
Survival Pathways of HIF-Deficient Tumour Cells: TCA Inhibition, Peroxisomal Fatty Acid Oxidation Activation and an AMPK-PGC-1α Hypoxia Sensor

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Method
2.1. Cell Culture
2.2. Metabolite and Enzyme Assays
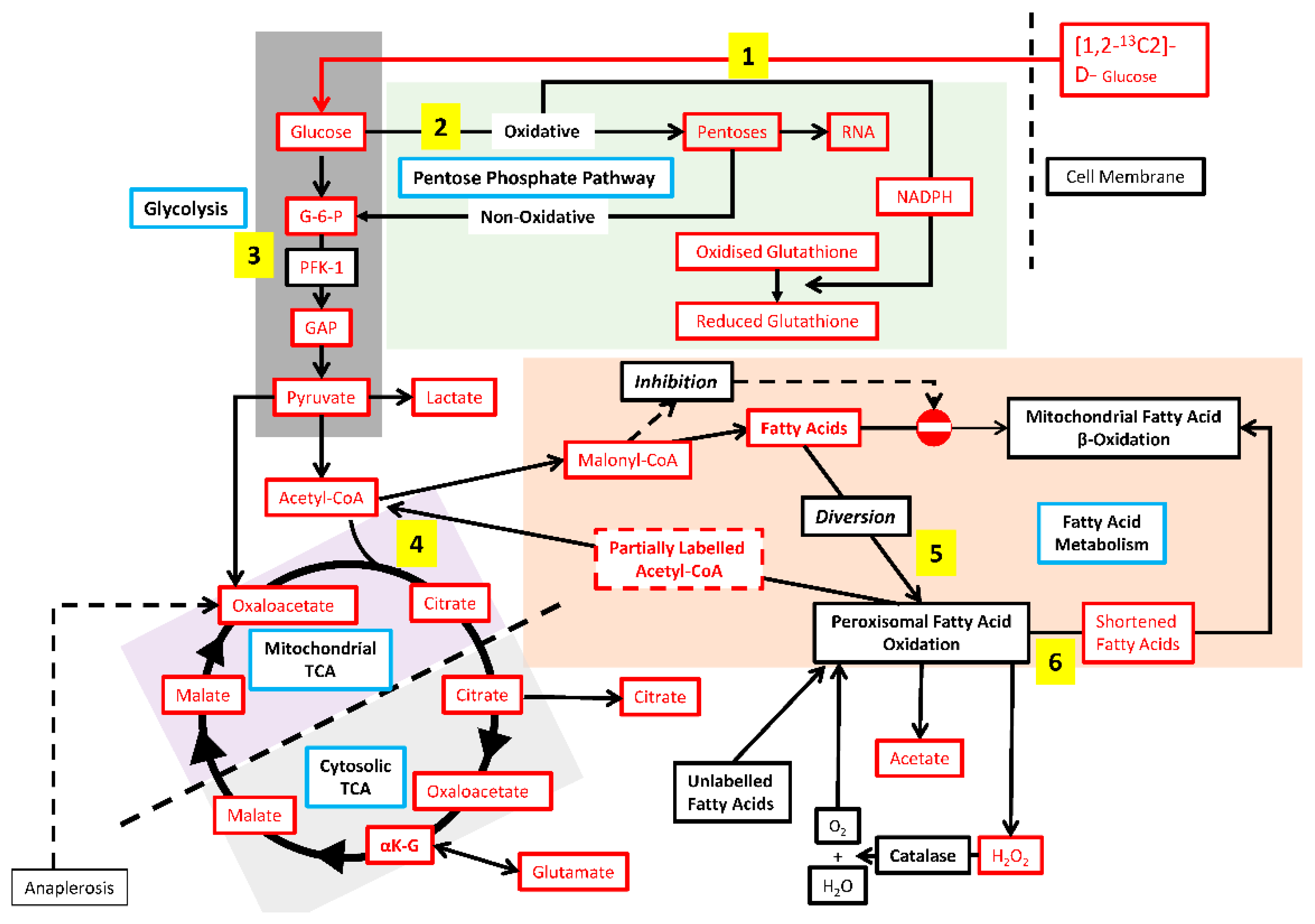
2.3. 13C tracer Studies
2.4. TaqMan Gene Expression and ELISA Measurements
2.5. Data Analysis and Statistical Methods
3. Results
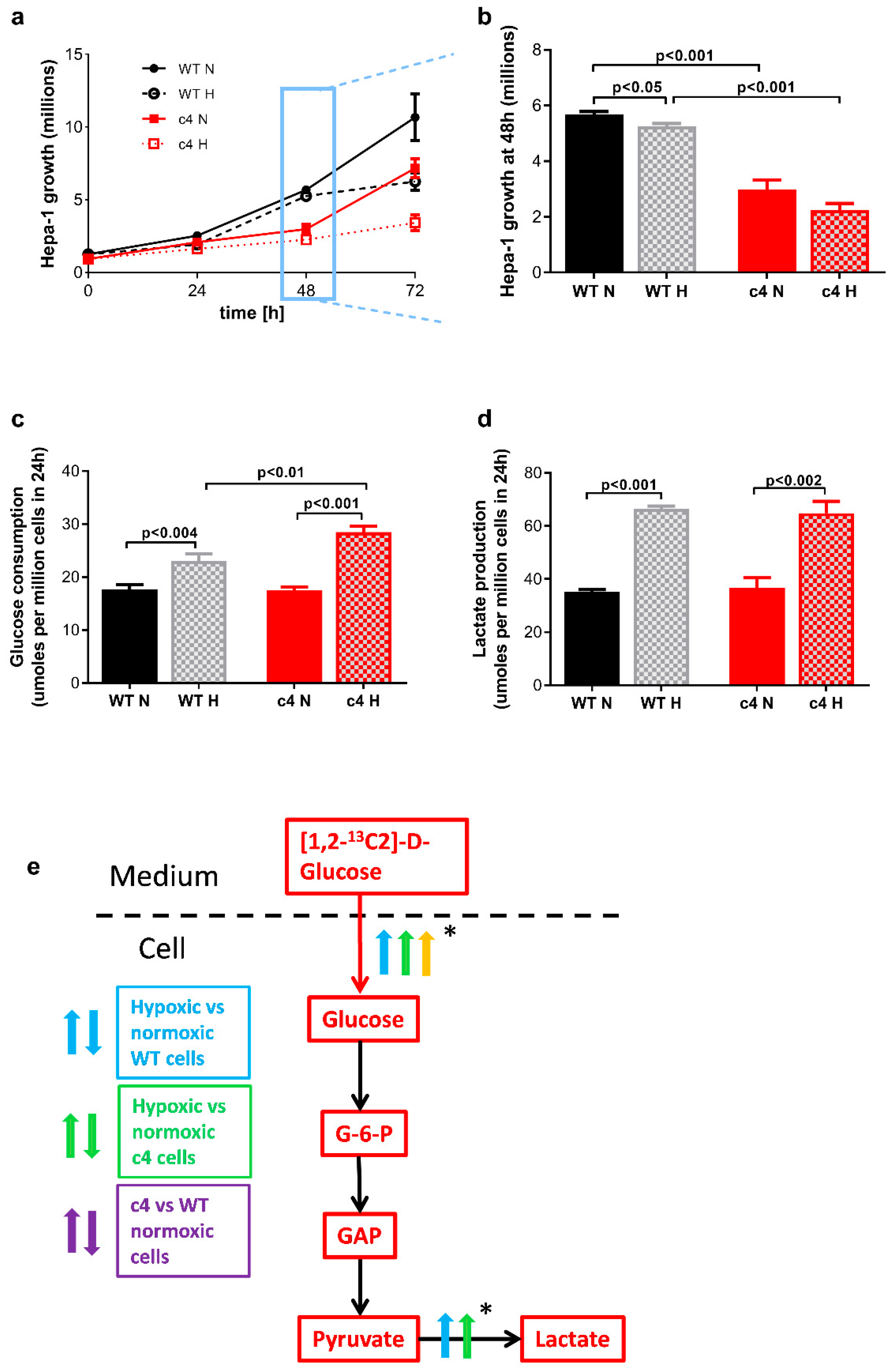
3.1. Hepa-1 HIF-1β-Deficient Cells Are Capable of Growth in Hypoxia
3.2. Glucose Consumption and Lactate Production Increases in Response to Hypoxia in HIF-1β-Deficient Hepa-1 Cells
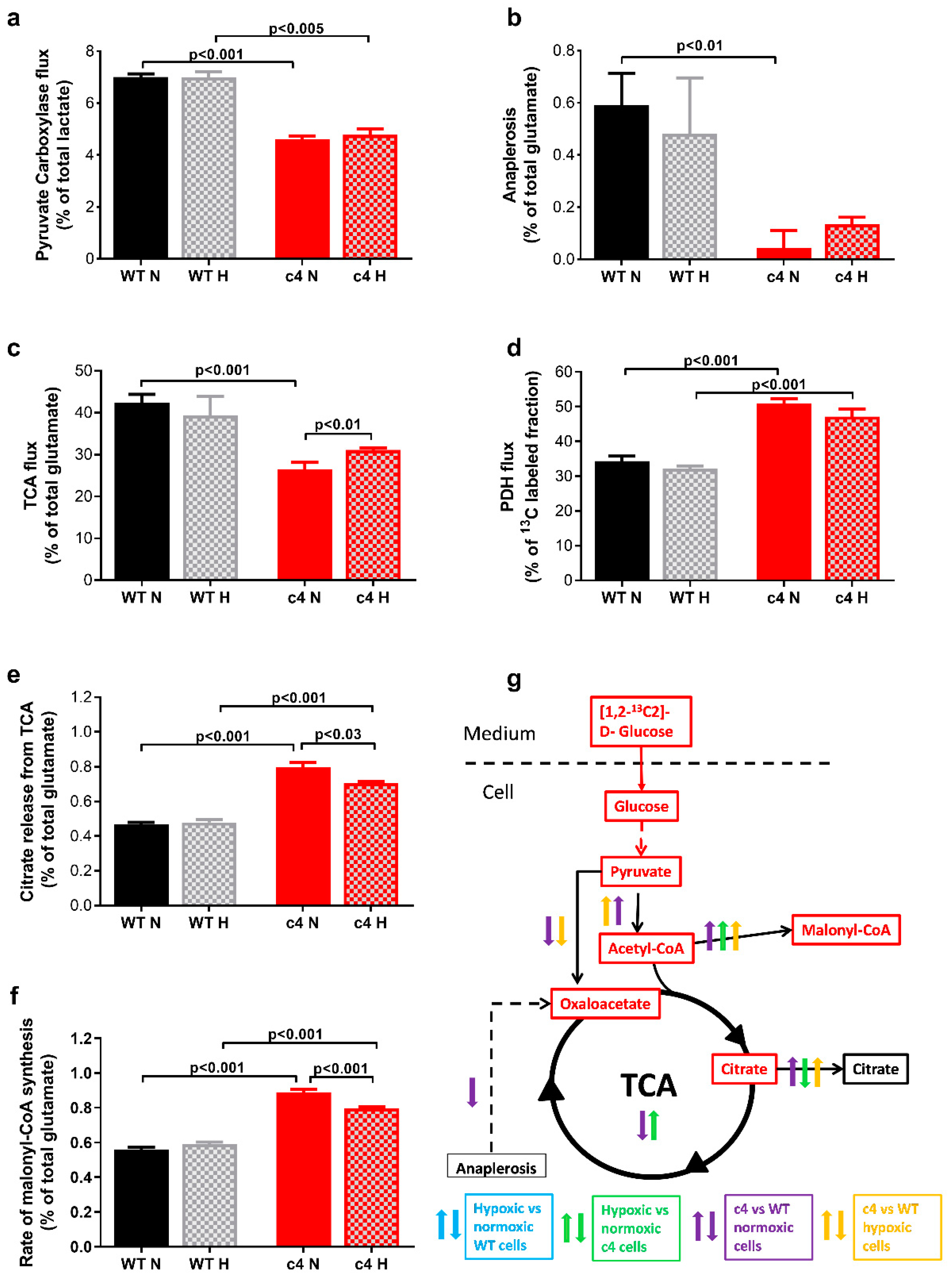
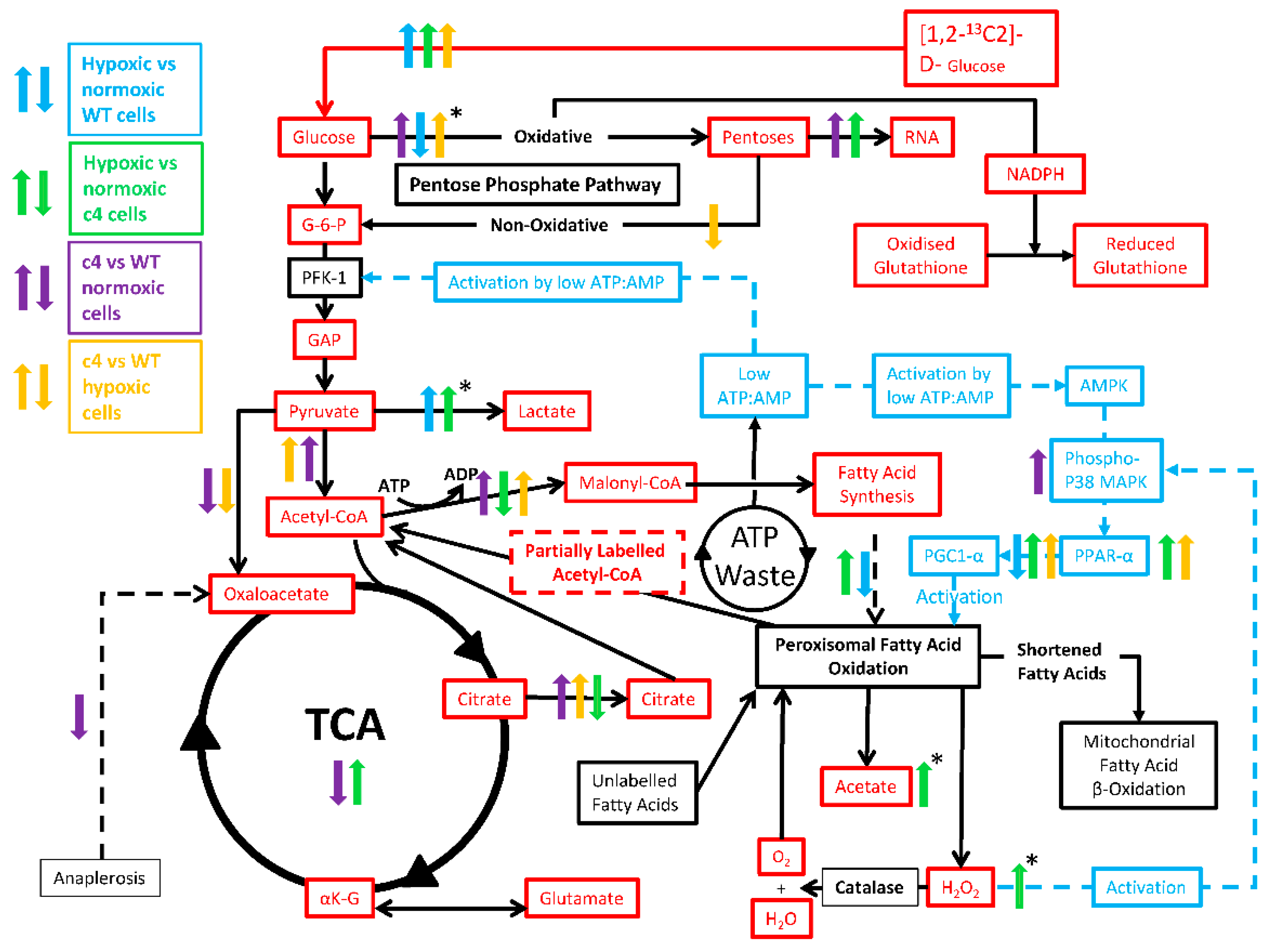
3.3. Hepa-1 HIF-1β-Deficient Cells Downregulate Their TCA Cycle Flux, Shuttle Citrate out, and Channel Carbons toward Malonyl-CoA Synthesis
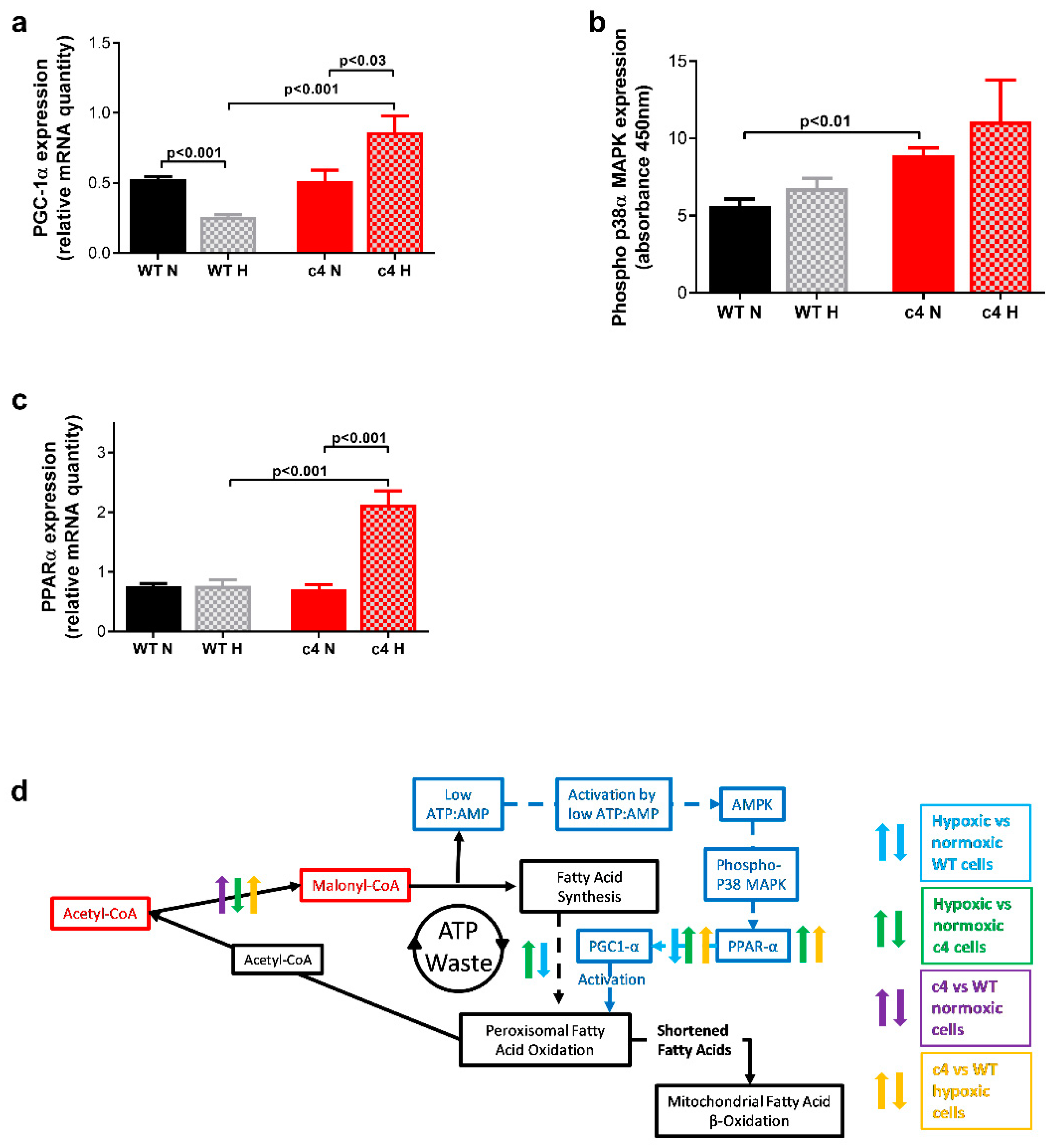
3.4. Hepa-1 HIF-1β-Deficient Cells Show Increased Signalling via AMPK-PPARα-PGC1α
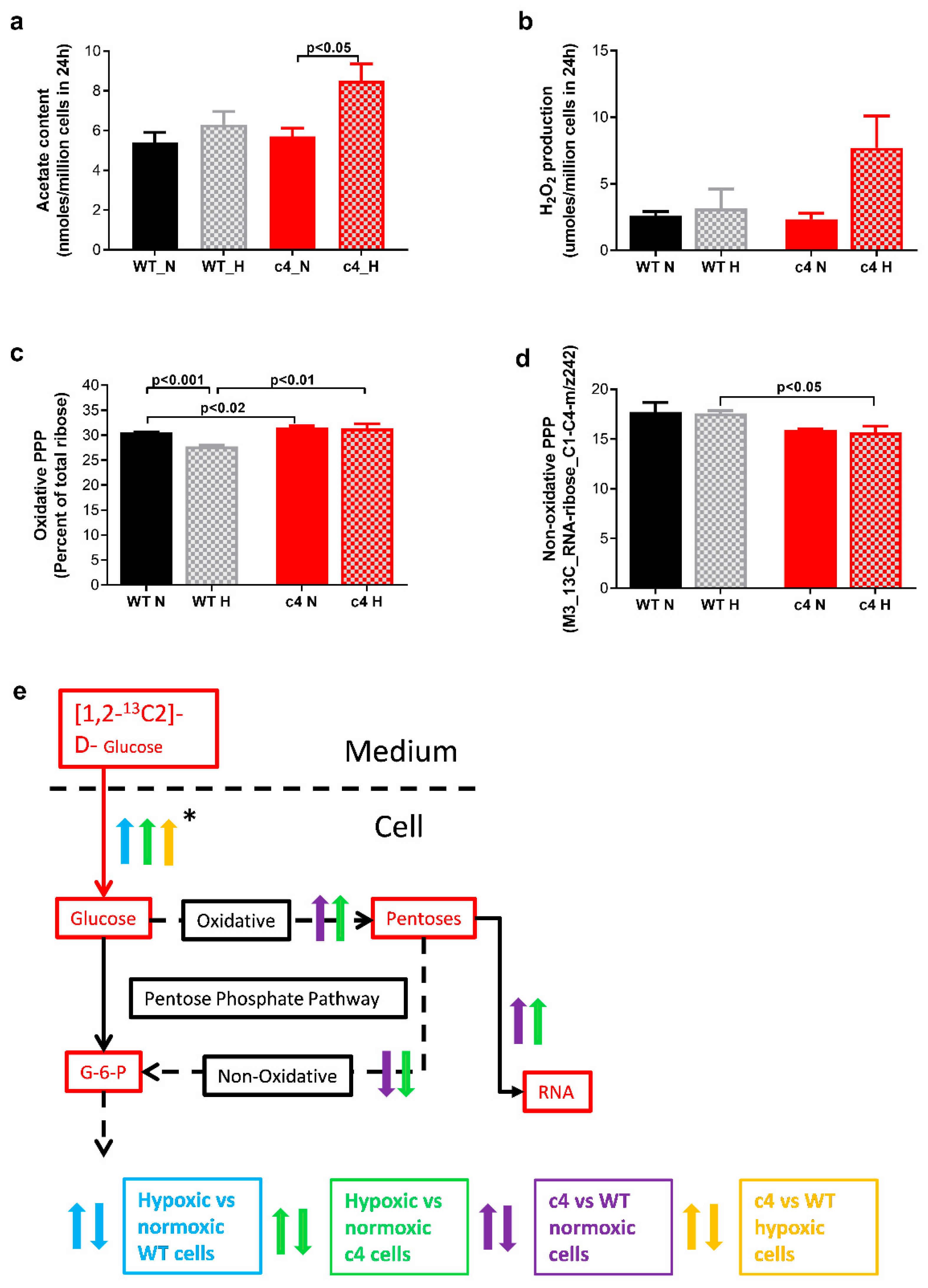
3.5. Hepa-1 c4 Cells Show Increases in Peroxisomal Markers and PPP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov. Today 2007, 12, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Dale, A.G.; Lombardo, C.M.; Valentine, H.; de la Fuente, M.; Schatzlein, A.; Neidle, S. Inhibition of the hypoxia-inducible factor pathway by a G-quadruplex binding small molecule. Sci. Rep. 2013, 3, 2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, Y.; Arakawa, T.; Nawata, Y.; Shimada, S.; Oishi, S.; Fujii, N.; Nishimura, S.; Hattori, A.; Kakeya, H. Design, synthesis, and structure-activity relationships of 1-ethylpyrazole-3-carboxamide compounds as novel hypoxia-inducible factor (HIF)-1 inhibitors. Bioorganic. Med. Chem. 2015, 23, 1776–1787. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Tang, B.; Sun, X. Development of inhibitors targeting hypoxia-inducible factor 1 and 2 for cancer therapy. Yonsei Med. J. 2017, 58, 489–496. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef]
