
Humoral Immunity to Allogeneic Immunoproteasome-Expressing Mesenchymal Stromal Cells Requires Efferocytosis by Endogenous Phagocytes

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Ethics
2.2. Cell Lines and Reagents
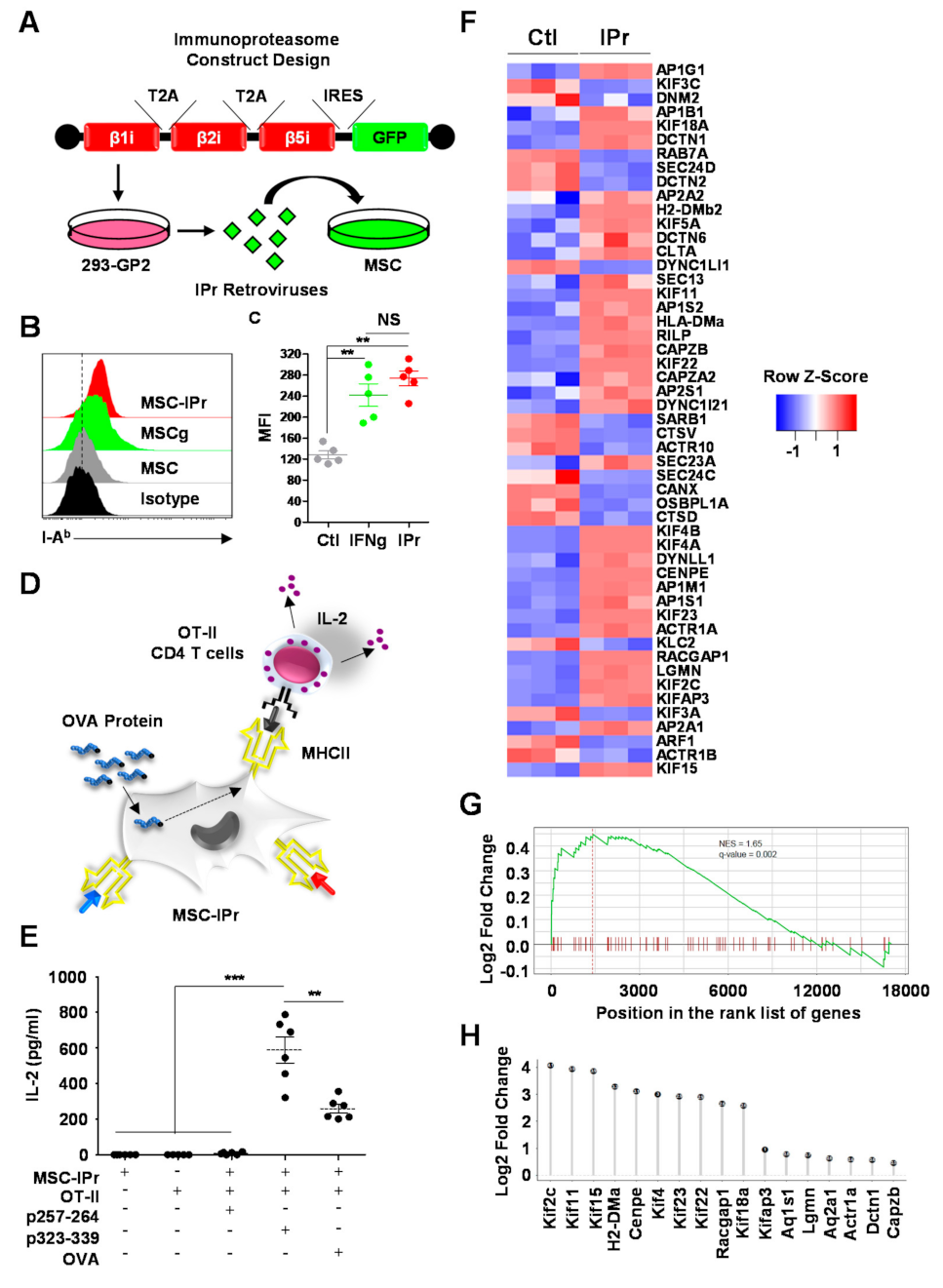
2.3. Generation of MSC-IPr Cells
2.4. Antigen Presentation Assay
2.5. RNA Sequencing and Bioinformatics Analysis
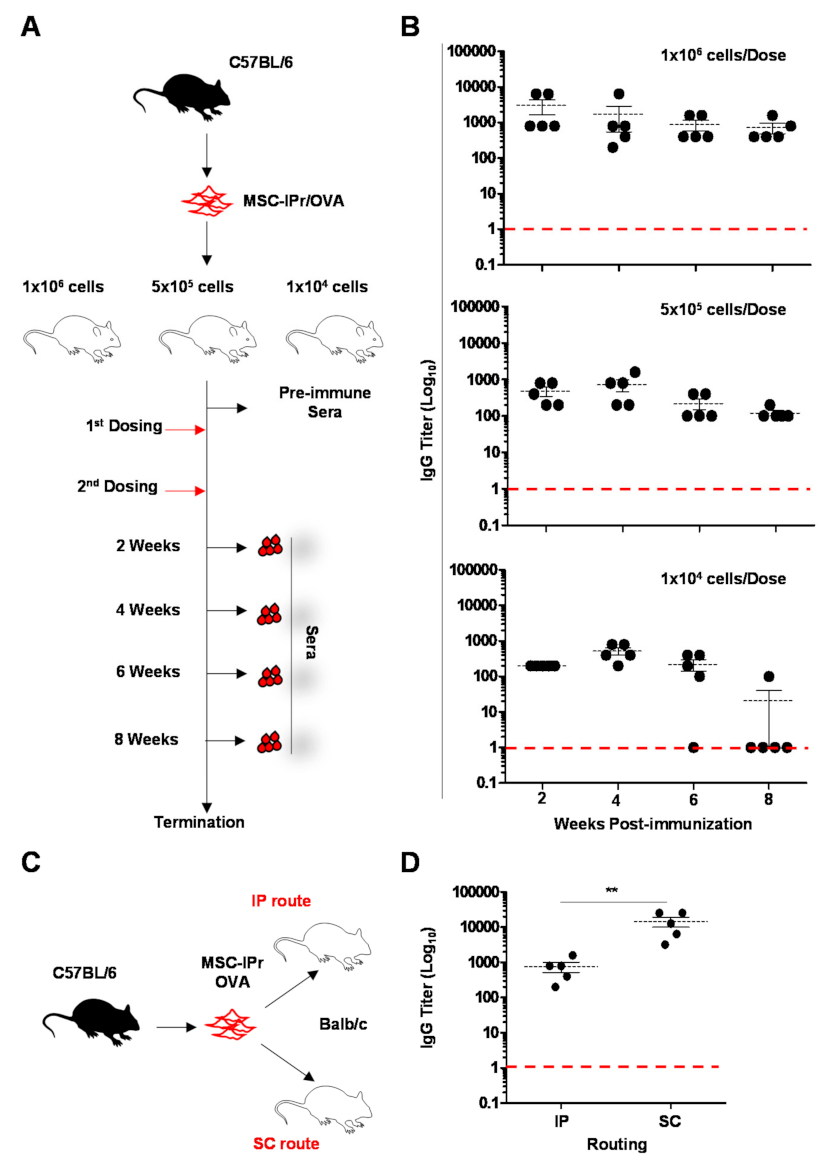
2.6. Immunization Studies
2.7. Antibody Titer Analysis by ELISA
2.8. Cytokine and Chemokine Analyses
2.9. Assessment of Cell Persistence Post-Injection
2.10. Tumor Neutralization Studies
2.11. Generation of Heat-Killed (HK) MSCs
2.12. Statistical Analysis
3. Results
3.1. MSC-IPr Can Effectively Present MHCII Peptides to CD4 T Cells
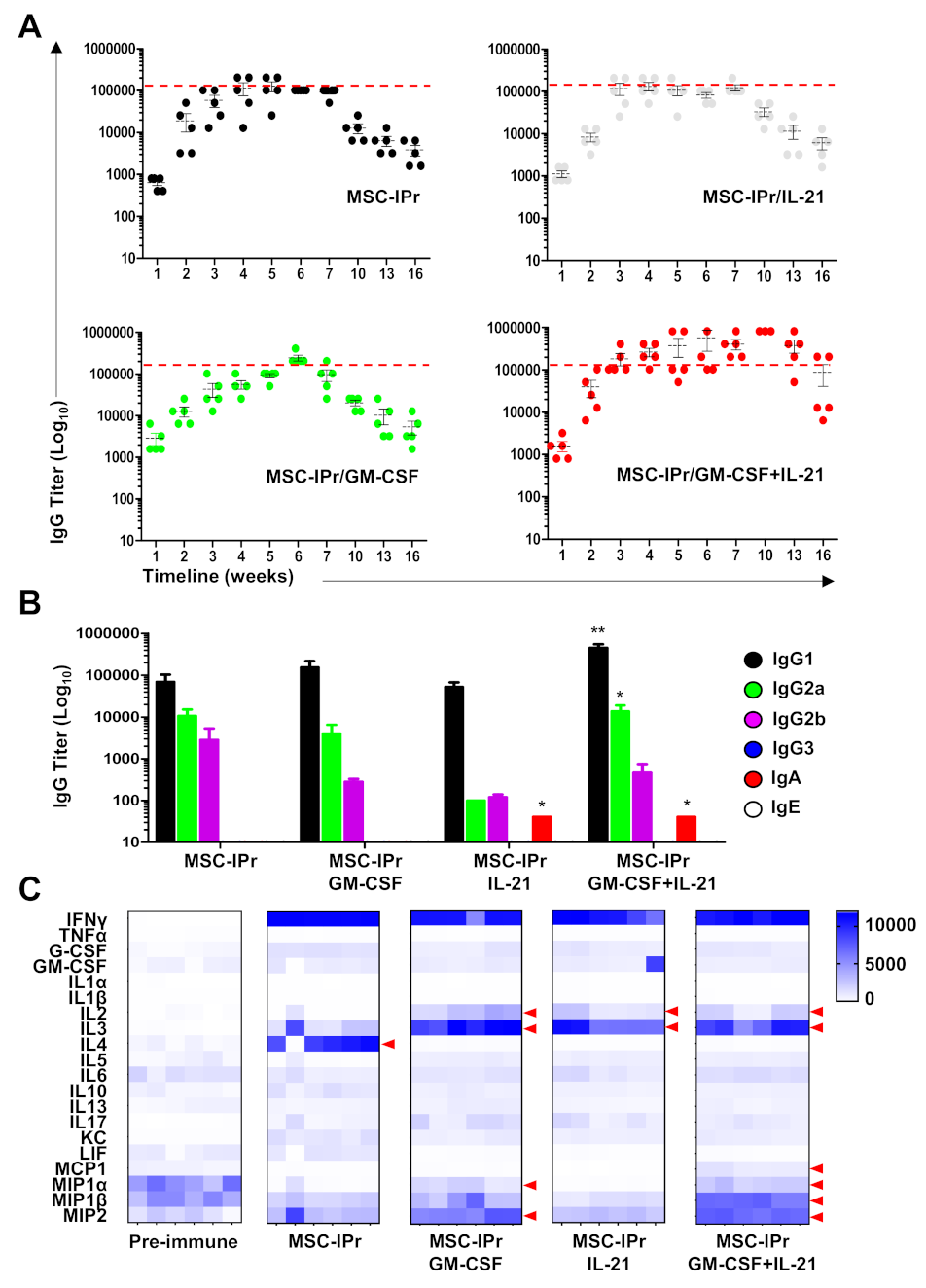
3.2. Humoral Responses Induced by Allogeneic MSC-IPr Are Both Dose- and Route-Dependent
3.3. Co-Administration of Pro-Inflammatory Cytokines Enhances Humoral Responses
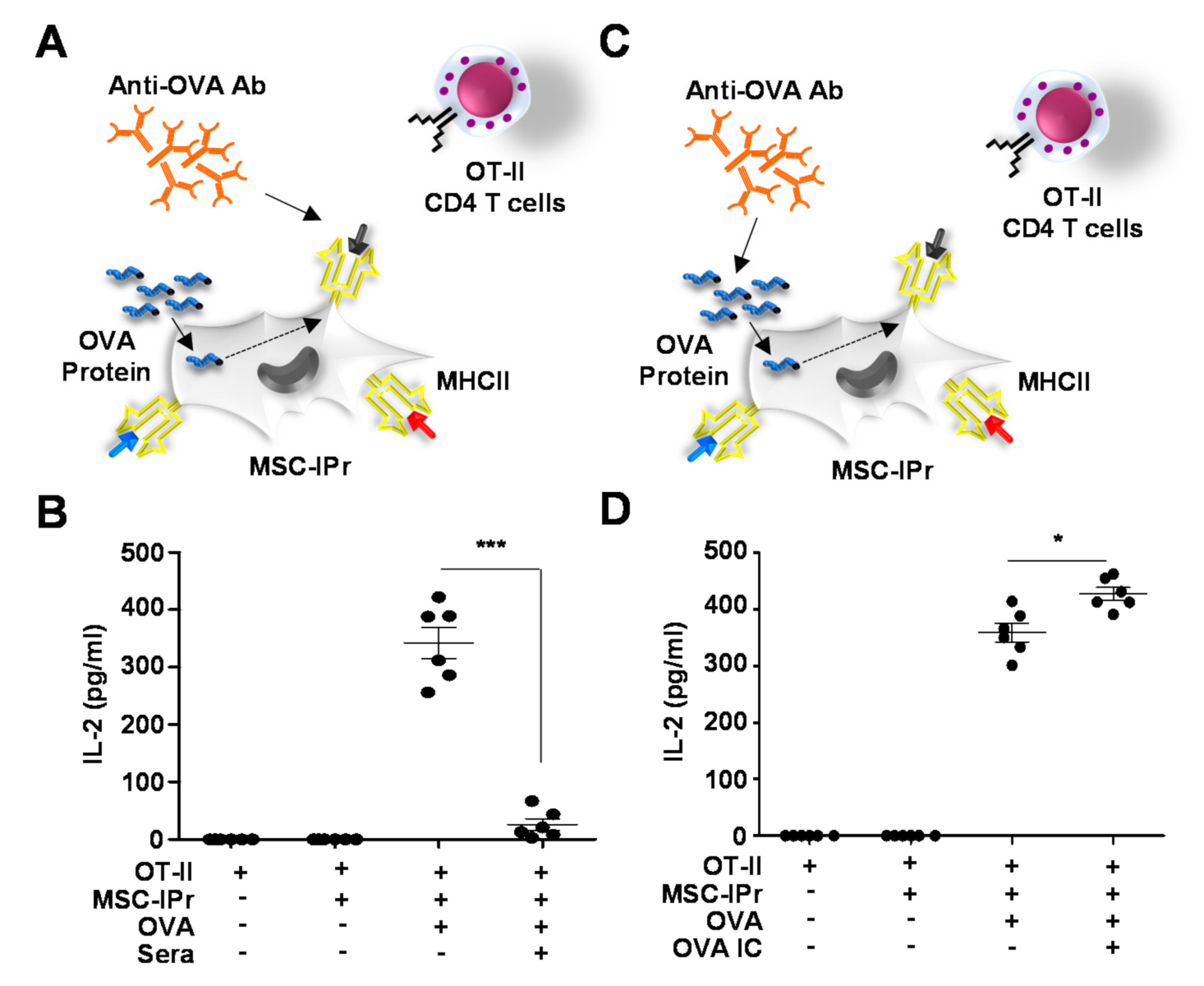
3.4. Generated Antibodies Neutralize OVA Peptide-MHCII Complexes but Enhance OVA-Antibody Complex Uptake by MSC-IPr
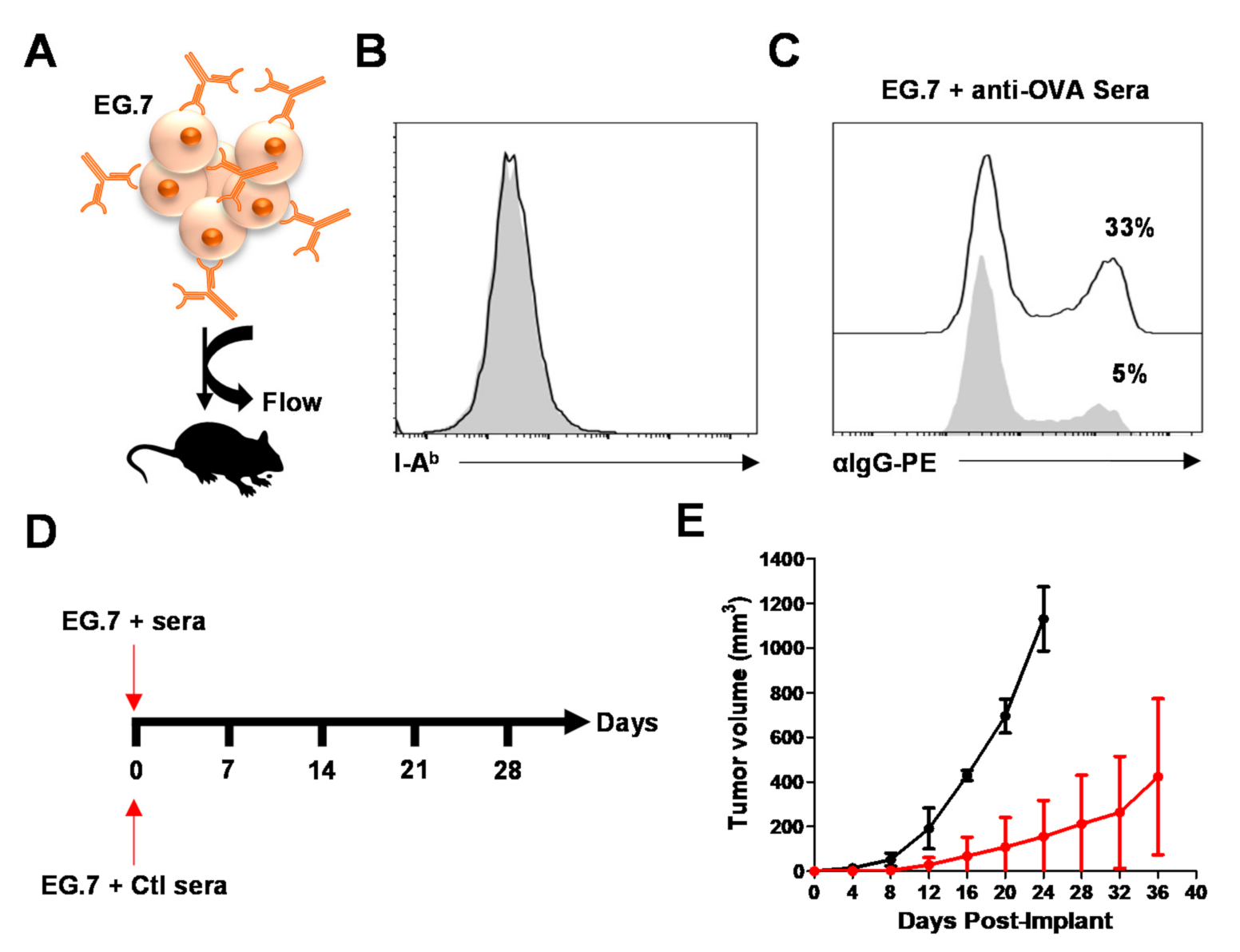
3.5. MSC-IPr-Generated Antibodies Can Impair Tumor Growth In Vivo
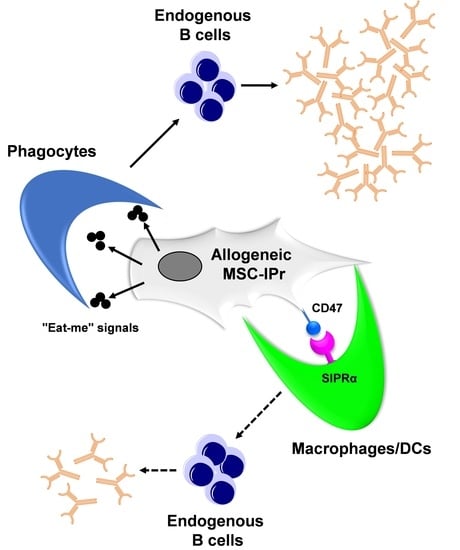
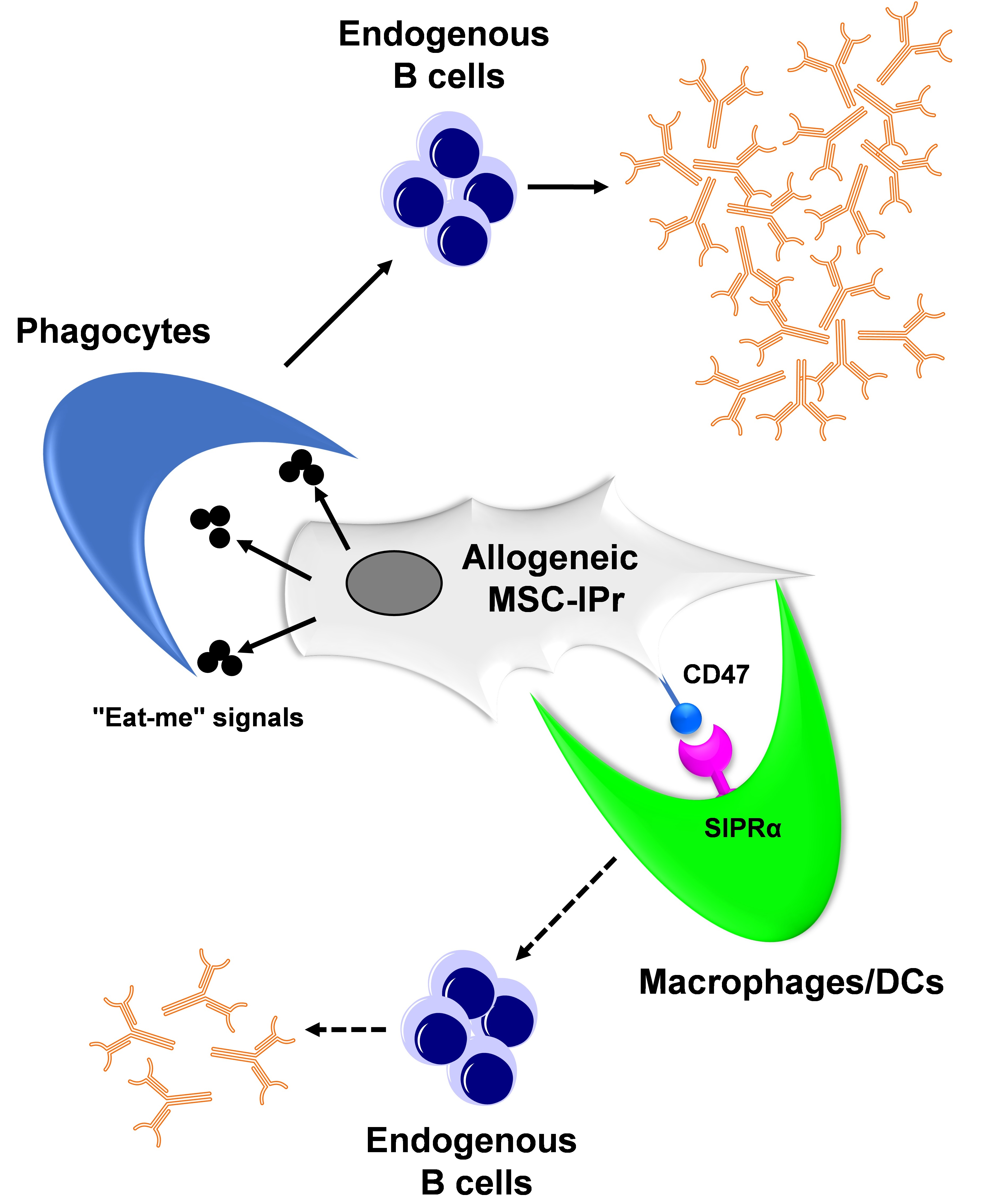
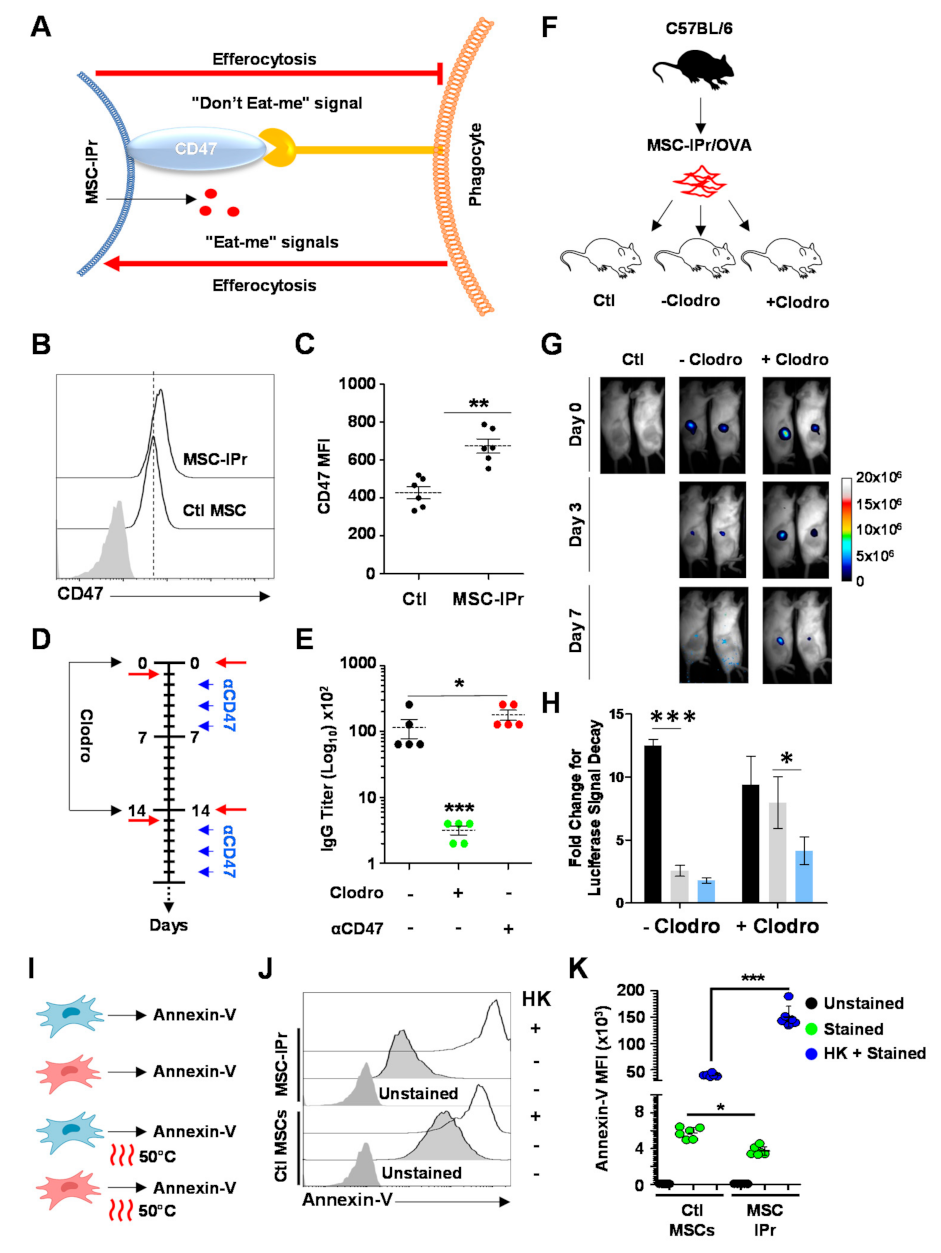
3.6. Humoral Responses Triggered in Response to Allogeneic MSC-IPr Require Efferocytosis
