
In Vitro Veritas: From 2D Cultures to Organ-on-a-Chip Models to Study Immunogenic Cell Death in the Tumor Microenvironment
 , , ,
, , , 
Abstract
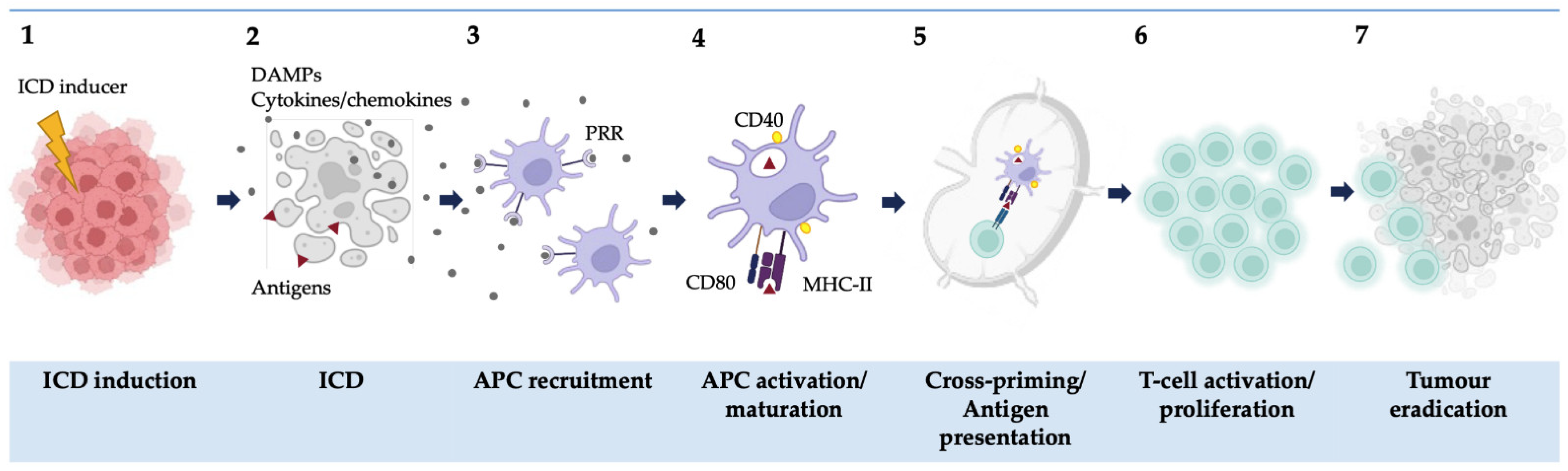
:1. Introduction
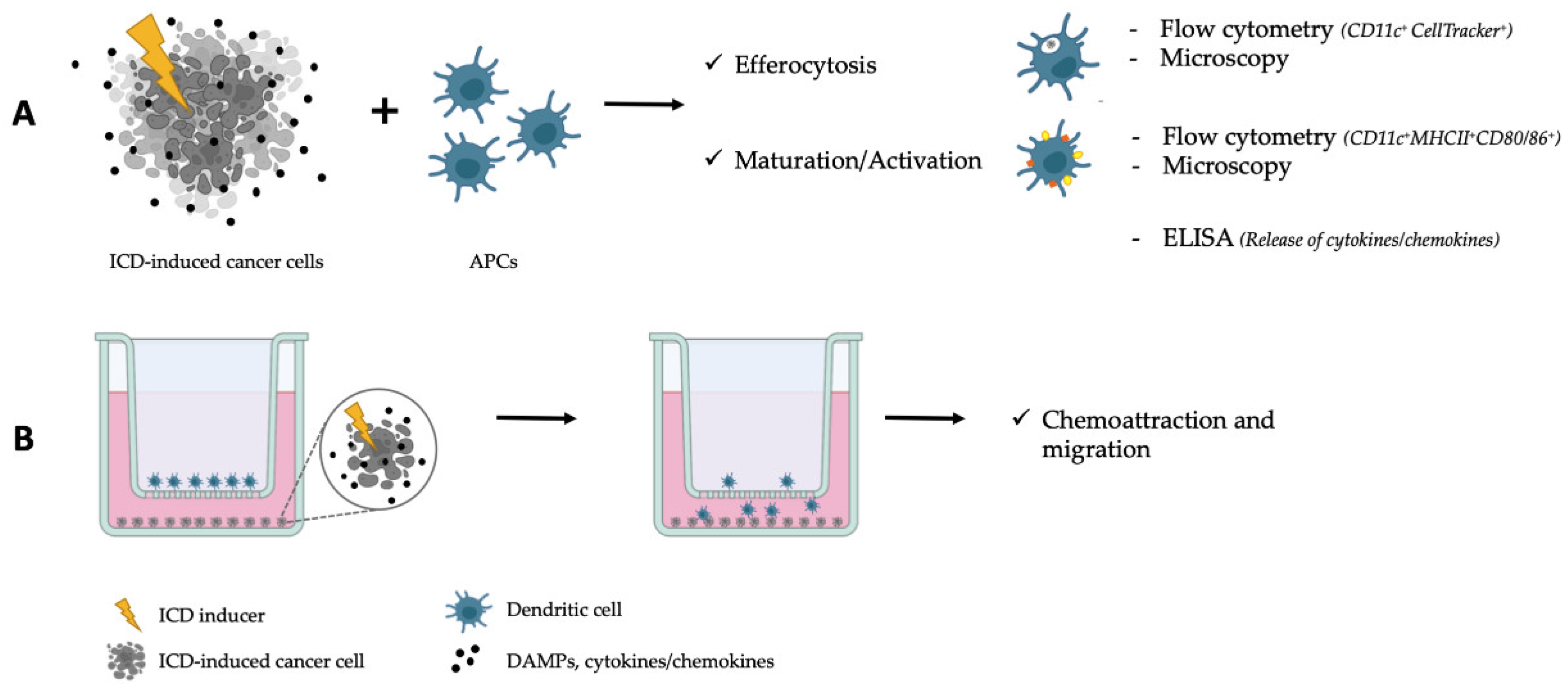
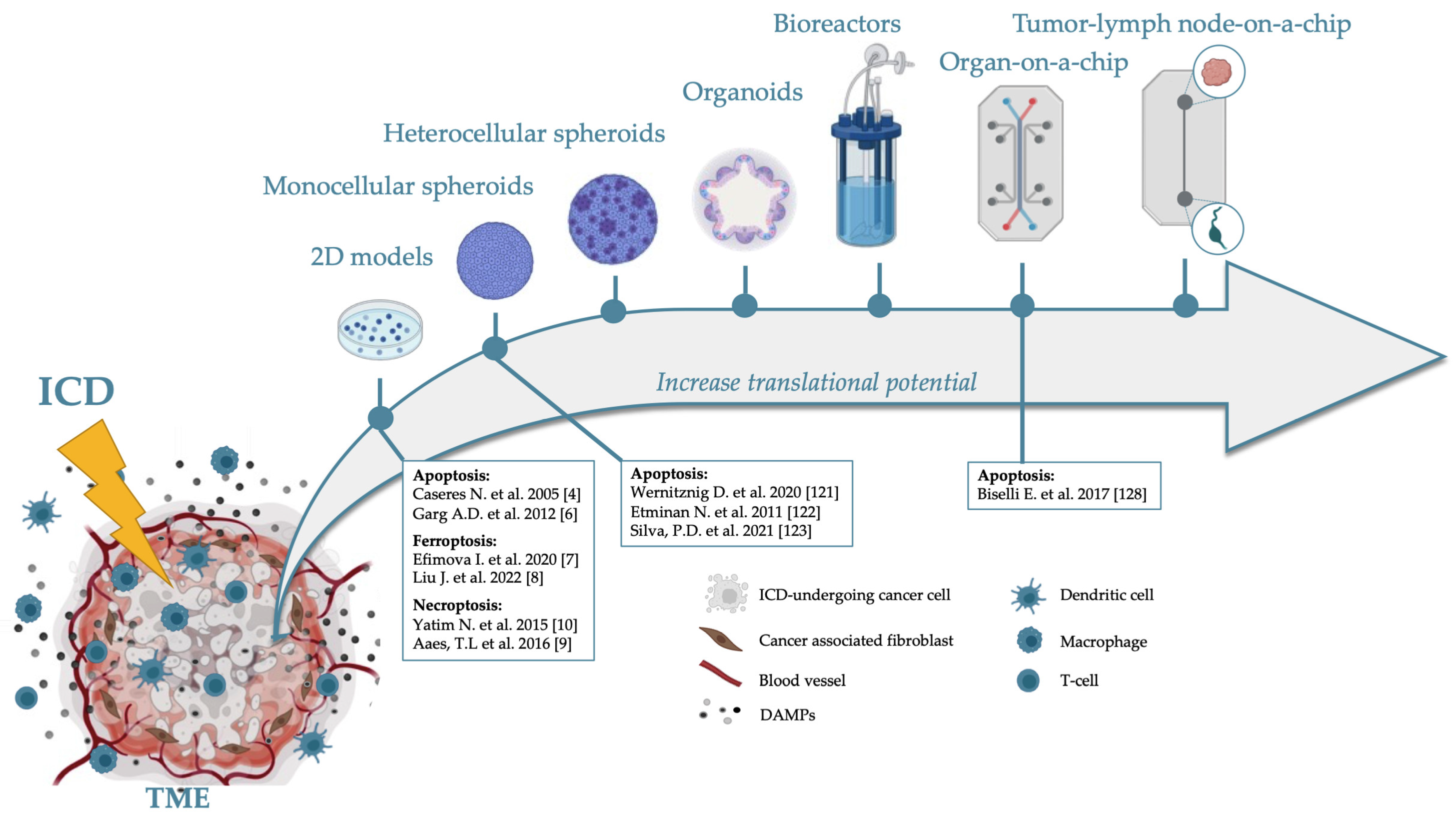
2. ICD in 2D Models
3. 3D Models
3.1. Spheroids
3.2. Complex 3D Models Which Include ECM

{kind=link}
{kind=link}
{kind=link}
| Biomaterial | Synthetic/Natural | (Non-)Reversible | (Dis)Advantages | References |
|---|---|---|---|---|
| Collagen | Natural | Reversible | +High density, natural −Batch-to-batch variation | [74,75] |
| GelMOD | Semi-synthetic | Reversible | +Biocompatible −Heterogenous network, shrinking during crosslinking | [77,84] |
| Matrigel | Natural | Non-reversible | +Mimics natural tumor ECM −Batch-to-batch variation, weaker material | [77,80] |
| Alginate | Natural | Non-reversible | +Stabilizes 3D structures −Difficult to work with | [82] |
| Poly-lactic acid | Synthetic | Reversible | +Strong material −Not natural | [83] |
3.2.1. Organoids
3.2.2. Perfusion Models
Perfusion Bioreactors
Tumor-on-a-Chip
4. Future Perspective and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic Cell Death and DAMPs in Cancer Therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of Immunogenic Cell Death and Its Relevance for Cancer Therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic Cell Stress and Death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.M.; et al. A Novel Pathway Combining Calreticulin Exposure and ATP Secretion in Immunogenic Cancer Cell Death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Efimova, I.; Catanzaro, E.; Van der Meeren, L.; Turubanova, V.D.; Hammad, H.; Mishchenko, T.A.; Vedunova, M.V.; Fimognari, C.; Bachert, C.; Coppieters, F.; et al. Vaccination with Early Ferroptotic Cancer Cells Induces Efficient Antitumor Immunity. J. Immunother. Cancer 2020, 8, e001369. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, S.; Zeng, L.; Li, J.; Klionsky, D.J.; Kroemer, G.; Jiang, J.; Tang, D.; Kang, R. DCN Released from Ferroptotic Cells Ignites AGER-Dependent Immune Responses. Autophagy 2022, 18, 2036–2049. [Google Scholar] [CrossRef]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-Tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [Green Version]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-ΚB Signaling in Dying Cells Determines Cross-Priming of CD8+ T Cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef]
- Turubanova, V.D.; Mishchenko, T.A.; Balalaeva, I.V.; Efimova, I.; Peskova, N.N.; Klapshina, L.G.; Lermontova, S.A.; Bachert, C.; Krysko, O.; Vedunova, M.V.; et al. Novel Porphyrazine-Based Photodynamic Anti-Cancer Therapy Induces Immunogenic Cell Death. Sci. Rep. 2021, 11, 7205. [Google Scholar] [CrossRef]
- Bloy, N.; Garcia, P.; Laumont, C.M.; Pitt, J.M.; Sistigu, A.; Stoll, G.; Yamazaki, T.; Bonneil, E.; Buqué, A.; Humeau, J.; et al. Immunogenic Stress and Death of Cancer Cells: Contribution of Antigenicity vs Adjuvanticity to Immunosurveillance. Immunol. Rev. 2017, 280, 165–174. [Google Scholar] [CrossRef]
- Galluzzi, L.; Petroni, G.; Kroemer, G. Immunogenicity of Cell Death Driven by Immune Effectors. J. Immunother. Cancer 2020, 8, e000802. [Google Scholar] [CrossRef] [Green Version]
- Rufo, N.; Garg, A.D.; Agostinis, P. The Unfolded Protein Response in Immunogenic Cell Death and Cancer Immunotherapy. Trends Cancer 2017, 3, 643–658. [Google Scholar] [CrossRef]
- Bezu, L.; Sauvat, A.; Humeau, J.; Gomes-da-Silva, L.C.; Iribarren, K.; Forveille, S.; Garcia, P.; Zhao, L.; Liu, P.; Zitvogel, L.; et al. EIF2α Phosphorylation Is Pathognomonic for Immunogenic Cell Death. Cell Death Differ. 2018, 25, 1375–1393. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Immunogenic Cell Death Induction by Anticancer Chemotherapeutics. Oncoimmunology 2017, 6, e1386829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demuynck, R.; Efimova, I.; Naessens, F.; Krysko, D.V. Immunogenic Ferroptosis and Where to Find It? J. Immunother. Cancer 2021, 9, e003430. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. CB 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Marzagalli, M.; Pelizzoni, G.; Fedi, A.; Vitale, C.; Fontana, F.; Bruno, S.; Poggi, A.; Dondero, A.; Aiello, M.; Castriconi, R.; et al. A Multi-Organ-on-Chip to Recapitulate the Infiltration and the Cytotoxic Activity of Circulating NK Cells in 3D Matrix-Based Tumor Model. Front. Bioeng. Biotechnol. 