima, T.; Piriz, J.; Duflot, S.; Fernandez, A.M.; Gaitan, G.; Gomez-Pinedo, U.; Verdugo, J.M.; Leroy, F.; Soya, H.; Nunez, A.; et al. Neuronal activity drives localized blood-brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron 2010, 67, 834–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, V.E.; Locatelli, V.; Rizzi, L. Neurotrophic and Neuroregenerative Effects of GH/IGF1. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, T.E.; Epa, V.C.; Garrett, T.P.; Ward, C.W. Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol. Life Sci. 2000, 57, 1050–1093. [Google Scholar] [CrossRef] [PubMed]
- Hakuno, F.; Takahashi, S.I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Choi, E.; Yu, H.; Bai, X.C. Structural basis of the activation of type 1 insulin-like growth factor receptor. Nat. Commun. 2019, 10, 4567. [Google Scholar] [CrossRef]
- Pal, M.M. Glutamate: The Master Neurotransmitter and Its Implications in Chronic Stress and Mood Disorders. Front. Hum. Neurosci. 2021, 15, 722323. [Google Scholar] [CrossRef]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, T.W.; Paul, B.D.; Parker, G.M.; Hester, L.D.; Snowman, A.M.; Taniguchi, Y.; Kamiya, A.; Snyder, S.H.; Sawa, A. The glutathione cycle shapes synaptic glutamate activity. Proc. Natl. Acad. Sci. USA 2019, 116, 2701–2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binvignat, O.; Olloquequi, J. Excitotoxicity as a Target Against Neurodegenerative Processes. Curr. Pharm. Des. 2020, 26, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Wollmuth, L.P. Ion permeation in ionotropic glutamate receptors: Still dynamic after all these years. Curr. Opin. Physiol. 2018, 2, 36–41. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Cavara, N.A.; Hollmann, M. Shuffling the deck anew: How NR3 tweaks NMDA receptor function. Mol. Neurobiol. 2008, 38, 16–26. [Google Scholar] [CrossRef]
- Zhou, X.; Hollern, D.; Liao, J.; Andrechek, E.; Wang, H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell. Death Dis. 2013, 4, e560. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Xu, J.; Kurup, P.; Zhang, Y.; Goebel-Goody, S.M.; Wu, P.H.; Hawasli, A.H.; Baum, M.L.; Bibb, J.A.; Lombroso, P.J. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J. Neurosci. 2009, 29, 9330–9343. [Google Scholar] [CrossRef] [PubMed]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Goncalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef] [PubMed]
- Rueda, C.B.; Llorente-Folch, I.; Traba, J.; Amigo, I.; Gonzalez-Sanchez, P.; Contreras, L.; Juaristi, I.; Martinez-Valero, P.; Pardo, B.; Del Arco, A.; et al. Glutamate excitotoxicity and Ca2+-regulation of respiration: Role of the Ca2+ activated mitochondrial transporters (CaMCs). Biochim. Biophys. Acta 2016, 1857, 1158–1166. [Google Scholar] [CrossRef]
- Mira, R.G.; Cerpa, W. Building a Bridge between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell. Mol. Neurobiol. 2021, 41, 1413–1430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Denisova, J.V.; Kang, K.S.; Fontes, J.D.; Zhu, B.T.; Belousov, A.B. Neuronal gap junctions are required for NMDA receptor-mediated excitotoxicity: Implications in ischemic stroke. J. Neurophysiol. 2010, 104, 3551–3556. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, C.; Yang, Q.; Jiao, M.; Qiu, S. Endocytosis of GluN2B-containing NMDA receptors mediates NMDA-induced excitotoxicity. Mol. Pain. 2017, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohr, G. NMDA receptor function: Subunit composition versus spatial distribution. Cell Tissue Res. 2006, 326, 439–446. [Google Scholar] [CrossRef]
- Wu, Q.J.; Tymianski, M. Targeting NMDA receptors in stroke: New hope in neuroprotection. Mol. Brain 2018, 11, 15. [Google Scholar] [CrossRef]