4. Discussion
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boylston, A. The origins of inoculation. J. R. Soc. Med. 2012, 105, 309–313. [Google Scholar] [CrossRef] [Green Version]
- Romo, M.R.; Pérez-Martínez, D.; Ferrer, C.C. Innate immunity in vertebrates: An overview. Immunology 2016, 148, 125–139. [Google Scholar] [CrossRef]
- Kurtz, J. Memory in the innate and adaptive immune systems. Microbes Infect. 2004, 6, 1410–1417. [Google Scholar] [CrossRef]
- Forthal, D.N. Functions of Antibodies. Microbiol. Spectr. 2014, 2, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hjelm, F.; Carlsson, F.; Getahun, A.; Heyman, B. Antibody-Mediated Regulation of the Immune Response. Scand. J. Immunol. 2006, 64, 177–184. [Google Scholar] [CrossRef]
- Uhr, J.W.; Möller, G. Regulatory Effect of Antibody on the Immune Response. Adv. Immunol. 1968, 8, 81–127. [Google Scholar] [CrossRef]
- Gasser, M.; Waaga-Gasser, A.M. Therapeutic Antibodies in Cancer Therapy. Adv. Exp. Med. Biol. 2016, 917, 95–120. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Onrust, S.V.; Lamb, H.M.; Balfour, J.A.B. Rituximab. Drugs 1999, 58, 79–88, discussion 89–90. [Google Scholar] [CrossRef]