2022, 10, 945149. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, A.A.; Li, E.; Weiner, L.M. 3D Culture Systems for Exploring Cancer Immunology. Cancers 2021, 13, 56. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Vandenabeele, P. Clearance of Dead Cells: Mechanisms, Immune Responses and Implication in the Development of Diseases. Apoptosis Int. J. Program. Cell Death 2010, 15, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Brouckaert, G.; Kalai, M.; Vandenabeele, P.; D’Herde, K. Mechanisms of Internalization of Apoptotic and Necrotic L929 Cells by a Macrophage Cell Line Studied by Electron Microscopy. J. Morphol. 2003, 258, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Denecker, G.; Festjens, N.; Gabriels, S.; Parthoens, E.; D’Herde, K.; Vandenabeele, P. Macrophages Use Different Internalization Mechanisms to Clear Apoptotic and Necrotic Cells. Cell Death Differ. 2006, 13, 2011–2022. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Xu, A.; Jin, J.; Zhang, M.; Lou, J.; Qian, C.; Zhu, J.; Wang, Y.; Yang, Z.; Li, X.; et al. MerTK-Mediated Efferocytosis Promotes Immune Tolerance and Tumor Progression in Osteosarcoma through Enhancing M2 Polarization and PD-L1 Expression. OncoImmunology 2022, 11, 2024941. [Google Scholar] [CrossRef]
- Werfel, T.A.; Elion, D.L.; Rahman, B.; Hicks, D.J.; Sanchez, V.; Gonzales-Ericsson, P.I.; Nixon, M.J.; James, J.L.; Balko, J.M.; Scherle, P.A.; et al. Treatment-Induced Tumor Cell Apoptosis and Secondary Necrosis Drive Tumor Progression in the Residual Tumor Microenvironment through MERTK and IDO1. Cancer Res. 2019, 79, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Tan, X.; Hu, X.; Zhou, M.; Yan, J.; Ding, C. Tumor Microenvironment Following Gemcitabine Treatment Favors Differentiation of Immunosuppressive Ly6Chigh Myeloid Cells. J. Immunol. 2020, 204, 212–223. [Google Scholar] [CrossRef]
- Petty, A.J.; Li, A.; Wang, X.; Dai, R.; Heyman, B.; Hsu, D.; Huang, X.; Yang, Y. Hedgehog Signaling Promotes Tumor-Associated Macrophage Polarization to Suppress Intratumoral CD8+ T Cell Recruitment. J. Clin. Investig. 2019, 129, 5151–5162. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ma, L.; Shen, S.; Guo, Y.; Cao, Q.; Cai, X.; Feng, J.; Yan, Y.; Hu, T.; Luo, S.; et al. Intestinal Dysbacteriosis-Induced IL-25 Promotes Development of HCC via Alternative Activation of Macrophages in Tumor Microenvironment. J. Exp. Clin. Cancer Res. 2019, 38, 303. [Google Scholar] [CrossRef]
- Chen, X.J.; Wu, S.; Yan, R.M.; Fan, L.S.; Yu, L.; Zhang, Y.M.; Wei, W.F.; Zhou, C.F.; Wu, X.G.; Zhong, M.; et al. The Role of the Hypoxia-Nrp-1 Axis in the Activation of M2-like Tumor-Associated Macrophages in the Tumor Microenvironment of Cervical Cancer. Mol. Carcinog. 2019, 58, 388–397. [Google Scholar] [CrossRef]
- Lee, H.E.; Lee, J.Y.; Yang, G.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Inhibition of NLRP3 Inflammasome in Tumor Microenvironment Leads to Suppression of Metastatic Potential of Cancer Cells. Sci. Rep. 2019, 9, 879. [Google Scholar] [CrossRef] [Green Version]
- Lauber, K.; Bohn, E.; Kröber, S.M.; Xiao, Y.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic Cells Induce Migration of Phagocytes via Caspase-3-Mediated Release of a Lipid Attraction Signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Andreucci, E.; Peppicelli, S.; Ruzzolini, J.; Bianchini, F.; Biagioni, A.; Papucci, L.; Magnelli, L.; Mazzanti, B.; Stecca, B.; Calorini, L. The Acidic Tumor Microenvironment Drives a Stem-like Phenotype in Melanoma Cells. J. Mol. Med. 2020, 98, 1431–1446. [Google Scholar] [CrossRef]
- Mathews, E.H.; Visagie, M.H.; Meyer, A.A.; Joubert, A.M.; Mathews, G.E. In Vitro Quantification: Long-Term Effect of Glucose Deprivation on Various Cancer Cell Lines. Nutrition 2020, 74, 110748. [Google Scholar] [CrossRef]
- Horsman, M.R.; Vaupel, P. Pathophysiological Basis for the Formation of the Tumor Microenvironment. Front. Oncol. 2016, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of Hypoxia in Cancer Therapy by Regulating the Tumor Microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [Green Version]