- Lee, S.-H.; Golinska, M.; Griffiths, J.R. HIF-1-Independent Mechanisms Regulating Metabolic Adaptation in Hypoxic Cancer Cells. Cells 2021, 10, 2371. [Google Scholar] [CrossRef]
- Lim, J.H.; Luo, C.; Vazquez, F.; Puigserver, P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res. 2014, 74, 3535–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankinson, O. Single-step selection of clones of a mouse hepatoma line deficient in aryl hydrocarbon hydroxylase. Proc. Natl. Acad. Sci. USA 1979, 76, 373–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numayama-Tsuruta, K.; Kobayashi, A.; Sogawa, K.; Fujii-Kuriyama, Y. A point mutation responsible for defective function of the aryl-hydrocarbon-receptor nuclear translocator in mutant Hepa-1c1c7 cells. Eur. J. Biochem. 1997, 246, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.M.; Gleadle, J.M.; Pugh, C.W.; Hankinson, O.; Ratcliffe, P.J. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression. Studies in ARNT-deficient cells. J. Biol. Chem. 1996, 271, 15117–15123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knaup, K.X.; Guenther, R.; Stoeckert, J.; Monti, J.M.; Eckardt, K.U.; Wiesener, M.S. HIF is not essential for suppression of experimental tumor growth by mTOR inhibition. J. Cancer 2017, 8, 1809–1817. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.J.; Telfer, B.A.; Airley, R.E.; Peters, H.P.W.; Sheridan, M.R.; van der Kogel, A.J.; Harris, A.L.; Stratford, I.J. A protective role for HIF-1 in response to redox manipulation and glucose deprivation: Implications for tumorigenesis. Oncogene 2002, 21, 282–290. [Google Scholar] [CrossRef]