- Stebegg, M.; Kumar, S.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the Germinal Center Response. Front. Immunol. 2018, 9, 2469. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Helper T Cells and Lymphocyte Activation. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Datta, J.; Terhune, J.H.; Lowenfeld, L.A.; Cintolo, J.; Xu, S.; Roses, R.; Czerniecki, B.J. Optimizing dendritic cell-based approaches for cancer immunotherapy. Yale J. Biol. Med. 2014, 87, 491–518. [Google Scholar]
- Tacken, P.J.; De Vries, I.J.M.; Torensma, R.; Figdor, C. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef]
- Moll, H. Dendritic Cell-Based Vaccination Strategies: Induction of Protective Immunity against Leishmaniasis. Immunobiology 2001, 204, 659–666. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [Green Version]
- Small, E.J.; Fratesi, P.; Reese, D.M.; Strang, G.; Laus, R.; Peshwa, M.V.; Valone, F.H. Immunotherapy of Hormone-Refractory Prostate Cancer With Antigen-Loaded Dendritic Cells. J. Clin. Oncol. 2000, 18, 3894–3903. [Google Scholar] [CrossRef]
- Guhathakurta, D.; Sheikh, N.A.; Fan, L.-Q.; Kandadi, H.; Meagher, T.C.; Hall, S.J.; Kantoff, P.; Higano, C.S.; Small, E.J.; Gardner, T.A.; et al. Humoral Immune Response against Nontargeted Tumor Antigens after Treatment with Sipuleucel-T and Its Association with Improved Clinical Outcome. Clin. Cancer Res. 2015, 21, 3619–3630. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.H.; Soleau, C.; Yu, E.Y. Improved disease markers suggest dual response in a patient with metastatic castration resistant prostate cancer and chronic lymphocytic leukemia following active cellular immunotherapy. J. Hematol. Oncol. 