- Sudhan, D.R.; Siemann, D.W. Cathepsin L Inhibition by the Small Molecule KGP94 Suppresses Tumor Microenvironment Enhanced Metastasis Associated Cell Functions of Prostate and Breast Cancer Cells. Clin. Exp. Metastasis 2013, 30, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Place, T.L.; Domann, F.E.; Case, A.J. Limitations of Oxygen Delivery to Cells in Culture: An Underappreciated Problem in Basic and Translational Research. Free Radic. Biol. Med. 2017, 113, 311–322. [Google Scholar] [CrossRef]
- Campillo, N.; Falcones, B.; Otero, J.; Colina, R.; Gozal, D.; Navajas, D.; Farré, R.; Almendros, I. Differential Oxygenation in Tumor Microenvironment Modulates Macrophage and Cancer Cell Crosstalk: Novel Experimental Settingand Proof of Concept. Front. Oncol. 2019, 9, 43. [Google Scholar] [CrossRef]
- Pavlacky, J.; Polak, J. Technical Feasibility and Physiological Relevance of Hypoxic Cell Culture Models. Front. Endocrinol. 2020, 11, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riemann, A.; Schneider, B.; Gündel, D.; Stock, C.; Gekle, M.; Thews, O. Acidosis Promotes Metastasis Formation by Enhancing Tumor Cell Motility. Adv. Exp. Med. Biol. 2016, 876, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Rohani, N.; Hao, L.; Alexis, M.S.; Joughin, B.A.; Krismer, K.; Moufarrej, M.N.; Soltis, A.R.; Lauffenburger, D.A.; Yaffe, M.B.; Burge, C.B.; et al. Acidification of Tumor at Stromal Boundaries Drives Transcriptome Alterations Associated with Aggressive Phenotypes. Cancer Res. 2019, 79, 1952–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauschner, M.; Hüsing, T.; Lange, L.; Jarosik, K.; Reime, S.; Riemann, A.; Thews, O. Role of Acidosis-Sensitive MicroRNAs in Gene Expression and Functional Parameters of Tumors in Vitro and in Vivo. Neoplasia 2021, 23, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Dierge, E.; Debock, E.; Guilbaud, C.; Corbet, C.; Mignolet, E.; Mignard, L.; Bastien, E.; Dessy, C.; Larondelle, Y.; Feron, O. Peroxidation of N-3 and n-6 Polyunsaturated Fatty Acids in the Acidic Tumor Environment Leads to Ferroptosis-Mediated Anticancer Effects. Cell Metab. 2021, 33, 1701–1715.e5. [Google Scholar] [CrossRef]
- Khajah, M.A.; Khushaish, S.; Luqmani, Y.A. Glucose Deprivation Reduces Proliferation and Motility, and Enhances the Anti-Proliferative Effects of Paclitaxel and Doxorubicin in Breast Cell Lines in Vitro. PLoS ONE 2022, 17, e0272449. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, T.; Sun, H.; Weng, W.; Zhang, Q.; Liu, C.; Han, Y.; Sheng, W. Pim1 Supports Human Colorectal Cancer Growth during Glucose Deprivation by Enhancing the Warburg Effect. Cancer Sci. 2018, 109, 1468–1479. [Google Scholar] [CrossRef] [Green Version]
- Demuynck, R.; Efimova, I.; Lin, A.; Declercq, H.; Krysko, D.V. A 3D Cell Death Assay to Quantitatively Determine Ferroptosis in Spheroids. Cells 2020, 9, E703. [Google Scholar] [CrossRef] [Green Version]
- Meyenberg Cunha-de Padua, M.; Noleto, G.R.; de Oliveira Petkowicz, C.L.; Cadena, S.M.S.C.; Bost, F.; Pouysségur, J.; Mazure, N.M. Hypoxia Protects against the Cell Death Triggered by Oxovanadium-Galactomannan Complexes in HepG2 Cells. Cell. Mol. Biol. Lett. 2019, 24, 18. [Google Scholar] [CrossRef] [Green Version]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef]
- Xu, S.; Yang, Y.; Dong, L.; Qiu, W.; Yang, L.; Wang, X.; Liu, L. Construction and Characteristics of an E-Cadherin-Related Three-Dimensional Suspension Growth Model of Ovarian Cancer. Sci. Rep. 2014, 4, 5646. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Lin, C.; Cheng, J.; Su, J.; Zhao, H.; Liu, T.; Wen, X.; Zhao, P. Generation of Multicellular Tumor Spheroids with Microwell-Based Agarose Scaffolds for Drug Testing. PLoS ONE 2015, 10, e0130348. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Sarvestani, S.K.; Moeinzadeh, S.; He, X.; Jabbari, E. Effect of CD44 Binding Peptide Conjugated to an Engineered Inert Matrix on Maintenance of Breast Cancer Stem Cells and Tumorsphere Formation. PLoS ONE 2013, 8, e59147. [Google Scholar] [CrossRef] [Green Version]
- Peirsman, A.; Blondeel, E.; Ahmed, T.; Anckaert, J.; Audenaert, D.; Boterberg, T.; Buzas, K.; Carragher, N.; Castellani, G.; Castro, F.; et al. MISpheroID: A Knowledgebase and Transparency Tool for Minimum Information in Spheroid Identity. Nat. Methods 2021, 18, 1294–1303. [Google Scholar] [CrossRef]