- Park, P.; Kang, H.; Sanderson, T.M.; Bortolotto, Z.A.; Georgiou, J.; Zhuo, M.; Kaang, B.K.; Collingridge, G.L. The Role of Calcium-Permeable AMPARs in Long-Term Potentiation at Principal Neurons in the Rodent Hippocampus. Front. Synaptic Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cull-Candy, S.G.; Farrant, M. Ca(2+) -permeable AMPA receptors and their auxiliary subunits in synaptic plasticity and disease. J. Physiol. 2021, 599, 2655–2671. [Google Scholar] [CrossRef]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Araujo, I.M.; Verdasca, M.J.; Leal, E.C.; Bahr, B.A.; Ambrosio, A.F.; Carvalho, A.P.; Carvalho, C.M. Early calpain-mediated proteolysis following AMPA receptor activation compromises neuronal survival in cultured hippocampal neurons. J. Neurochem. 2004, 91, 1322–1331. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Li, S.; Zhang, H.; Pei, L.; Zou, S.; Lee, F.J.; Wang, Y.T.; Liu, F. Direct interaction between GluR2 and GAPDH regulates AMPAR-mediated excitotoxicity. Mol. Brain 2012, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrier, J.; Luscher, C.; Pascoli, V. Cell-Type Specific Insertion of GluA2-Lacking AMPARs with Cocaine Exposure Leading to Sensitization, Cue-Induced Seeking, and Incubation of Craving. Neuropsychopharmacology 2016, 41, 1779–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, R.; Dravid, S.M.; Yuan, H.; Traynelis, S.F. AMPA Receptors: Molecular Biology and Pharmacology. Neurosci. Biobehav. Psychol. 2017, 10. [Google Scholar] [CrossRef]
- Walia, A.; Lee, C.; Hartsock, J.; Goodman, S.S.; Dolle, R.; Salt, A.N.; Lichtenhan, J.T.; Rutherford, M.A. Reducing Auditory Nerve Excitability by Acute Antagonism of Ca(2+)-Permeable AMPA Receptors. Front. Synaptic Neurosci. 2021, 13, 680621. [Google Scholar] [CrossRef] [PubMed]
- Gratacos-Batlle, E.; Yefimenko, N.; Cascos-Garcia, H.; Soto, D. AMPAR interacting protein CPT1C enhances surface expression of GluA1-containing receptors. Front. Cell. Neurosci. 2014, 8, 469. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Alonso, J.; Morishita, W.; Incontro, S.; Simms, J.; Holtzman, J.; Gill, M.; Mucke, L.; Malenka, R.C.; Nicoll, R.A. Long-term potentiation is independent of the C-tail of the GluA1 AMPA receptor subunit. Elife 2020, 9. [Google Scholar] [CrossRef]
- Guo, C.; Ma, Y.Y. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circuits 2021, 15, 711564. [Google Scholar] [CrossRef]
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol. Ther. 1999, 81, 163–221. [Google Scholar] [CrossRef]
- Sendrowski, K.; Rusak, M.; Sobaniec, P.; Ilendo, E.; Dabrowska, M.; Bockowski, L.; Koput, A.; Sobaniec, W. Study of the protective effect of calcium channel blockers against neuronal damage induced by glutamate in cultured hippocampal neurons. Pharmacol. Rep. 2013, 65, 730–736. [Google Scholar] [CrossRef]
- Stanika, R.I.; Villanueva, I.; Kazanina, G.; Andrews, S.B.; Pivovarova, N.B. Comparative impact of voltage-gated calcium channels and NMDA receptors on mitochondria-mediated neuronal injury. J. Neurosci. 2012, 32, 6642–6650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of voltage-gated calcium channels. Neuron 2000, 25, 533–535. [Google Scholar] [CrossRef] [Green Version]
- Dolphin, A.C. Voltage-gated calcium channel alpha 2delta subunits: An assessment of proposed novel roles. F1000Res. 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tottene, A.; Urbani, A.; Pietrobon, D. Role of different voltage-gated Ca2+ channels in cortical spreading depression: Specific requirement of P/Q-type Ca2+ channels. Channels 2011, 5, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, A.S.; Cardozo, P.L.; Silva, F.R.; de Souza, J.M.; Olmo, I.G.; Cruz, J.S.; Gomez, M.V.; Ribeiro, F.M.; Vieira, L.B. Alterations of Calcium Channels in a Mouse Model of Huntington’s Disease and Neuroprotection by Blockage of CaV1 Channels. ASN Neuro. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.H.; Clemens, J.A.; Bondy, C.A. Insulin-like growth factors in the response to cerebral ischemia. Mol. Cell. Neurosci. 1992, 3, 36–43. [Google Scholar] [CrossRef]