2015, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Abusarah, J.; Khodayarian, F.; El-Hachem, N.; Salame, N.; Olivier, M.; Balood, M.; Roversi, K.; Talbot, S.; Bikorimana, J.-P.; Chen, J.; et al. Engineering immunoproteasome-expressing mesenchymal stromal cells: A potent cellular vaccine for lymphoma and melanoma in mice. Cell Rep. Med. 2021; in press. [Google Scholar] [CrossRef]
- Eliopoulos, N.; Francois, M.; Boivin, M.-N.; Martineau, D.; Galipeau, J. Neo-Organoid of Marrow Mesenchymal Stromal Cells Secreting Interleukin-12 for Breast Cancer Therapy. Cancer Res. 2008, 68, 4810–4818. [Google Scholar] [CrossRef] [Green Version]
- Bikorimana, J.-P.; El-Hachem, N.; El-Kadiry, A.E.-H.; Abusarah, J.; Salame, N.; Shamma, R.; Rafei, M. Thymoproteasome-Expressing Mesenchymal Stromal Cells Confer Protective Anti-Tumor Immunity via Cross-Priming of Endogenous Dendritic Cells. Front. Immunol. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Tomchuck, S.L.; Norton, E.B.; Garry, R.F.; Bunnell, B.A.; Morris, C.A.; Freytag, L.C.; Clements, J.D. Mesenchymal stem cells as a novel vaccine platform. Front. Cell. Infect. Microbiol. 2012, 2, 140. [Google Scholar] [CrossRef] [Green Version]
- Grasse, M.; Meryk, A.; Miggitsch, C.; Grubeck-Loebenstein, B. GM-CSF improves the immune response to the diphtheria-component in a multivalent vaccine. Vaccine 2018, 36, 4672–4680. [Google Scholar] [CrossRef]
