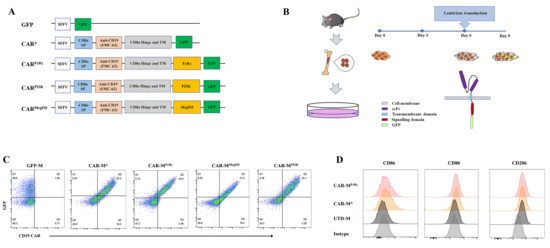
3.1. Generation of Three Different CAR-Ms and Comparison of their Phagocytic and Killing Functions
The choice of intracellular domain is of great importance when designing CAR-M. The validation of potent signaling domain on primary macrophage is an essential step towards a possible therapeutic option. Therefore, we sought to screen the best intracellular domain on primary murine macrophages, BMDM.
To this end, we first optimized the culture conditions to obtain a high number of BMDM. The concentrations of FBS and murine M-CSF were used as main determining factors for optimization. The results showed that RPMI-1640 medium with 20% FBS and 20 ng/mL M-CSF was the best medium condition, in which the largest number of BMDM was harvested (
Figure S1A,B). The purity of BMDM was around 99% as determined by F4/80 and CD11b expression (
Figure S1C). To identify the best cytoplasmic domain capable of promoting phagocytosis and cytotoxicity, three effective phagocytic receptor intracellular domains were selected as components of CARs, which are CAR
FcRγ, CAR
Megf10, and the cytoplasmic domain that recruits the p85 subunit of PI3K (CAR
PI3K). All the three selected intracellular domains have been previously validated to promote phagocytosis of antigen-ligated beads on CAR-macrophage cell lines [
3]. CD3ζ intracellular domain was used on CAR-M in the clinical trial. We did not include CD3ζ in our comparation list, since CD3ζ and FcRγ have been demonstrated to be functionally similar on human primary macrophage [
5], and CD3ζ is not naturally present in macrophage. The three selected intracellular domains were fused with a CD8α signal peptide, FMC63 scFv specifically binding to human CD19, CD8α hinge and transmembrane domain and GFP tag. Additionally, a construct without any intracellular domain (CAR
Δ) and a construct with only a GFP tag were generated as controls (
Figure 1A). All the CARs can be highly expressed on BMDM by means of the established CAR-M production platform (
Figure 1B,C). Then, a CAR positive rate of 30% on CAR-M was set as a cutoff for further activity test. In addition, macrophage polarization M1 and M2 surface markers (CD86, CD80, CD206) were analyzed for macrophages with or without lentivirus transduction. The data showed that the BMDM differentiated and matured using M-CSF expressed CD206, which was consistent with the report that M-CSF stimulation induces an M2-like phenotype in macrophages [
16]. Compared with untransduced macrophage (UTD-M), CAR-M transduced by lentivirus possessed more M1 features with more CD86 and CD80 expression (
Figure 1D;
Figure S1D). It implies that the lentiviral vector used to transduce macrophages with CAR probably induce CAR-M M1 phenotype polarization.
The phagocytic and killing capacity of CAR-Ms to antigen-positive tumor cells are the most direct and convincing indicators for comparing different CARs. Microscopy- and FACS-based phagocytosis assessment of CAR-Ms towards cancerous Raji cells that express high levels of CD19 was performed. CAR-M cells normalized for transduction efficiency were used in these assays. All three CAR-Ms were able to trigger engulfment of Raji cells. CAR-M
FcRγ and CAR-M
PI3K could engulf more target cells than CAR-M
Megf10, while CAR-M
FcRγ and CAR-M
PI3K showed comparable phagocytic capacity (
Figure 2A–D). Cytotoxicity of CAR-Ms against Raji was performed using the luciferase-based and FACS counting-based killing assay. The three CAR-Ms also showed potent cytotoxicity against Raji, and CAR-M
FcRγ was demonstrated to be the most effective (
Figure 2E,F). CAR-M
FcRγ performed better than CAR-M
PI3K in terms of killing capacity, though they had comparable phagocytic capacity. CAR-M can initiate both whole cell eating and trogocytosis leading to cancer cell elimination [
3], and our killing assays do not distinguish between whole cell engulfment or death following trogocytosis.