- Troy, H.; Chung, Y.-L.; Mayr, M.; Ly, L.; Williams, K.; Stratford, I.; Harris, A.; Griffiths, J.; Stubbs, M. Metabolic profiling of hypoxia-inducible factor-1β-deficient and wild type Hepa-1 cells: Effects of hypoxia measured by 1H magnetic resonance spectroscopy. Metabolomics 2005, 1, 293–303. [Google Scholar] [CrossRef]
- Golinska, M.; Troy, H.; Chung, Y.L.; McSheehy, P.M.; Mayr, M.; Yin, X.; Ly, L.; Williams, K.J.; E Airley, R.; Harris, A.L.; et al. Adaptation to HIF-1 deficiency by upregulation of the AMP/ATP ratio and phosphofructokinase activation in hepatomas. BMC Cancer 2011, 11, 198. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, J.R.; McSheehy, P.M.J.; Robinson, S.P.; Troy, H.; Chung, Y.-L.; Leek, R.D.; Williams, K.J.; Stratford, I.J.; Harris, A.L.; Stubbs, M. Metabolic changes detected by in vivo magnetic resonance studies of HEPA-1 wild-type tumors and tumors deficient in hypoxia-inducible factor-1beta (HIF-1beta): Evidence of an anabolic role for the HIF-1 pathway. Cancer Res. 2002, 62, 688–695. [Google Scholar]
- Lee, W.N.; Boros, L.G.; Puigjaner, J.; Bassilian, S.; Lim, S.; Cascante, M. Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2-13C2]glucose. Am. J. Physiol. 1998, 274, E843–E851. [Google Scholar] [CrossRef]
- Boros, L.G.; Brackett, D.J.; Harrigan, G.G. Metabolic biomarker and kinase drug target discovery in cancer using stable isotope-based dynamic metabolic profiling (SIDMAP). Curr. Cancer Drug Targets 2003, 3, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, G.G.; Colca, J.; Szalma, S.; Boros, L.G. PNU-91325 increases fatty acid synthesis from glucose and mitochondrial long chain fatty acid degradation: A comparative tracer-based metabolomics study with rosiglitazone and pioglitazone in HepG2 cells. Metabolomics 2006, 2, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizan, P.; Boros, L.G.; Figueras, A.; Capella, G.; Mangues, R.; Bassilian, S.; Lim, S.; Lee, W.N.; Cascante, M. K-ras codon-specific mutations produce