- Beilharz, E.J.; Russo, V.C.; Butler, G.; Baker, N.L.; Connor, B.; Sirimanne, E.S.; Dragunow, M.; Werther, G.A.; Gluckman, P.D.; Williams, C.E.; et al. Co-ordinated and cellular specific induction of the components of the IGF/IGFBP axis in the rat brain following hypoxic-ischemic injury. Brain Res. Mol. Brain Res. 1998, 59, 119–134. [Google Scholar] [CrossRef]
- Herrera, M.L.; Bandín, S.; Champarini, L.G.; Hereñú, C.B.; Bellini, M.J. Intramuscular insulin-like growth factor-1 gene therapy modulates reactive microglia after traumatic brain injury. Brain Res. Bull. 2021, 175, 196–204. [Google Scholar] [CrossRef]
- Walter, H.J.; Berry, M.; Hill, D.J.; Logan, A. Spatial and temporal changes in the insulin-like growth factor (IGF) axis indicate autocrine/paracrine actions of IGF-I within wounds of the rat brain. Endocrinology 1997, 138, 3024–3034. [Google Scholar] [CrossRef]
- Rubovitch, V.; Edut, S.; Sarfstein, R.; Werner, H.; Pick, C.G. The intricate involvement of the Insulin-like growth factor receptor signaling in mild traumatic brain injury in mice. Neurobiol. Dis. 2010, 38, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Saatman, K.E.; Contreras, P.C.; Smith, D.H.; Raghupathi, R.; McDermott, K.L.; Fernandez, S.C.; Sanderson, K.L.; Voddi, M.; McIntosh, T.K. Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp. Neurol. 1997, 147, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Williams, C.; Gunning, M.; Mallard, C.; Gluckman, P. The effects of IGF-1 treatment after hypoxic-ischemic brain injury in adult rats. J. Cereb. Blood Flow Metab. 1993, 13, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassil, F.; Fernagut, P.O.; Bezard, E.; Meissner, W.G. Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: Targets for disease modification? Prog. Neurobiol. 2014, 118, 1–18. [Google Scholar] [CrossRef]
- Farias Quipilidor, G.E.; Mao, K.; Hu, Z.; Novaj, A.; Cui, M.-H.; Gulinello, M.; Branch, C.A.; Gubbi, S.; Patel, K.; Moellering, D.L.; et al. Central IGF-1 protects against features of cognitive and sensorimotor decline with aging in male mice. GeroScience 2019, 41, 185–208. [Google Scholar] [CrossRef]
- Saez, J.M. Possible usefulness of growth hormone/insulin-like growth factor-I axis in Alzheimer’s disease treatment. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 274–286. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Li, D.; Li, M.; Wang, P.; Wen, J.; Liang, M.; Su, B.; Yin, Y. IGF-1 alleviates NMDA-induced excitotoxicity in cultured hippocampal neurons against autophagy via the NR2B/PI3K-AKT-mTOR pathway. J. Cell. Physiol. 2014, 229, 1618–1629. [Google Scholar] [CrossRef]
- Wang, H.; Liao, S.; Geng, R.; Zheng, Y.; Liao, R.; Yan, F.; Thrimawithana, T.; Little, P.J.; Feng, Z.P.; Lazarovici, P.; et al. IGF-1 signaling via the PI3K/Akt pathway confers neuroprotection in human retinal pigment epithelial cells exposed to sodium nitroprusside insult. J. Mol. Neurosci. 2015, 55, 931–940. [Google Scholar] [CrossRef]
- Williams, R.H.; Vazquez-DeRose, J.; Thomas, A.M.; Piquet, J.; Cauli, B.; Kilduff, T.S. Cortical nNOS/NK1 Receptor Neurons are Regulated by Cholinergic Projections From the Basal Forebrain. Cereb. Cortex 2018, 28, 1959–1979. [Google Scholar] [CrossRef]
- Sun, X.J.; Rothenberg, P.; Kahn, C.R.; Backer, J.M.; Araki, E.; Wilden, P.A.; Cahill, D.A.; Goldstein, B.J.; White, M.F. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 1991, 352, 73–77. [Google Scholar] [CrossRef]