After investigation of CAR constructs with different intracellular domains, we found that CAR-M
FcRγ has the most potent phagocytic and killing capacity to Raji cells among the tested three CAR-Ms. To validate that CAR-M
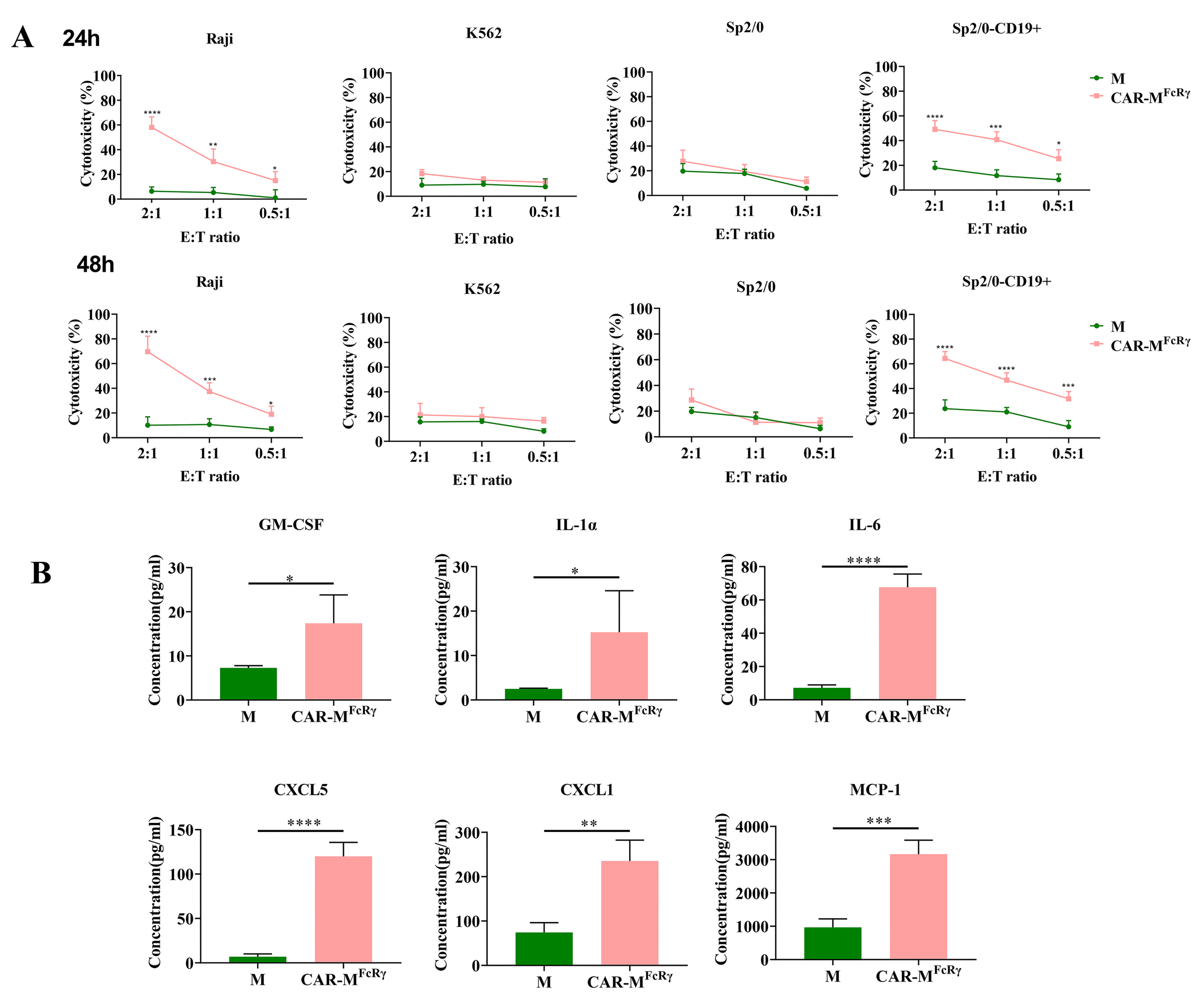
FcRγ triggers antigen-specific cellular killing effects, Raji, human CD19-negative tumor cell K562, murine cancer cell Sp2/0, and human CD19-overexpressed Sp2/0 (Sp2/0-CD19+) cells were used as target cells for cytotoxicity assessment. The killing assay was performed at different time points (24 h and 48 h) and different E:T ratios (2, 1 or 0.5). The data indicate that CAR-M
FcRγ exerted potent specific cytotoxicity function in a dose- and time-dependent manner against CD19-positive cells compared with the GFP-M (M) control (
Figure 3A). Additionally, a panel of 18 cytokine/chemokine (GM-CSF, IFN-γ, IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-10, IL-12(p70), IL-13, CXCL5, IL-17A, CXCL1, MCP-1, MIP-2, TNF-α) analysis was performed for the supernatant of M control or CAR-M
FcRγ and Raji coculture system. The levels of GM-CSF, IL-1α, IL-6, CXCL5, CXCL1 and MCP-1 in the CAR-M
FcRγ group were significantly higher than the M control in various degrees (
Figure 3B), while the other cytokines/chemokines did not display significant difference (data not shown). These data indicate that CAR-M
FcRγ were activated by Raji cells and induced an inflammatory response. The significantly increased proinflammatory cytokines and chemokines could be beneficial for antitumor immunity. Moreover, Klichinsky et al. [
5] have shown that human CAR-M with a CD3ζ intracellular domain generated by an Ad5-F35 vector can reprogram the tumor microenvironment (TME) by releasing proinflammatory cytokines that can activate innate immune cells and achieve a remarkable antitumor effect in vivo.
3.2. CAR-M and CAR-T Cells Demonstrated Synergistic Cytotoxicity