distinctive metabolic phenotypes in NIH3T3 mice [corrected] fibroblasts. Cancer Res. 2005, 65, 5512–5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, E.; Hagmüller, E.; Staedt, U.; Schlickeiser, G.; Günther, H.J.; Leweling, H.; Tokus, M.; Kollmar, H.B. Substrate balances across colonic carcinomas in humans. Cancer Res. 1995, 55, 1373–1378. [Google Scholar] [PubMed]
- Kallinowski, F.; Schlenger, K.H.; Runkel, S.; Kloes, M.; Stohrer, M.; Okunieff, P.; Vaupel, P. Blood flow, metabolism, cellular microenvironment, and growth rate of human tumor xenografts. Cancer Res. 1989, 49, 3759–3764. [Google Scholar] [PubMed]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.-L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, T.B.; Serrao, E.M.; Kennedy, B.W.C.; Hu, D.-E.; Kettunen, M.I.; Brindle, K.M. Magnetic resonance imaging of tumor glycolysis using hyperpolarized 13C-labeled glucose. Nat. Med. 2014, 20, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Arany, Z.; Foo, S.-Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef] [Green Version]
- Shoag, J.; Arany, Z. Regulation of hypoxia-inducible genes by PGC-1 alpha. Arterioscler. Thromb Vasc. Biol. 2010, 30, 662–666. [Google Scholar] [CrossRef] [Green Version]
- Boros, L.G.; Cascante, M.; Lee, W.N.P. Metabolic profiling of cell growth and death in cancer: Applications in drug discovery. Drug. Discov. Today 2002, 7, 364–372. [Google Scholar] [CrossRef]
- Lee, W.N.; Edmond, J.; Bassilian, S.; Morrow, J.W. Mass isotopomer study of glutamine oxidation and synthesis in primary culture of astrocytes. Dev. Neurosci. 1996, 18, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Leimer, K.R.; Rice, R.H.; Gehrke, C.W. Complete mass spectra of N-trifluoroacetyl-n-butyl esters of amino acids. J. Chromatogr. A 1977, 141, 121–144. [Google Scholar] [CrossRef]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Li, C.F.; Han, F.; Liu, C.; Zhang, A.; Hsu, C.C.; Peng, D.; Zhang, X.; Jin, G.; Rezaeian, A.