- Ryu, N.-E.; Lee, S.-H.; Park, H. Spheroid Culture System Methods and Applications for Mesenchymal Stem Cells. Cells 2019, 8, E1620. [Google Scholar] [CrossRef] [Green Version]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.-R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D Pancreatic Carcinoma Spheroids Induce a Matrix-Rich, Chemoresistant Phenotype Offering a Better Model for Drug Testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Xiu, J.; Liu, Y.; Zhang, T.; Pan, W.; Zheng, X.; Zhang, X. A 3D Printed Hanging Drop Dripper for Tumor Spheroids Analysis Without Recovery. Sci. Rep. 2019, 9, 19717. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.-Y.; Lee, J.-H.; Shin, Y.; Chung, S.; Kuh, H.-J. Co-Culture of Tumor Spheroids and Fibroblasts in a Collagen Matrix-Incorporated Microfluidic Chip Mimics Reciprocal Activation in Solid Tumor Microenvironment. PLoS ONE 2016, 11, e0159013. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.M.; Camões, S.P.; Filipe, E.; Cipriano, M.; Barcia, R.N.; Filipe, M.; Teixeira, M.; Simões, S.; Gaspar, M.; Mosqueira, D.; et al. Three-Dimensional Spheroid Cell Culture of Umbilical Cord Tissue-Derived Mesenchymal Stromal Cells Leads to Enhanced Paracrine Induction of Wound Healing. Stem Cell Res. Ther. 2015, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- De Moor, L.; Merovci, I.; Baetens, S.; Verstraeten, J.; Kowalska, P.; Krysko, D.V.; De Vos, W.H.; Declercq, H. High-Throughput Fabrication of Vascularized Spheroids for Bioprinting. Biofabrication 2018, 10, 035009. [Google Scholar] [CrossRef]
- Stadler, M.; Scherzer, M.; Walter, S.; Holzner, S.; Pudelko, K.; Riedl, A.; Unger, C.; Kramer, N.; Weil, B.; Neesen, J.; et al. Exclusion from Spheroid Formation Identifies Loss of Essential Cell-Cell Adhesion Molecules in Colon Cancer Cells. Sci. Rep. 2018, 8, 1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, C.; Yang, M.; Fan, Z.; Li, S.; Gao, T.; Fang, Z. Associations of Chemo- and Radio-Resistant Phenotypes with the Gap Junction, Adhesion and Extracellular Matrix in a Three-Dimensional Culture Model of Soft Sarcoma. J. Exp. Clin. Cancer Res. 2015, 34, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulin, A.-L.; Broekgaarden, M.; Simeone, D.; Hasan, T. Low Dose Photodynamic Therapy Harmonizes with Radiation Therapy to Induce Beneficial Effects on Pancreatic Heterocellular Spheroids. Oncotarget 2019, 10, 2625–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebelo, S.P.; Pinto, C.; Martins, T.R.; Harrer, N.; Estrada, M.F.; Loza-Alvarez, P.; Cabeçadas, J.; Alves, P.M.; Gualda, E.J.; Sommergruber, W.; et al. 3D-3-Culture: A Tool to Unveil Macrophage Plasticity in the Tumour Microenvironment. Biomaterials 2018, 163, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.B.; Kim, E.M.; Byun, H.; Chang, H.; Jeong, K.; Aman, Z.M.; Choi, Y.S.; Park, J.; Shin, H. Engineering Spheroids Potentiating Cell-Cell and Cell-ECM Interactions by Self-Assembly of Stem Cell Microlayer. Biomaterials 2018, 165, 105–120. [Google Scholar] [CrossRef]
- Aoudjit, F.; Vuori, K. Integrin Signaling in Cancer Cell Survival and Chemoresistance. Chemother. Res. Pract. 2012, 2012, 283181. [Google Scholar] [CrossRef] [Green Version]
- Chang, Q.; Ornatsky, O.I.; Siddiqui, I.; Straus, R.; Baranov, V.I.; Hedley, D.W. Biodistribution of Cisplatin Revealed by Imaging Mass Cytometry Identifies Extensive Collagen Binding in Tumor and Normal Tissues. Sci. Rep. 2016, 6, 36641. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Tajik, A.; Chen, J.; Jia, Q.; Chowdhury, F.; Wang, L.; Chen, J.; Zhang, S.; Hong, Y.; Yi, H.; et al. Matrix Softness Regulates Plasticity of Tumour-Repopulating Cells via H3K9 Demethylation and Sox2 Expression. Nat. Commun. 2014, 5, 4619. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Koh, I.; Lee, J.E.; Lim, J.Y.; Cheong, J.-H.; Kim, P. Increased Extracellular Matrix Density Disrupts E-Cadherin/β-Catenin Complex in Gastric Cancer Cells. Biomater. Sci. 2018, 6, 2704–2713. [Google Scholar] [CrossRef]
- Pang, M.-F.; Siedlik, M.J.; Han, S.; Stallings-Mann, M.; Radisky, D.C.; Nelson, C.M. Tissue Stiffness and Hypoxia Modulate the Integrin-Linked Kinase ILK to Control Breast Cancer Stem-like Cells. Cancer Res. 2016, 76, 5277–5287. [Google Scholar] [CrossRef]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; LLeonart, M.E. Insights into New Mechanisms and Models of Cancer Stem Cell Multidrug Resistance. Semin. Cancer Biol. 2020, 60, 166–180. [Google Scholar] [CrossRef]