Despite the effective tumoricidal activity of CAR-M, given the complexity and difficulty of treating cancer, rationally combining CAR-M therapy with other complementary immunotherapy is a potential strategy in combating cancer [
17]. The use of combination therapy to simultaneously target different mechanisms of action has proven to be a viable approach to treat cancer [
18]. CAR-M has potential advantages in homing and infiltrating to solid tumor and can release proinflammatory cytokines to improve TME. Despite rapid advances in CAR-T immunotherapy, clinical responses in solid tumors have been limited. The major obstacles of CAR-T cell therapy to treat solid tumors include limited infiltration into the dense extracellular matrix of tumor and exhaustion in immunosuppressive TME [
19]. It is generally accepted that CAR-M could leverage the natural tumor-homing ability of myeloid cells to enter solid tumors, which is superior to CAR-T [
20]. Though the comparison of the homing capacity between CAR-M and CAR-T has not been confirmed experimentally, it is foreseen in future studies. Moreover, the macrophage has the potential to promote T cell activation. Therefore, it is likely that CAR-M and CAR-T can complement each other and significantly augment tumor responses. Considering the very limited number of tumor-specific antigens clinically, the combination of CAR-M and CAR-T with CAR targeting the same tumor antigen is realistic. On the other hand, trogocytosis occurs during CAR-T killing target tumor cells, in which the target antigen is transferred to CAR-T cells, thereby abating T cell activity by promoting fratricide CAR-T-cell killing [
21]. It is uncertain whether CAR-M would attack antigen-positive CAR-T cells and lessen the overall tumoricidal activity when CAR-M and CAR-T were combined. Therefore, the real experiment needs to be performed to investigate whether the combination of CAR-T and CAR-M targeting the same tumor antigen have a synergistic, additive, or antagonistic effect against tumors.