-H.; et al. Phosphorylation of PDHA by AMPK Drives TCA Cycle to Promote Cancer Metastasis. Mol. Cell 2020, 80, 263–278.e7. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Foxworthy, P.; Siesky, A.; Ficorilli, J.V.; Gao, H.; Li, S.; Christe, M.; Ryan, T.; Cao, G.; Eacho, P.; et al. Hepatic peroxisomal fatty acid β-oxidation is regulated by liver X receptor α. Endocrinology 2005, 146, 5380–5387. [Google Scholar] [CrossRef] [Green Version]
- Chien, D.; Dean, D.; Saha, A.K.; Flatt, J.P.; Ruderman, N.B. Malonyl-CoA content and fatty acid oxidation in rat muscle and liver in vivo. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E259–E265. [Google Scholar] [CrossRef]
- Wolfgang, M.J.; Kurama, T.; Dai, Y.; Suwa, A.; Asaumi, M.; Matsumoto, S.-I.; Cha, S.H.; Shimokawa, T.; Lane, M.D. The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 7282–7287. [Google Scholar] [CrossRef] [Green Version]
- Hardwick, J.P. Cytochrome P450 omega hydroxylase (CYP4) function in fatty acid metabolism and metabolic diseases. Biochem. Pharmacol. 2008, 75, 2263–2275. [Google Scholar] [CrossRef] [PubMed]
- Leighton, F.; Bergseth, S.; Rortveit, T.; Christiansen, E.N.; Bremer, J. Free acetate production by rat hepatocytes during peroxisomal fatty acid and dicarboxylic acid oxidation. J. Biol. Chem. 1989, 264, 10347–10350. [Google Scholar] [CrossRef]
- Gladden, L.B. Lactate metabolism: A new paradigm for the third millennium. J. Physiol. 2004, 558, 5–30. [Google Scholar] [CrossRef] [PubMed]
- Hempel, N.; Bartling, T.R.; Mian, B.; Melendez, J.A. Acquisition of the metastatic phenotype is accompanied by H2O2-dependent activation of the p130Cas signaling complex. Mol. Cancer Res. 2013, 11, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Lin, S.-H.; Ren, F.; Li, J.-T.; Chen, J.-J.; Yao, C.-B.; Yang, H.-B.; Jiang, S.-X.; Yan, G.-Q.; Wang, D.; et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7, 11960. [Google Scholar] [CrossRef] [Green Version]