- Chen, B.S.; Roche, K.W. Growth factor-dependent trafficking of cerebellar NMDA receptors via protein kinase B/Akt phosphorylation of NR2C. Neuron 2009, 62, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, E.D.; Sanchez-Perez, A.; Trejo, J.L.; Martin-Aldana, J.A.; Cano Jaimez, M.; Pons, S.; Acosta Umanzor, C.; Menes, L.; White, M.F.; Burks, D.J. IRS-2 Deficiency impairs NMDA receptor-dependent long-term potentiation. Cereb. Cortex 2012, 22, 1717–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sun, W.; Han, S.; Li, J.; Ding, S.; Wang, W.; Yin, Y. IGF-1-Involved Negative Feedback of NR2B NMDA Subunits Protects Cultured Hippocampal Neurons Against NMDA-Induced Excitotoxicity. Mol. Neurobiol. 2017, 54, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Han, C.; Zeng, Z.; Liu, L.; Wang, H.; Xu, J.; Feng, Z.P.; Little, P.J.; Quirion, R.; Zheng, W. Glutamate Attenuates the Survival Property of IGFR through NR2B Containing N-Methyl-D-aspartate Receptors in Cortical Neurons. Oxid. Med. Cell. Longev. 2020, 2020, 5173184. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, F.; Prisco, M.; Dews, M.; Salomoni, P.; Grassilli, E.; Romano, G.; Calabretta, B.; Baserga, R. Multiple signaling pathways of the insulin-like growth factor 1 receptor in protection from apoptosis. Mol. Cell. Biol. 1999, 19, 7203–7215. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Meng, Q.; Zhang, L.; Wang, H.; Quirion, R.; Zheng, W. Glutamate attenuates IGF-1 receptor tyrosine phosphorylation in mouse brain: Possible significance in ischemic brain damage. Neurosci. Res. 2012, 74, 290–297. [Google Scholar] [CrossRef]
- Zeng, Z.; Wang, D.; Gaur, U.; Rifang, L.; Wang, H.; Zheng, W. H2O2 attenuates IGF-1R tyrosine phosphorylation and its survival signaling properties in neuronal cells via NR2B containing NMDA receptor. Oncotarget 2017, 8, 65313–65328. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.H.; Quirion, R. Glutamate acting on N-methyl-D-aspartate receptors attenuates insulin-like growth factor-1 receptor tyrosine phosphorylation and its survival signaling properties in rat hippocampal neurons. J. Biol. Chem. 2009, 284, 855–861. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Galloway, E.; Arango, C.; Pons, S.; Torres-Aleman, I. Glutamate excitotoxicity attenuates insulin-like growth factor-I prosurvival signaling. Mol. Cell. Neurosci. 2003, 24, 1027–1037. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.J.; Sun, Q.; Zhang, W.H.; Huo, Y.J.; Xu, C.J.; Liu, J.F. Specific activation of mGlu2 induced IGF-1R transactivation in vitro through FAK phosphorylation. Acta Pharmacol. Sin. 2019, 40, 460–467. [Google Scholar] [CrossRef]
- Prabhu, D.; Khan, S.M.; Blackburn, K.; Marshall, J.P.; Ashpole, N.M. Loss of insulin-like growth factor-1 signaling in astrocytes disrupts glutamate handling. J. Neurochem. 2019, 151, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Nanou, E.; Catterall, W.A. Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron. 2018, 98, 466–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magee, J.C.; Grienberger, C. Synaptic Plasticity Forms and Functions. Annu. Rev. Neurosci. 2020, 43, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Aberg, N.D.; Brywe, K.G.; Isgaard, J. Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. Science 2006, 6, 53–80. [Google Scholar] [CrossRef]
- Chin, P.C.; Majdzadeh, N.; D’Mello, S.R. Inhibition of GSK3beta is a common event in neuroprotection by different survival factors. Brain Res. Mol. Brain Res. 2005, 137, 193–201. [Google Scholar] [CrossRef]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Vaynman, S.; Akhavan, M.; Ying, Z.; Gomez-Pinilla, F. Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience 2006, 140, 823–833. [Google Scholar] [CrossRef]
- Marshall, J.; Dolan, B.M.; Garcia, E.P.; Sathe, S.; Tang, X.; Mao, Z.; Blair, L.A. Calcium channel and NMDA receptor activities differentially regulate nuclear C/EBPbeta levels to control neuronal survival. Neuron 2003, 39, 625–639. [Google Scholar] [CrossRef] [Green Version]
- Ogundele, O.M.; Ebenezer, P.J.; Lee, C.C.; Francis, J. Stress-altered synaptic plasticity and DAMP signaling in the hippocampus-PFC axis; elucidating the significance of IGF-1/IGF-1R/CaMKIIalpha expression in neural changes associated with a prolonged exposure therapy. Neuroscience 2017, 353, 147–165. [Google Scholar] [CrossRef]