Murine CD19 CAR-Ts with a classic second-generation CAR structure (
Figure S2) were prepared for the combinational study with CAR-M
FcRγ against Raji cells. Untransduced T and GFP-M (M) cells were employed as controls. Immune effector cells (T, CAR-T, M and CAR-M
FcRγ) were added either alone or in combination with the counterpart effector cells to kill Raji cells. Of interest, CAR-T showed more potent cytotoxicity than CAR-M
FcRγ at the same E:T ratio (E:T = 1). CAR-T + CAR-M
FcRγ combination significantly augmented the cytotoxicity compared with CAR-T or CAR-M
FcRγ alone, in which this increase exceeded simple additive effects. Of note, the 0.5 (CAR-M
FcRγ + CAR-T) group, which combined half the dose of CAR-T alone and CAR-M
FcRγ alone group (0.5 + 0.5), demonstrated significantly greater killing ability than CAR-T or CAR-M
FcRγ alone as well (
Figure 4A). This greater-than-expected tumor killing indicates that CAR-M
FcRγ and CAR-T have synergistic effect against Raji cells. In addition to CAR-M
FcRγ, the combination of CAR-T with other CAR-Ms (namely CAR-M
Megf10 and CAR-M
PI3K) were assessed as well. CAR-T also synergized with other CAR-Ms in terms of killing Raji using the luciferase-based cytotoxicity assay (
Figure S3A). Moreover, synergistic effect occurred similarly after the coculture with the murine cancer cell, Sp2/0-CD19+ (
Figure S3B). Taken together, these data demonstrate for the first time that CAR-M synergize with CAR-T therapy against tumor cells. The combination approach offers a novel strategy for treating cancer.
3.3. The Mechanism Underlying the Synergistic Effect of CAR-M and CAR-T
Clarifying the mechanism underlying the synergistic effect of CAR-M and CAR-T is beneficial for the further development of the combination therapy. We hypothesized that the cytokines released by CAR-T or CAR-M may be involved in the synergy. To explore the possible mechanism, the supernatants of T or CAR-T with the Raji cells coculture system after 48 h of killing, hereafter referred to as (T)s or (CAR-T)s, were collected to supplement the coculture system of M or CAR-M
FcRγ killing Raji. Similarly, the supernatants of M or CAR-M
FcRγ with Raji cells coculture system after 48 h of killing, hereafter referred to as (M)s or (CAR-M
FcRγ)s, were collected to supplement the coculture system of T or CAR-T killing Raji. The cytotoxicity of (CAR-T)s and (CAR-M
FcRγ)s alone were also performed to exclude their possible direct killing ability against Raji. After killing for 48 h, the results showed that CAR-M
FcRγ exposing to (CAR-T)s achieved more potent activity than CAR-M
FcRγ alone, while CAR-T exposing to (CAR-M
FcRγ)s did not improve the killing ability. Moreover, (CAR-T)s or (CAR-M
FcRγ)s alone showed no potent direct cytotoxicity (
Figure 4B). To further validate the effect of (CAR-T)s on CAR-M
FcRγ, M or CAR-M
FcRγ were exposed to (T)s or (CAR-T)s for 24 h followed by the removal of cell culture supernatants. The cytotoxicity assay was performed at different E:T ratios. CAR-M
FcRγ exposed to (CAR-T)s demonstrated more potent cytotoxicity than untreated and (T)s treated CAR-M
FcRγ. M treated by (CAR-T)s also showed slightly enhanced cytotoxicity, but to a less extent than CAR-M (
Figure 4C). This also indicates that (CAR-T)s pretreatment enhanced the killing ability of M, while a CAR can amplify this effect. The similar phenomenon was also observed using the murine cancer cell Sp2/0-CD19+ as the target cell (
Figure S3C). Taken together, CAR-T-derived cytokines can enhance the killing activity of CAR-M, which contributes to the synergistic effect of CAR-M and CAR-T.