- Kulisz, A.; Chen, N.; Chandel, N.S.; Shao, Z.; Schumacker, P.T. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am. J. Physiol. Cell Mol. Physiol. 2002, 282, L1324–L1329. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.K.; Vavvas, D.; Kurowski, T.G.; Apazidis, A.; Witters, L.A.; Shafrir, E.; Ruderman, N.B. Malonyl-CoA regulation in skeletal muscle: Its link to cell citrate and the glucose-fatty acid cycle. Am. J. Physiol. 1997, 272, E641–E648. [Google Scholar] [CrossRef]
- Chakravarthy, M.V.; Pan, Z.; Zhu, Y.; Tordjman, K.; Schneider, J.G.; Coleman, T.; Turk, J.; Semenkovich, C.F. ‘New’ hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 2005, 1, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Knight, B.L.; Hebbachi, A.; Hauton, D.; Brown, A.-M.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. A role for PPARalpha in the control of SREBP activity and lipid synthesis in the liver. Biochem. J. 2005, 389, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Hebbachi, A.M.; Knight, B.L.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. Peroxisome proliferator-activated receptor alpha deficiency abolishes the response of lipogenic gene expression to re-feeding: Restoration of the normal response by activation of liver X receptor alpha. J. Biol. Chem. 2008, 283, 4866–4876. [Google Scholar] [CrossRef] [Green Version]
- Narravula, S.; Colgan, S.P. Hypoxia-Inducible Factor 1-Mediated Inhibition of Peroxisome Proliferator-Activated Receptor α Expression during Hypoxia. J. Immunol. 2001, 166, 7543–7548. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, S.; Xue, J.; Avery, J.; Wu, J.; Lind, S.E.; Ding, W.Q. Activation of peroxisome proliferator-activated receptor α (PPARα) suppresses hypoxia-inducible factor-1α (HIF-1α) signaling in cancer cells. J. Biol. Chem. 2012, 287, 35161–35169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [CrossRef]