- Huang, Q.; Zou, Y.; Arno, M.C.; Chen, S.; Wang, T.; Gao, J.; Dove, A.P.; Du, J. Hydrogel Scaffolds for Differentiation of Adipose-Derived Stem Cells. Chem. Soc. Rev. 2017, 46, 6255–6275. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The Collagen Family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [Green Version]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.-L.; Carretta, M.; Kalvisa, A.; Siersbæk, M.S.; Simões, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen Density Regulates the Activity of Tumor-Infiltrating T Cells. J. Immunother. Cancer 2019, 7, 68. [Google Scholar] [CrossRef] [Green Version]
- Van Hoorick, J.; Gruber, P.; Markovic, M.; Tromayer, M.; Van Erps, J.; Thienpont, H.; Liska, R.; Ovsianikov, A.; Dubruel, P.; Van Vlierberghe, S. Cross-Linkable Gelatins with Superior Mechanical Properties Through Carboxylic Acid Modification: Increasing the Two-Photon Polymerization Potential. Biomacromolecules 2017, 18, 3260–3272. [Google Scholar] [CrossRef] [Green Version]
- Augustine, R.; Zahid, A.A.; Mraiche, F.; Alam, K.; Al Moustafa, A.-E.; Hasan, A. Gelatin-Methacryloyl Hydrogel Based in Vitro Blood-Brain Barrier Model for Studying Breast Cancer-Associated Brain Metastasis. Pharm. Dev. Technol. 2021, 26, 490–500. [Google Scholar] [CrossRef]
- Hughes, C.S.; Postovit, L.M.; Lajoie, G.A. Matrigel: A Complex Protein Mixture Required for Optimal Growth of Cell Culture. Proteomics 2010, 10, 1886–1890. [Google Scholar] [CrossRef]
- Keeratichamroen, S.; Lirdprapamongkol, K.; Svasti, J. Mechanism of ECM-Induced Dormancy and Chemoresistance in A549 Human Lung Carcinoma Cells. Oncol. Rep. 2018, 39, 1765–1774. [Google Scholar] [CrossRef] [Green Version]
- Edmondson, R.; Adcock, A.F.; Yang, L. Influence of Matrices on 3D-Cultured Prostate Cancer Cells’ Drug Response and Expression of Drug-Action Associated Proteins. PLoS ONE 2016, 11, e0158116. [Google Scholar] [CrossRef] [Green Version]
- Sodek, K.L.; Brown, T.J.; Ringuette, M.J. Collagen I but Not Matrigel Matrices Provide an MMP-Dependent Barrier to Ovarian Cancer Cell Penetration. BMC Cancer 2008, 8, 223. [Google Scholar] [CrossRef]
- Anguiano, M.; Morales, X.; Castilla, C.; Pena, A.R.; Ederra, C.; Martínez, M.; Ariz, M.; Esparza, M.; Amaveda, H.; Mora, M.; et al. The Use of Mixed Collagen-Matrigel Matrices of Increasing Complexity Recapitulates the Biphasic Role of Cell Adhesion in Cancer Cell Migration: ECM Sensing, Remodeling and Forces at the Leading Edge of Cancer Invasion. PLoS ONE 2020, 15, e0220019. [Google Scholar] [CrossRef] [PubMed]
- Cavo, M.; Caria, M.; Pulsoni, I.; Beltrame, F.; Fato, M.; Scaglione, S. A New Cell-Laden 3D Alginate-Matrigel Hydrogel Resembles Human Breast Cancer Cell Malignant Morphology, Spread and Invasion Capability Observed “In Vivo”. Sci. Rep. 2018, 8, 5333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jaeghere, E.; De Vlieghere, E.; Van Hoorick, J.; Van Vlierberghe, S.; Wagemans, G.; Pieters, L.; Melsens, E.; Praet, M.; Van Dorpe, J.; Boone, M.N.; et al. Heterocellular 3D Scaffolds as Biomimetic to Recapitulate the Tumor Microenvironment of Peritoneal Metastases in Vitro and in Vivo. Biomaterials 2018, 158, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Nanki, Y.; Chiyoda, T.; Hirasawa, A.; Ookubo, A.; Itoh, M.; Ueno, M.; Akahane, T.; Kameyama, K.; Yamagami, W.; Kataoka, F.; et al. Patient-Derived Ovarian Cancer Organoids Capture the Genomic Profiles of Primary Tumours Applicable for Drug Sensitivity and Resistance Testing. Sci. Rep. 2020, 10, 12581. [Google Scholar] [CrossRef] [PubMed]
- Múnera, J.O.; Wells, J.M. Generation of Gastrointestinal Organoids from Human Pluripotent Stem Cells. Methods Mol. Biol. Clifton 2017, 1597, 167–177. [Google Scholar] [CrossRef]
- Voabil, P.; de Bruijn, M.; Roelofsen, L.M.; Hendriks, S.H.; Brokamp, S.; van den Braber, M.; Broeks, A.; Sanders, J.; Herzig, P.; Zippelius, A.; et al. An Ex Vivo Tumor Fragment Platform to Dissect Response to PD-1 Blockade in Cancer. Nat. Med. 2021, 27, 1250–1261. [Google Scholar] [CrossRef]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897.e11. [Google Scholar] [CrossRef] [Green Version]