Additionally, the polarization phenotype of CAR-M from killing assay in combination with CAR-T was analyzed. The expression of CD86 and CD80 on CAR-M
FcRγ was significantly upregulated after the coculture with CAR-T and Raji for 48 h, while CD206 was downregulated (
Figure 4D;
Figure S4A). However, Raji cells alone could not significantly trigger the upregulation of CD80 or CD86 on CAR-M
FcRγ. The similar trend was observed when CAR-M were treated with (CAR-T)s for 24 h (
Figure S4B,C). This suggests that CAR-M were polarized to a more M1 phenotype by CAR-T cell-derived cytokines. Inflammatory stimuli (e.g., IFN-γ, GM-CSF) have been shown to polarize macrophages toward a M1 phenotype, that favors the elimination of tumor cells [
22]. We also observed a high level of IFN-γ and GM-CSF secreted by CAR-T during killing Raji cells (
Figure 4E). These data reveal that the inflammatory cytokines (including but not limited to IFN-γ and GM-CSF) released by CAR-T during killing tumor cells probably polarize CAR-M toward a more M1 phenotype, which augments the cytotoxicity of CAR-M.
Given that inflammatory stimuli augment the ability of macrophages to phagocytose and destroy cancer cells, we sought to take advantage of the principle to design more potent CAR-M. A very recent mechanism study demonstrated that CD18 of macrophage is crucial for the increased phagocytosis and antitumor activity in inflammatory settings [
22]. Therefore, we sought to improve the killing capacity of CAR
FcRγ by assembling the intracellular domain of CD18 with FcRγ in a tandem array to construct CAR
FcRγ-CD18 (
Figure S5A). The cytotoxicity of CAR-M
FcRγ-CD18 and CAR-M
FcRγ were compared in parallel. However, an additional CD18 intracellular domain failed to augment the cytotoxicity (
Figure S5B). The transmembrane domain and/or extracellular domain of CD18 may be necessary for the improved phagocytic capacity of inflammatory macrophages. Further investigation is required to determine the precise cause of this unexpected outcome.
CD86 and CD80 on the macrophage are the ligands of costimulatory molecule CD28 on CAR-T cells, which contribute a positive costimulatory signal during T cell priming and activation [
23]. The upregulated CD86 and CD80 on CAR-M induced by CAR-T secreted cytokines may amplify the magnitude of CAR-driven signals and mediate a set of signaling events that influence CAR-T cell killing ability in turn. Furthermore, the cytokine/chemokine analysis showed that IFN-γ, IL-1β, CXCL1, MIP-2, IL-6 and MCP-1 were significantly increased when CAR-T and CAR-M
FcRγ were combined (
Figure 4F), indicating that the interaction of CAR-M and CAR-T may encourage mutual activation and can form an inflammatory TME. On the other hand, attention should be paid to the possible cytokine storm resulted from the combination of CAR-M and CAR-T in the clinic in the future. It has been shown that the main cytokine responsible for Cytokine Release Syndrome (CRS) in recipients of CAR-T is IL-6 produced by recipient macrophages cells [
24]. Maybe the only way to evaluate a potential beneficial or detrimental effect of CAR-M cytokine secretion will be with in vivo testing.
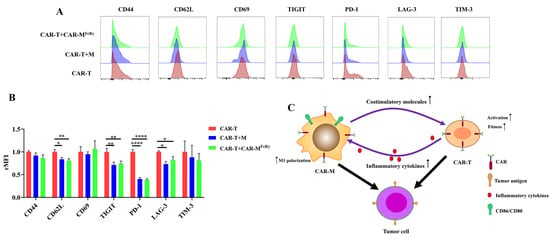
To further explore the impact of CAR-M on CAR-T cells in detail, activation/memory markers (CD44, CD62L, and CD69) and exhaustion markers (PD-1, TIM-3, TIGIT, and LAG-3) on CAR-T cells were analyzed after exposure to Raji alone or in the presence of CAR-M
FcRγ or M. The expression of CD62L of CAR-T in the presence of CAR-M
FcRγ or M significantly decreased (
Figure 5A,B). CD62L has be used as a marker of T cell activation [
25]. It suggests that CAR-M or M may facilitate the activation of CAR-T cells. In addition, the presence of CAR-M
FcRγ or M decreased the expression of exhaustion markers PD-1, TIGIT, and LAG-3 of CAR-T cells, which indicates that the fitness of CAR-T was improved by CAR-M
FcRγ after exposure to Raji in vitro (
Figure 5A,B). Taken together, the presence of CAR-M
FcRγ is beneficial for CAR-T to kill tumor cells, through promoting the level of activation and fitness of CAR-T after exposure to target tumor cells. It could be due to the upregulated CD28 ligands, CD86 and CD80, on the macrophage [
23]. The detailed mechanism underlying this phenomenon awaits further study.