- Cao, A.T.; Yao, S.; Gong, B.; Nurieva, R.I.; Elson, C.O.; Cong, Y. Interleukin (IL)-21 promotes intestinal IgA response to microbiota. Mucosal Immunol. 2015, 8, 1072–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchen, S.; Robbins, R.; Sims, G.P.; Sheng, C.; Phillips, T.M.; Lipsky, P.E.; Ettinger, R. Essential Role of IL-21 in B Cell Activation, Expansion, and Plasma Cell Generation during CD4+ T Cell-B Cell Collaboration. J. Immunol. 2007, 179, 5886–5896. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Zhang, L.; Zhao, X.; Xu, G.; Zhang, Y.; Roberts, A.I.; Zhao, R.C.; Shi, Y. Mesenchymal Stem Cell-Mediated Immunosuppression Occurs via Concerted Action of Chemokines and Nitric Oxide. Cell Stem Cell 2008, 2, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, L.; Yang, J.; Yu, Y.; Chai, J.; Wang, L.; Ma, L.; Yin, H. Exosome Derived From Human Umbilical Cord Mesenchymal Stem Cell Mediates MiR-181c Attenuating Burn-induced Excessive Inflammation. EBioMedicine 2016, 8, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Su, J.; Roberts, A.I.; Shou, P.; Rabson, A.B.; Ren, G. How mesenchymal stem cells interact with tissue immune responses. Trends Immunol. 2012, 33, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Galleu, A.; Riffo-Vasquez, Y.; Trento, C.; Lomas, C.; Dolcetti, L.; Cheung, T.S.; von Bonin, M.; Barbieri, L.; Halai, K.; Ward, S.; et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci. Transl. Med. 2017, 9, eaam7828. [Google Scholar] [CrossRef] [Green Version]