- Kucab, J.E.; Hollstein, M.; Arlt, V.M.; Phillips, D.H. Nutlin-3a Selects for Cells Harbouring TP53 Mutations. Int. J. Cancer 2017, 140, 877–887. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e17. [Google Scholar] [CrossRef] [Green Version]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef]
- Shiihara, M.; Furukawa, T. Application of Patient-Derived Cancer Organoids to Personalized Medicine. J. Pers. Med. 2022, 12, 789. [Google Scholar] [CrossRef]
- Cattaneo, C.M.; Dijkstra, K.K.; Fanchi, L.F.; Kelderman, S.; Kaing, S.; van Rooij, N.; van den Brink, S.; Schumacher, T.N.; Voest, E.E. Tumor Organoid-T-Cell Coculture Systems. Nat. Protoc. 2020, 15, 15–39. [Google Scholar] [CrossRef]
- Valančiūtė, A.; Mathieson, L.; O’Connor, R.A.; Scott, J.I.; Vendrell, M.; Dorward, D.A.; Akram, A.R.; Dhaliwal, K. Phototherapeutic Induction of Immunogenic Cell Death and CD8+ T Cell-Granzyme B Mediated Cytolysis in Human Lung Cancer Cells and Organoids. Cancers 2022, 14, 4119. [Google Scholar] [CrossRef]
- Duval, K.; Grover, H.; Han, L.-H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiol. Bethesda Md 2017, 32, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Muraro, M.G.; Muenst, S.; Mele, V.; Quagliata, L.; Iezzi, G.; Tzankov, A.; Weber, W.P.; Spagnoli, G.C.; Soysal, S.D. Ex-Vivo Assessment of Drug Response on Breast Cancer Primary Tissue with Preserved Microenvironments. Oncoimmunology 2017, 6, e1331798. [Google Scholar] [CrossRef] [Green Version]
- Hirt, C.; Papadimitropoulos, A.; Muraro, M.G.; Mele, V.; Panopoulos, E.; Cremonesi, E.; Ivanek, R.; Schultz-Thater, E.; Droeser, R.A.; Mengus, C.; et al. Bioreactor-Engineered Cancer Tissue-like Structures Mimic Phenotypes, Gene Expression Profiles and Drug Resistance Patterns Observed “In Vivo”. Biomaterials 2015, 62, 138–146. [Google Scholar] [CrossRef]
- Manfredonia, C.; Muraro, M.G.; Hirt, C.; Mele, V.; Governa, V.; Papadimitropoulos, A.; Däster, S.; Soysal, S.D.; Droeser, R.A.; Mechera, R.; et al. Maintenance of Primary Human Colorectal Cancer Microenvironment Using a Perfusion Bioreactor-Based 3D Culture System. Adv. Biosyst. 2019, 3, e1800300. [Google Scholar] [CrossRef]
- Goliwas, K.F.; Marshall, L.E.; Ransaw, E.L.; Berry, J.L.; Frost, A.R. A Recapitulative Three-Dimensional Model of Breast Carcinoma Requires Perfusion for Multi-Week Growth. J. Tissue Eng. 2016, 7, 2041731416660739. [Google Scholar] [CrossRef]
- Pasini, A.; Lovecchio, J.; Cortesi, M.; Liverani, C.; Spadazzi, C.; Mercatali, L.; Ibrahim, T.; Giordano, E. Perfusion Flow Enhances Viability and Migratory Phenotype in 3D-Cultured Breast Cancer Cells. Ann. Biomed. Eng. 2021, 49, 2103–2113. [Google Scholar] [CrossRef]
- Rafaeva, M.; Horton, E.R.; Jensen, A.R.D.; Madsen, C.D.; Reuten, R.; Willacy, O.; Brøchner, C.B.; Jensen, T.H.; Zornhagen, K.W.; Crespo, M.; et al. Modeling Metastatic Colonization in a Decellularized Organ Scaffold-Based Perfusion Bioreactor. Adv. Healthc. Mater. 2022, 11, e2100684. [Google Scholar] [CrossRef]
- Martinez, A.; Buckley, M.S.; Scalise, C.B.; Wang, D.; Katre, A.A.; Birrer, M.J.; Berry, J.L.; Arend, R.C. Utilization of a 3-D Tissue Engineered Model to Investigate the Effects of Perfusion on Gynecologic Cancer Biology. J. Tissue Eng. 2021, 12, 20417314211055016. [Google Scholar] [CrossRef] [PubMed]
- Jasuja, H.; Kar, S.; Katti, D.R.; Katti, K.S. Perfusion Bioreactor Enabled Fluid-Derived Shear Stress Conditions for Novel Bone Metastatic Prostate Cancer Testbed. Biofabrication 2021, 13, 035004. [Google Scholar] [CrossRef] [PubMed]
- Trachtenberg, J.E.; Santoro, M.; Williams, C.; Piard, C.M.; Smith, B.T.; Placone, J.K.; Menegaz, B.A.; Molina, E.R.; Lamhamedi-Cherradi, S.-E.; Ludwig, J.A.; et al. Effects of Shear Stress Gradients on Ewing Sarcoma Cells Using 3D Printed Scaffolds and Flow Perfusion. ACS Biomater. Sci. Eng. 2018, 4, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Lamhamedi-Cherradi, S.-E.; Menegaz, B.A.; Ludwig, J.A.; Mikos, A.G. Flow Perfusion Effects on Three-Dimensional Culture and Drug Sensitivity of Ewing Sarcoma. Proc. Natl. Acad. Sci. USA 2015, 112, 10304–10309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usuludin, S.B.M.; Cao, X.; Lim, M. Co-Culture of Stromal and Erythroleukemia Cells in a Perfused Hollow Fiber Bioreactor System as an in Vitro Bone Marrow Model for Myeloid Leukemia. Biotechnol. Bioeng. 2012, 109, 1248–1258. [Google Scholar] [CrossRef]