- Ramsey, M.M.; Weiner, J.L.; Moore, T.P.; Carter, C.S.; Sonntag, W.E. Growth hormone treatment attenuates age-related changes in hippocampal short-term plasticity and spatial learning. Neuroscience 2004, 129, 119–127. [Google Scholar] [CrossRef]
- Calamandrei, G.; Alleva, E. Neuronal growth factors, neurotrophins and memory deficiency. Behav. Brain Res. 1995, 66, 129–132. [Google Scholar] [CrossRef]
- Ceprian, M.; Fulton, D. Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivakumar, V.; Ling, E.A.; Lu, J.; Kaur, C. Role of glutamate and its receptors and insulin-like growth factors in hypoxia induced periventricular white matter injury. Glia 2010, 58, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Bozdagi, O.; Tavassoli, T.; Buxbaum, J.D. Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol. Autism 2013, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutin, E.; Sakkaki, S.; Compan, V.; Bouquier, N.; Giona, F.; Areias, J.; Goyet, E.; Hemonnot-Girard, A.L.; Seube, V.; Glasson, B.; et al. Restoring glutamate receptosome dynamics at synapses rescues autism-like deficits in Shank3-deficient mice. Mol. Psychiatry 2021. [Google Scholar] [CrossRef]
- Blair, L.A.; Marshall, J. IGF-1 modulates N and L calcium channels in a PI 3-kinase-dependent manner. Neuron 1997, 19, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Blair, L.A.; Salinas, G.D.; Needleman, L.A.; Marshall, J. Insulin-like growth factor-1 modulation of CaV1.3 calcium channels depends on Ca2+ release from IP3-sensitive stores and calcium/calmodulin kinase II phosphorylation of the alpha1 subunit EF hand. J. Neurosci. 2006, 26, 6259–6268. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.C.; Lopez-Zapata, D.F.; Francis, L.; De Los Reyes, L. Effects of estradiol and IGF-1 on the sodium calcium exchanger in rat cultured cortical neurons. Cell. Mol. Neurobiol. 2011, 31, 619–627. [Google Scholar] [CrossRef]
- Russo, V.C.; Gluckman, P.D.; Feldman, E.L.; Werther, G.A. The insulin-like growth factor system and its pleiotropic functions in brain. Endocr. Rev. 2005, 26, 916–943. [Google Scholar] [CrossRef] [Green Version]
- Costales, J.; Kolevzon, A. The therapeutic potential of insulin-like growth factor-1 in central nervous system disorders. Neurosci. Biobehav. Rev. 2016, 63, 207–222. [Google Scholar] [CrossRef] [Green Version]
- Serhan, A.; Boddeke, E.; Kooijman, R. Insulin-Like Growth Factor-1 Is Neuroprotective in Aged Rats With Ischemic Stroke. Front. Aging Neurosci. 2019, 11, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevigny, J.J.; Ryan, J.M.; van Dyck, C.H.; Peng, Y.; Lines, C.R.; Nessly, M.L.; Group, M.K.P.S. Growth hormone secretagogue MK-677: No clinical effect on AD progression in a randomized trial. Neurology 2008, 71, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Duron, E.; Funalot, B.; Brunel, N.; Coste, J.; Quinquis, L.; Viollet, C.; Belmin, J.; Jouanny, P.; Pasquier, F.; Treluyer, J.M.; et al. Insulin-like growth factor-I and insulin-like growth factor binding protein-3 in Alzheimer’s disease. J. Clin. Endocrinol. Metab. 2012, 97, 4673–4681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrowski, P.P.; Barszczyk, A.; Forstenpointner, J.; Zheng, W.; Feng, Z.P. Meta-Analysis of Serum Insulin-Like Growth Factor 1 in Alzheimer’s Disease. PLoS ONE 2016, 11, e0155733. [Google Scholar] [CrossRef] [Green Version]
- Frank, Y. The Neurological Manifestations of Phelan-McDermid Syndrome. Pediatr. Neurol. 2021, 122, 59–64. [Google Scholar] [CrossRef]