- Pang, S.H.M.; D’rozario, J.; Mendonca, S.; Bhuvan, T.; Payne, N.L.; Zheng, D.; Hisana, A.; Wallis, G.; Barugahare, A.; Powell, D.; et al. Mesenchymal stromal cell apoptosis is required for their therapeutic function. Nat. Commun. 2021, 12, 6495. [Google Scholar] [CrossRef]
- Russ, A.; Hua, A.B.; Montfort, W.R.; Rahman, B.; Bin Riaz, I.; Khalid, M.U.; Carew, J.S.; Nawrocki, S.T.; Persky, D.; Anwer, F. Blocking “don’t eat me” signal of CD47-SIRPα in hematological malignancies, an in-depth review. Blood Rev. 2018, 32, 480–489. [Google Scholar] [CrossRef]
- Ravichandran, K.S. Find-me and eat-me signals in apoptotic cell clearance: Progress and conundrums. J. Exp. Med. 2010, 207, 1807–1817. [Google Scholar] [CrossRef]
- Azuma, Y.; Nakagawa, H.; Dote, K.; Higai, K.; Matsumoto, K. Decreases in CD31 and CD47 Levels on the Cell Surface during Etoposide-Induced Jurkat Cell Apoptosis. Biol. Pharm. Bull. 2011, 34, 1828–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.; Oguin, T.H.; Martinez, J. The clearance of dying cells: Table for two. Cell Death Differ. 2016, 23, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Burnham, A.J.; Foppiani, E.M.; Horwitz, E.M. Key Metabolic Pathways in MSC-Mediated Immunomodulation: Implications for the Prophylaxis and Treatment of Graft Versus Host Disease. Front. Immunol. 2020, 11, 609277. [Google Scholar] [CrossRef] [PubMed]