Overall, the synergistic effect of CAR-M and CAR-T against tumor cells probably depend on a feedback loop triggered by the activation of CAR-T. The inflammatory factors secreted by CAR-T increase the expression of costimulatory ligands (CD86 and CD80) on CAR-M and augment the cytotoxicity of CAR-M by inducing macrophage to M1 polarization. The upregulated costimulatory ligands may promote the fitness and activation of CAR-T cells in turn, ultimately achieving significantly enhanced cytotoxicity (
Figure 5C). Considering the mechanism underlying the synergistic effect of CAR-T and CAR-M in our study, the use of CAR-M and other second generation CAR-T cells (e.g., 41BB-CD3ζ CAR-T) could show the similar synergistic effect. The presence of the costimulatory domain of CAR-T probably play an important role in the synergistic effect, since the costimulatory domain contributes to CAR-T cells persistence and the sustained high level of inflammatory factors released by CAR-T during the killing of tumor cells [
26].
In this study, we revealed that CAR-M and CAR-T cells synergistically kill tumor cells in vitro and provided rationale for the combination of CAR-M with CAR-T to treat cancer. Despite the achievements made, as the tumor microenvironment in vivo is highly intricate, further studies are required to evaluate the synergistic effect of CAR-M and CAR-T in vivo. Moreover, the detailed analysis of the tumor immune microenvironment after the CAR-T and CAR-M combinational treatment in a syngeneic immunocompetent animal model can deepen our understanding to adoptive immune-cell therapy, which may aid in developing more effective therapies. Additionally, the combination of CAR-M and CAR-T targeting different tumor antigens probably demonstrate a more profound synergistic effect, in which tumor cells with either of two different antigens could be directly killed and the fratricidal killing due to CAR-T cell trogocytosis may be limited [
21]. The potential drawback of the combination of CAR-M and CAR-T targeting different tumor antigens in the clinic could be the safety issue, given the limited number of tumor specific antigens.
Although CAR-M therapy has shown its effective antitumor ability in animal experiments, it also has many shortcomings to be overcome. Therefore, future effort should be given to maximize the effectiveness and safety of CAR-M in clinical treatment. First, the previously reported achievements, including our study, are all based on the first generation of CAR-M, in which the intracellular signaling is mediated by a single effector domain [
3,
5,
6,
7]. Therefore, CAR structure can be optimized by incorporating tandem activation domains or proinflammatory cytokines (e.g., IFN-γ, GM-CSF) to enhance its effectiveness [
27]. Multiantigen logic gates or drug-sensitive modules can also be designed to engineer CAR-M to improve its safety. Additionally, considering the laborious and costly process of ex vivo autologous CAR-M cell production, in vivo programming of the macrophage into CAR-M using nonviral delivery of CAR-encoding DNA or mRNA is a promising strategy [
28,
29]. Lastly, combining CAR-M therapy with other therapeutics will be of great benefit to patients, especially those with high tumor burden. In addition to CAR-T therapy in this study, chemotherapies, CD47/SIRPa antibodies, T cell checkpoint inhibitors, or radiation therapy can also be carefully evaluated for the possible synergy with CAR-M, aiming to significantly improve the overall therapeutic efficacy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