- Deacon, R.M.; Glass, L.; Snape, M.; Hurley, M.J.; Altimiras, F.J.; Biekofsky, R.R.; Cogram, P. NNZ-2566, a novel analog of (1-3) IGF-1, as a potential therapeutic agent for fragile X syndrome. Neuromolecular Med. 2015, 17, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Kolevzon, A.; Bush, L.; Wang, A.T.; Halpern, D.; Frank, Y.; Grodberg, D.; Rapaport, R.; Tavassoli, T.; Chaplin, W.; Soorya, L.; et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol. Autism 2014, 5, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry-Kravis, E.; Horrigan, J.P.; Tartaglia, N.; Hagerman, R.; Kolevzon, A.; Erickson, C.A.; Hatti, S.; Snape, M.; Yaroshinsky, A.; Stoms, G.; et al. A Double-Blind, Randomized, Placebo-Controlled Clinical Study of Trofinetide in the Treatment of Fragile X Syndrome. Pediatr. Neurol. 2020, 110, 30–41. [Google Scholar] [CrossRef]
- Castro, J.; Garcia, R.I.; Kwok, S.; Banerjee, A.; Petravicz, J.; Woodson, J.; Mellios, N.; Tropea, D.; Sur, M. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 9941–9946. [Google Scholar] [CrossRef] [Green Version]
- Glaze, D.G.; Neul, J.L.; Percy, A.; Feyma, T.; Beisang, A.; Yaroshinsky, A.; Stoms, G.; Zuchero, D.; Horrigan, J.; Glass, L.; et al. A Double-Blind, Randomized, Placebo-Controlled Clinical Study of Trofinetide in the Treatment of Rett Syndrome. Pediatr. Neurol. 2017, 76, 37–46. [Google Scholar] [CrossRef]
- Franco, C.; Fernandez, S.; Torres-Aleman, I. Frataxin deficiency unveils cell-context dependent actions of insulin-like growth factor I on neurons. Mol. Neurodegener. 2012, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Franco, C.; Genis, L.; Navarro, J.A.; Perez-Domper, P.; Fernandez, A.M.; Schneuwly, S.; Torres Aleman, I. A role for astrocytes in cerebellar deficits in frataxin deficiency: Protection by insulin-like growth factor I. Mol. Cell. Neurosci. 2017, 80, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Gallego, I.; Torres-Aleman, I.; Arpa, J. IGF-1 in Friedreich’s Ataxia–proof-of-concept trial. Cerebellum Ataxias 2014, 1, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, C.; Ribeiro, M.; Duarte, A.I.; Humbert, S.; Saudou, F.; Pereira de Almeida, L.; Hayden, M.; Rego, A.C. IGF-1 intranasal administration rescues Huntington’s disease phenotypes in YAC128 mice. Mol. Neurobiol. 2014, 49, 1126–1142. [Google Scholar] [CrossRef]
- Saleh, N.; Moutereau, S.; Durr, A.; Krystkowiak, P.; Azulay, J.P.; Tranchant, C.; Broussolle, E.; Morin, F.; Bachoud-Levi, A.C.; Maison, P. Neuroendocrine disturbances in Huntington’s disease. PLoS ONE 2009, 4, e4962. [Google Scholar] [CrossRef] [Green Version]
- Miltiadous, P.; Stamatakis, A.; Koutsoudaki, P.N.; Tiniakos, D.G.; Stylianopoulou, F. IGF-I ameliorates hippocampal neurodegeneration and protects against cognitive deficits in an animal model of temporal lobe epilepsy. Exp. Neurol. 2011, 231, 223–235. [Google Scholar] [CrossRef]
- Chen, S.F.; Jou, S.B.; Chen, N.C.; Chuang, H.Y.; Huang, C.R.; Tsai, M.H.; Tan, T.Y.; Tsai, W.C.; Chang, C.C.; Chuang, Y.C. Serum Levels of Brain-Derived Neurotrophic Factor and Insulin-Like Growth Factor 1 Are Associated With Autonomic Dysfunction and Impaired Cerebral Autoregulation in Patients With Epilepsy. Front. Neurol. 2018, 9, 969. [Google Scholar] [CrossRef]
- Lu, X.C.; Si, Y.; Williams, A.J.; Hartings, J.A.; Gryder, D.; Tortella, F.C. NNZ-2566, a glypromate analog, attenuates brain ischemia-induced non-convulsive seizures in rats. J. Cereb. Blood Flow Metab. 2009, 29, 1924–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamato, D.; Mitra, P.; Davis, F.; Osman, N.; Chaplin, R.; Cabot, P.J.; Afroz, R.; Thomas, W.; Zheng, W.; Kaur, H.; et al. Gaq proteins: Molecular pharmacology and therapeutic potential. Cell. Mol. Life Sci. 2017, 74, 1379–1390. [Google Scholar] [CrossRef]
- Frater, J.; Lie, D.; Bartlett, P.; McGrath, J.J. Insulin-like Growth Factor 1 (IGF-1) as a marker of cognitive decline in normal ageing: A review. Ageing Res. Rev. 2018, 42, 14–27. [Google Scholar] [CrossRef]