- Guillamat-Prats, R. The Role of MSC in Wound Healing, Scarring and Regeneration. Cells 2021, 10, 1729. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; An, N.; Ren, Y.; Yang, C.; Zhang, X.; Li, L. Immunosuppressive Effects of Mesenchymal Stem Cells-derived Exosomes. Stem Cell Rev. Rep. 2021, 17, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Rezakhani, L.; Kelishadrokhi, A.F.; Soleimanizadeh, A.; Rahmati, S. Mesenchymal stem cell (MSC)-derived exosomes as a cell-free therapy for patients Infected with COVID-19: Real opportunities and range of promises. Chem. Phys. Lipids 2021, 234, 105009. [Google Scholar] [CrossRef]
- Wechsler, M.E.; Rao, V.V.; Borelli, A.N.; Anseth, K.S. Engineering the MSC Secretome: A Hydrogel Focused Approach. Adv. Healthc. Mater. 2021, 10, 2001948. [Google Scholar] [CrossRef]
- Salame, N.; Bikorimana, J.-P.; El-Hachem, N.; Saad, W.; Kurdi, M.; Zhao, J.; Eliopoulos, N.; Shammaa, R.; Rafei, M. UM171A-induced ROS promote antigen cross-presentation of immunogenic peptides by bone marrow-derived mesenchymal stromal cells. Stem Cell Res. Ther. 2021; in press. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.; Bouchentouf, M.; Rafei, M.; Romieu-Mourez, R.; Hsieh, J.; Boivin, M.