- Lelliott, C.; Vidal-Puig, A.J. Lipotoxicity, an imbalance between lipogenesis de novo and fatty acid oxidation. Int. J. Obes. Relat Metab. Disord 2004, 28, S22–S28. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P. Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-alpha. Int. J. Obes. 2005, 29, S5–S9. [Google Scholar] [CrossRef] [Green Version]
- Koo, S.-H.; Satoh, H.; Herzig, S.; Lee, C.-H.; Hedrick, S.; Kulkarni, R.; Evans, R.M.; Olefsky, J.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef]
- Reddy, J.K.; Hashimoto, T. P Eroxisomal β-O Xidation and P Eroxisome P Roliferator—A Ctivated R Eceptor α: An adaptive Metabolic System. Annu. Rev. Nutr. 2001, 21, 193–230. [Google Scholar] [CrossRef]
- Lustig, Y.; Ruas, J.L.; Estall, J.L.; Lo, J.C.; Devarakonda, S.; Laznik, D.; Choi, J.H.; Ono, H.; Olsen, J.V.; Spiegelman, B.M. Separation of the gluconeogenic and mitochondrial functions of PGC-1{alpha} through S6 kinase. Genes. Dev. 2011, 25, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Mazzio, E.; Badisa, R.; Mack, N.; Cassim, S.; Zdralevic, M.; Pouyssegur, J.; Soliman, K.F. Whole-transcriptome analysis of fully viable energy efficient glycolytic-null cancer cells established by double genetic knockout of lactate dehydrogenase A/B or glucose-6-phosphate isomerase. Cancer Genom. Proteom. 2020, 17, 469–497. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.K.; Jackson, B.T.; Paras, K.I.; Brunner, J.S.; Hart, M.L.; Newsom, O.J.; Alibeckoff, S.P.; Endress, J.; Drill, E.; Sullivan, L.B.; et al. A non-canonical tricarboxylic acid cycle underlies cellular identity. Nature 2022, 603, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pursiheimo, J.-P.; Rantanen, K.; Heikkinen, P.T.; Johansen, T.; Jaakkola, P.M. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene 2009, 28, 334–344. [Google Scholar] [CrossRef] [Green Version]