- Green, C.J.; Holly, J.M.; Bayer, A.; Fish, M.; Ebrahim, S.; Gallacher, J.; Ben-Shlomo, Y. The role of IGF-I, IGF-II, and IGFBP-3 in male cognitive aging and dementia risk: The Caerphilly Prospective Study. J. Alzheimers Dis. 2014, 41, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cai, H.; Zhang, P.; Liu, H.; Li, Z. Activation of ERK1/2 and PI3K/Akt by IGF-1 on GAP-43 expression in DRG neurons with excitotoxicity induced by glutamate in vitro. Cell. Mol. Neurobiol. 2012, 32, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, L.; Sui, G.; Yang, C.; Guo, M.; Xiong, X.; Chen, Z.; Lei, P. IGF-1 Alleviates Mitochondrial Apoptosis through the GSK3β/NF-κB/NLRP3 Signaling Pathway in LPS-Treated PC-12 Cells. J. Mol. Neurosci. 2021, 71, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; He, B.; Tong, W.; Zeng, W.; Zheng, P. Astrocytic Insulin-Like Growth Factor-1 Protects Neurons Against Excitotoxicity. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, K.F.S.; Bent, R.J.; Meese-Tamuri, S.; Ali, A.; Forder, J.P.; Aarts, M.M. Calmodulin kinase IV-dependent CREB activation is required for neuroprotection via NMDA receptor-PSD95 disruption. J. Neurochem. 2013, 126, 274–287. [Google Scholar] [CrossRef]
- Takahashi, S.; Hisatsune, A.; Kurauchi, Y.; Seki, T.; Katsuki, H. Insulin-like growth factor 1 specifically up-regulates expression of modifier subunit of glutamate-cysteine ligase and enhances glutathione synthesis in SH-SY5Y cells. Eur. J. Pharmacol. 2016, 15, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dong, H.; Li, J.; Liu, H.; Liu, Z.; Li, Z. Neuroprotective effect of insulin-like growth factor-1: Effects on tyrosine kinase receptor (Trk) expression in dorsal root ganglion neurons with glutamate-induced excitotoxicity in vitro. Brain Res. Bull. 2013, 97, 86–95. [Google Scholar] [CrossRef]
- Velikanov, I.I. The dynamics of the rheoencephalographic indices in patients with combined cerebrovascular and coronary insufficiency and cervical osteochondrosis under the influence of carbonate baths, impulse currents and cervical spinal traction. Vopr. Kurortol. Fizioter. Lech. Fiz. Kult. 1991, 3, 15–18. [Google Scholar]
- Morel, G.M.; López León, M.; Uriarte, M.; Reggiani, P.C.; Goya, R.G. Therapeutic potential of IGF-I on hippocampal neurogenesis and function during aging. Neurogenesis 2016, 4, e1259709. [Google Scholar] [CrossRef] [Green Version]
- Rosenbloom, A.L.; Rivkees, S.A. Off-label use of recombinant igf-I to promote growth: Is it appropriate? J. Clin. Endocrinol. Metab. 2010, F95, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Yamahara, K.; Asaka, N.; Kita, T.; Kishimoto, I.; Matsunaga, M.; Yamamoto, N.; Omori, K.; Nakagawa, T. Insulin-like growth factor 1 promotes cochlear synapse regeneration after excitotoxic trauma in vitro. Hear. Res. 2019, 374, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Alkan, S.; Orenstein, J.M. Bacillary peliosis hepatis. N. Engl. J. Med. 1991, 324, 1513–1514. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Animal Models | Human Patients | Reference |
|---|---|---|---|
| Ischemic Stroke | Attenuated infarct size with IGF-1 treatment in MCAO and improved post-stroke neurological behaviors. | Inverse correlation between circulating IGF-1 levels and stroke incidence; The levels of IGF-1in the serum is also inversely associated with the neurological deficits following stroke. | [5,111] |
| Traumatic brain injury (TBI) | IGF-1 is neuroprotective. Functional neurological improvement of motor and cognitive functions in different TBI models. | IGF-1 clinical trials in TBI demonstrate that IGF-1 administration either alone or in combination with GH was safe to humans and successful in improving metabolic parameters in moderate-to-severe TBI patients. | [82] |
| Amyotrophic Lateral Sclerosis (ALS) | In mouse models of ALS rhIGF-1 delayed disease onset, reduced muscle atrophy, promoted peripheral motor nerve regeneration, and extended life. | Randomized, double-blind, placebo-controlled, phase two and three clinical trials reaffirmed that rhIGF-1 administration was safe and well tolerated in most subjects but efficacy was not statistically significant. | [110] |