-N.; Yuan, S.; Forner, K.A.; Birman, E.; Galipeau, J. A Dendritic Cell Population Generated by a Fusion of GM-CSF and IL-21 Induces Tumor-Antigen–Specific Immunity. J. Immunol. 2010, 185, 7358–7366. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.; Rafei, M.; Bouchentouf, M.; Raven, J.; Yuan, S.; Cuerquis, J.; Forner, K.A.; Birman, E.; Galipeau, J. A Fusion of GMCSF and IL-21 Initiates Hypersignaling through the IL-21Rα Chain with Immune Activating and Tumoricidal Effects in vivo. Mol. Ther. 2010, 18, 1293–1301. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bikorimana, J.-P.; Abusarah, J.; Salame, N.; El-Hachem, N.; Shammaa, R.; Rafei, M. Humoral Immunity to Allogeneic Immunoproteasome-Expressing Mesenchymal Stromal Cells Requires Efferocytosis by Endogenous Phagocytes. Cells 2022, 11, 596. https://doi.org/10.3390/cells11040596
Bikorimana J-P, Abusarah J, Salame N, El-Hachem N, Shammaa R, Rafei M. Humoral Immunity to Allogeneic Immunoproteasome-Expressing Mesenchymal Stromal Cells Requires Efferocytosis by Endogenous Phagocytes. Cells. 2022; 11(4):596. https://doi.org/10.3390/cells11040596
Chicago/Turabian StyleBikorimana, Jean-Pierre, Jamilah Abusarah, Natasha Salame, Nehme El-Hachem, Riam Shammaa, and Moutih Rafei. 2022. "Humoral Immunity to Allogeneic Immunoproteasome-Expressing Mesenchymal Stromal Cells Requires Efferocytosis by Endogenous Phagocytes" Cells 11, no. 4: 596. https://doi.org/10.3390/cells11040596