- Kučera, J.; Netušilová, J.; Sladeček, S.; Lánová, M.; Vašíček, O.; Štefková, K.; Navrátilová, J.; Kubala, L.; Pacherník, J. Hypoxia Downregulates MAPK/ERK but Not STAT3 Signaling in ROS-Dependent and HIF-1-Independent Manners in Mouse Embryonic Stem Cells. Oxidative Med. Cell. Longev. 2017, 2017, 4386947. [Google Scholar] [CrossRef] [Green Version]
- Vara-Ciruelos, D.; Russell, F.M.; Grahame Hardie, D. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol. 2019, 9, 190099. [Google Scholar] [CrossRef] [Green Version]
- Viollet, B.; Horman, S.; Leclerc, J.; Lantier, L.; Foretz, M.; Billaud, M.; Giri, S.; Andreelli, F. AMPK inhibition in health and disease. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 276. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golinska, M.A.; Stubbs, M.; Harris, A.L.; Boros, L.G.; Basetti, M.; McIntyre, D.J.O.; Griffiths, J.R. Survival Pathways of HIF-Deficient Tumour Cells: TCA Inhibition, Peroxisomal Fatty Acid Oxidation Activation and an AMPK-PGC-1α Hypoxia Sensor. Cells 2022, 11, 3595. https://doi.org/10.3390/cells11223595
Golinska MA, Stubbs M, Harris AL, Boros LG, Basetti M, McIntyre DJO, Griffiths JR. Survival Pathways of HIF-Deficient Tumour Cells: TCA Inhibition, Peroxisomal Fatty Acid Oxidation Activation and an AMPK-PGC-1α Hypoxia Sensor. Cells. 2022; 11(22):3595. https://doi.org/10.3390/cells11223595
Chicago/Turabian StyleGolinska, Monika A., Marion Stubbs, Adrian L. Harris, Laszlo G. Boros, Madhu Basetti, Dominick J. O. McIntyre, and John R. Griffiths. 2022. "Survival Pathways of HIF-Deficient Tumour Cells: TCA Inhibition, Peroxisomal Fatty Acid Oxidation Activation and an AMPK-PGC-1α Hypoxia Sensor" Cells 11, no. 22: 3595. https://doi.org/10.3390/cells11223595