- García-García, A.; Klein, T.; Born, G.; Hilpert, M.; Scherberich, A.; Lengerke, C.; Skoda, R.C.; Bourgine, P.E.; Martin, I. Culturing Patient-Derived Malignant Hematopoietic Stem Cells in Engineered and Fully Humanized 3D Niches. Proc. Natl. Acad. Sci. USA 2021, 118, e2114227118. [Google Scholar] [CrossRef]
- Shekarian, T.; Zinner, C.P.; Bartoszek, E.M.; Duchemin, W.; Wachnowicz, A.T.; Hogan, S.; Etter, M.M.; Flammer, J.; Paganetti, C.; Martins, T.A.; et al. Immunotherapy of Glioblastoma Explants Induces Interferon-γ Responses and Spatial Immune Cell Rearrangements in Tumor Center, but Not Periphery. Sci. Adv. 2022, 8, eabn9440. [Google Scholar] [CrossRef]
- Di Maggio, N.; Piccinini, E.; Jaworski, M.; Trumpp, A.; Wendt, D.J.; Martin, I. Toward Modeling the Bone Marrow Niche Using Scaffold-Based 3D Culture Systems. Biomaterials 2011, 32, 321–329. [Google Scholar] [CrossRef]
- Guller, A.E.; Grebenyuk, P.N.; Shekhter, A.B.; Zvyagin, A.V.; Deyev, S.M. Bioreactor-Based Tumor Tissue Engineering. Acta Naturae 2016, 8, 44–58. [Google Scholar] [CrossRef]
- Leung, C.M.; de Haan, P.; Ronaldson-Bouchard, K.; Kim, G.-A.; Ko, J.; Rho, H.S.; Chen, Z.; Habibovic, P.; Jeon, N.L.; Takayama, S.; et al. A Guide to the Organ-on-a-Chip. Nat. Rev. Methods Primer 2022, 2, 1–29. [Google Scholar] [CrossRef]
- Yoon, P.S.; Del Piccolo, N.; Shirure, V.S.; Peng, Y.; Kirane, A.; Canter, R.J.; Fields, R.C.; George, S.C.; Gholami, S. Advances in Modeling the Immune Microenvironment of Colorectal Cancer. Front. Immunol. 2020, 11, 614300. [Google Scholar] [CrossRef]
- Nguyen, M.; De Ninno, A.; Mencattini, A.; Mermet-Meillon, F.; Fornabaio, G.; Evans, S.S.; Cossutta, M.; Khira, Y.; Han, W.; Sirven, P.; et al. Dissecting Effects of Anti-Cancer Drugs and Cancer-Associated Fibroblasts by On-Chip Reconstitution of Immunocompetent Tumor Microenvironments. Cell Rep. 2018, 25, 3884–3893.e3. [Google Scholar] [CrossRef] [Green Version]
- Shirure, V.S.; Bi, Y.; Curtis, M.B.; Lezia, A.; Goedegebuure, M.M.; Goedegebuure, S.P.; Aft, R.; Fields, R.C.; George, S.C. Tumor-on-a-Chip Platform to Investigate Progression and Drug Sensitivity in Cell Lines and Patient-Derived Organoids. Lab. Chip 2018, 18, 3687–3702. [Google Scholar] [CrossRef]
- Bi, Y.; Shirure, V.S.; Liu, R.; Cunningham, C.; Ding, L.; Meacham, J.M.; Goedegebuure, S.P.; George, S.C.; Fields, R.C. Tumor-on-a-Chip Platform to Interrogate the Role of Macrophages in Tumor Progression. Integr. Biol. 2020, 12, 221–232. [Google Scholar] [CrossRef]
- Parlato, S.; De Ninno, A.; Molfetta, R.; Toschi, E.; Salerno, D.; Mencattini, A.; Romagnoli, G.; Fragale, A.; Roccazzello, L.; Buoncervello, M.; et al. 3D Microfluidic Model for Evaluating Immunotherapy Efficacy by Tracking Dendritic Cell Behaviour toward Tumor Cells. Sci. Rep. 2017, 7, 1093. [Google Scholar] [CrossRef] [Green Version]
- Ayuso, J.M.; Virumbrales-Muñoz, M.; Lacueva, A.; Lanuza, P.M.; Checa-Chavarria, E.; Botella, P.; Fernández, E.; Doblare, M.; Allison, S.J.; Phillips, R.M.; et al. Development and Characterization of a Microfluidic Model of the Tumour Microenvironment. Sci. Rep. 2016, 6, 36086. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-Cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Aref, A.R.; Campisi, M.; Ivanova, E.; Portell, A.; Larios, D.; Piel, B.P.; Mathur, N.; Zhou, C.; Coakley, R.V.; Bartels, A.; et al. 3D Microfluidic Ex Vivo Culture of Organotypic Tumor Spheroids to Model Immune Checkpoint Blockade. Lab. Chip 2018, 18, 3129–3143. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of Clinical Trial Success Rates and Related Parameters. Biostat. Oxf. Engl. 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Wernitznig, D.; Meier-Menches, M.S.; Cseh, K.; Theiner, S.; Wenisch, D.; Schweikert, A.; Jakupec, A.M.; Koellensperger, G.; Wernitznig, A.; Sommergruber, W.; et al. Plecstatin-1 Induces an Immunogenic Cell Death Signature in Colorectal Tumour Spheroids. Metallomics 2020, 12, 2121–2133. [Google Scholar] [CrossRef] [PubMed]