| Alzheimer’s Disease (AD) | In mice with increased cerebral beta-amyloid plaques serum IGF-1 modulated brain levels of beta-amyloid and prevented premature death | Multicenter, cross-sectional study to assess the relationship between IGF-1 and cognitive decline indicated that serum IGF-IGFBP-3 levels were implicated in men with AD. However, a double-blind, multicenter study using growth hormone secretagogue MK-677 which stimulates upregulation and circulation of IGF-1, failed to show efficacy in slowing disease progression. | [109,112,113,114] |
| Autism spectrum disorder (ASD)- Phelan-McDermid Syndrome (PMS) | I.p. injection of rhIGF-1 in Shank3-deficient mice at clinically approved doses of 0.24 mg/kg/day for 2 weeks reversed the electro-physiological deficits and demonstrated reduced AMPAR-mediated transmission and showed normal LTP comparable to the wild type control mice | A clinical trial using 0.24 mg/kg/day of rhIGF-1 in divided doses, in nine children with PMS (Shank3 deficient) demonstrated safety, tolerability, and efficacy. | [104,115,117] |
| ASD- Fragile X Syndrome (FXS) | In Fmr1 knockout mice characterized by reduced excitatory synaptic currents, enhanced glutamate receptor dependent-LTD, 100 mg/kg i.p. injection of IGF-1 analog Trofinetide (NNZ-2566) resulted with reduced hyperactivity, improved LSTM and LTP, and normalized social recognition and behaviors. | Phase II randomized, double-blind, placebo-controlled, parallel-group, confirmed the safety, tolerability and efficacy at the high dose of treatment with oral administration of Trofinetide at 35 or 70 mg/kg twice daily, in 72 adolescent or adult males with FXS. | [119,120] |
| Friedreich’s ataxia (FRDA) | IGF-I in FRDA-like transgenic mice (YG8R mice) conferred neuroprotection and normalized motor coordination. | In a clinical proof of concept pilot study, patients were treated s.c. with IGF-1 therapy with 50 μg/kg twice a day for 12 months and tolerability and decrease in the progression of neurological symptoms was measured, together with long-term stability of cardiac function. | [121,122,123] |
| Huntington’s disease (HD) | IGF-1 intranasal delivery rescues HD phenotype in YAC128 mice. | In 219 patients with genetically documented HD and in 71 sex- and age-matched controls, IGF-1 serum levels were significantly higher in patients than in controls, indicating somatotropic axis is overactive to confer neuroprotection. | [124,125] |
| Epilepsy | IGF-I ameliorated hippocampal neurodegeneration and protected against cognitive deficits in an animal model of temporal lobe epilepsy. | 57 patients with focal epilepsy and 35 healthy controls were evaluated for IGF-1 level; reduced serum levels of IGF-1were found to correlate with age and cardiovagal function, a parameter of cerebral autoregulation (the breath-hold index). Patients with a longer history of epilepsy, presented higher seizure frequency, and temporal lobe epilepsy and had lower serum levels of IGF-1. | [126,127] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, L.; Liu, S.; Rubin, L.; Lazarovici, P.; Zheng, W. Research Progress on Neuroprotection of Insulin-like Growth Factor-1 towards Glutamate-Induced Neurotoxicity. Cells 2022, 11, 666. https://doi.org/10.3390/cells11040666
Ge L, Liu S, Rubin L, Lazarovici P, Zheng W. Research Progress on Neuroprotection of Insulin-like Growth Factor-1 towards Glutamate-Induced Neurotoxicity. Cells. 2022; 11(4):666. https://doi.org/10.3390/cells11040666
Chicago/Turabian StyleGe, Lijun, Shuyuan Liu, Limor Rubin, Philip Lazarovici, and Wenhua Zheng. 2022. "Research Progress on Neuroprotection of Insulin-like Growth Factor-1 towards Glutamate-Induced Neurotoxicity" Cells 11, no. 4: 666. https://doi.org/10.3390/cells11040666