- Etminan, N.; Peters, C.; Lakbir, D.; Bünemann, E.; Börger, V.; Sabel, M.C.; Hänggi, D.; Steiger, H.-J.; Stummer, W.; Sorg, R.V. Heat-Shock Protein 70-Dependent Dendritic Cell Activation by 5-Aminolevulinic Acid-Mediated Photodynamic Treatment of Human Glioblastoma Spheroids in Vitro. Br. J. Cancer 2011, 105, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.D.; Bano, S.; Pogue, B.W.; Wang, K.K.; Maytin, E.V.; Hasan, T. Photodynamic Priming with Triple-Receptor Targeted Nanoconjugates That Trigger T Cell-Mediated Immune Responses in a 3D in Vitro Heterocellular Model of Pancreatic Cancer. Nanophotonics 2021, 10, 3199–3214. [Google Scholar] [CrossRef]
- Sebrell, T.A.; Hashimi, M.; Sidar, B.; Wilkinson, R.A.; Kirpotina, L.; Quinn, M.T.; Malkoç, Z.; Taylor, P.J.; Wilking, J.N.; Bimczok, D. A Novel Gastric Spheroid Co-Culture Model Reveals Chemokine-Dependent Recruitment of Human Dendritic Cells to the Gastric Epithelium. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 157–171.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyga, A.; Neves, J.; Stamati, K.; Loizidou, M.; Emberton, M.; Cheema, U. The next Level of 3D Tumour Models: Immunocompetence. Drug Discov. Today 2016, 21, 1421–1428. [Google Scholar] [CrossRef]
- Weydert, Z.; Lal-Nag, M.; Mathews-Greiner, L.; Thiel, C.; Cordes, H.; Küpfer, L.; Guye, P.; Kelm, J.M.; Ferrer, M. A 3D Heterotypic Multicellular Tumor Spheroid Assay Platform to Discriminate Drug Effects on Stroma versus Cancer Cells. SLAS Discov. 2020, 25, 265–276. [Google Scholar] [CrossRef]
- Grönholm, M.; Feodoroff, M.; Antignani, G.; Martins, B.; Hamdan, F.; Cerullo, V. Patient-Derived Organoids for Precision Cancer Immunotherapy. Cancer Res. 2021, 81, 3149–3155. [Google Scholar] [CrossRef]
- Biselli, E.; Agliari, E.; Barra, A.; Bertani, F.R.; Gerardino, A.; De Ninno, A.; Mencattini, A.; Di Giuseppe, D.; Mattei, F.; Schiavoni, G.; et al. Organs on Chip Approach: A Tool to Evaluate Cancer -Immune Cells Interactions. Sci. Rep. 2017, 7, 12737. [Google Scholar] [CrossRef]
- Tumor and Lymph Node on Chip for Cancer Studies | Tumor-LN-OC Project | Fact Sheet | H2020. Available online: https://cordis.europa.eu/project/id/953234 (accessed on 7 October 2022).
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus Guidelines for the Definition, Detection and Interpretation of Immunogenic Cell Death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [Green Version]
- Aaes, T.L.; Verschuere, H.; Kaczmarek, A.; Heyndrickx, L.; Wiernicki, B.; Delrue, I.; De Craene, B.; Taminau, J.; Delvaeye, T.; Bertrand, M.J.M.; et al. Immunodominant AH1 Antigen-Deficient Necroptotic, but Not Apoptotic, Murine Cancer Cells Induce Antitumor Protection. J. Immunol. 2020, 204, 775–787. [Google Scholar] [CrossRef]



| Model | Advantages | Disadvantages |
|---|---|---|
| Phagocytosis (efferocytosis) | Direct evidence of interaction and/or dying/dead cancer cell engulfment | Limited (to the surface area and cell variety) and artificial environment of the phagocytosis assay (culture media concentrated with SNs of the dying/dead cancer cells). Highly dependent on the ratios of dead/dying cells towards the APCs and time of their co-culture. |
| Maturation/activation of APCs Polarization of macrophages | Direct evidence (on morphological, genetic, protein, and physiological levels) of immune response towards the (treated) dying/dead cancer cells | Similar to the phagocytosis. |
| Alteration of cancer cell metabolism | Closer recreation of TME conditions (low glucose, hypoxia, etc.) | Time- (chronic exposure of cells is needed to switch to hypoxic/acidic metabolism) and labor-intensive (special amino and fatty acids with deprived or conditioned media). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krysko, D.V.; Demuynck, R.; Efimova, I.; Naessens, F.; Krysko, O.; Catanzaro, E. In Vitro Veritas: From 2D Cultures to Organ-on-a-Chip Models to Study Immunogenic Cell Death in the Tumor Microenvironment. Cells 2022, 11, 3705. https://doi.org/10.3390/cells11223705
Krysko DV, Demuynck R, Efimova I, Naessens F, Krysko O, Catanzaro E. In Vitro Veritas: From 2D Cultures to Organ-on-a-Chip Models to Study Immunogenic Cell Death in the Tumor Microenvironment. Cells. 2022; 11(22):3705. https://doi.org/10.3390/cells11223705
Chicago/Turabian StyleKrysko, Dmitri V., Robin Demuynck, Iuliia Efimova, Faye Naessens, Olga Krysko, and Elena Catanzaro. 2022. "In Vitro Veritas: From 2D Cultures to Organ-on-a-Chip Models to Study Immunogenic Cell Death in the Tumor Microenvironment" Cells 11, no. 22: 3705. https://doi.org/10.3390/cells11223705