
Microglia–Neutrophil Interactions Drive Dry AMD-like Pathology in a Mouse Model
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Animals
2.3. RPE Explant Culture
2.4. Microglia Culture
2.5. Neutrophil Culture
2.6. Immunophenotyping of SRS
2.7. Evaluation of Cell Surface Receptor Expression
2.8. Cytometry Bead Array (CBA)
2.9. Single Cell (sc) RNA Sequencing and Bioinformatics
2.10. Subretinal Injection
2.11. RPE Flatmount and Phalloidin Staining
2.12. Retinal Cryosections and Immunostaining
2.13. Western Blotting
2.14. Enzyme-Linked Immunosorbent Assay (ELISA)
2.15. Statistical Analysis
3. Results
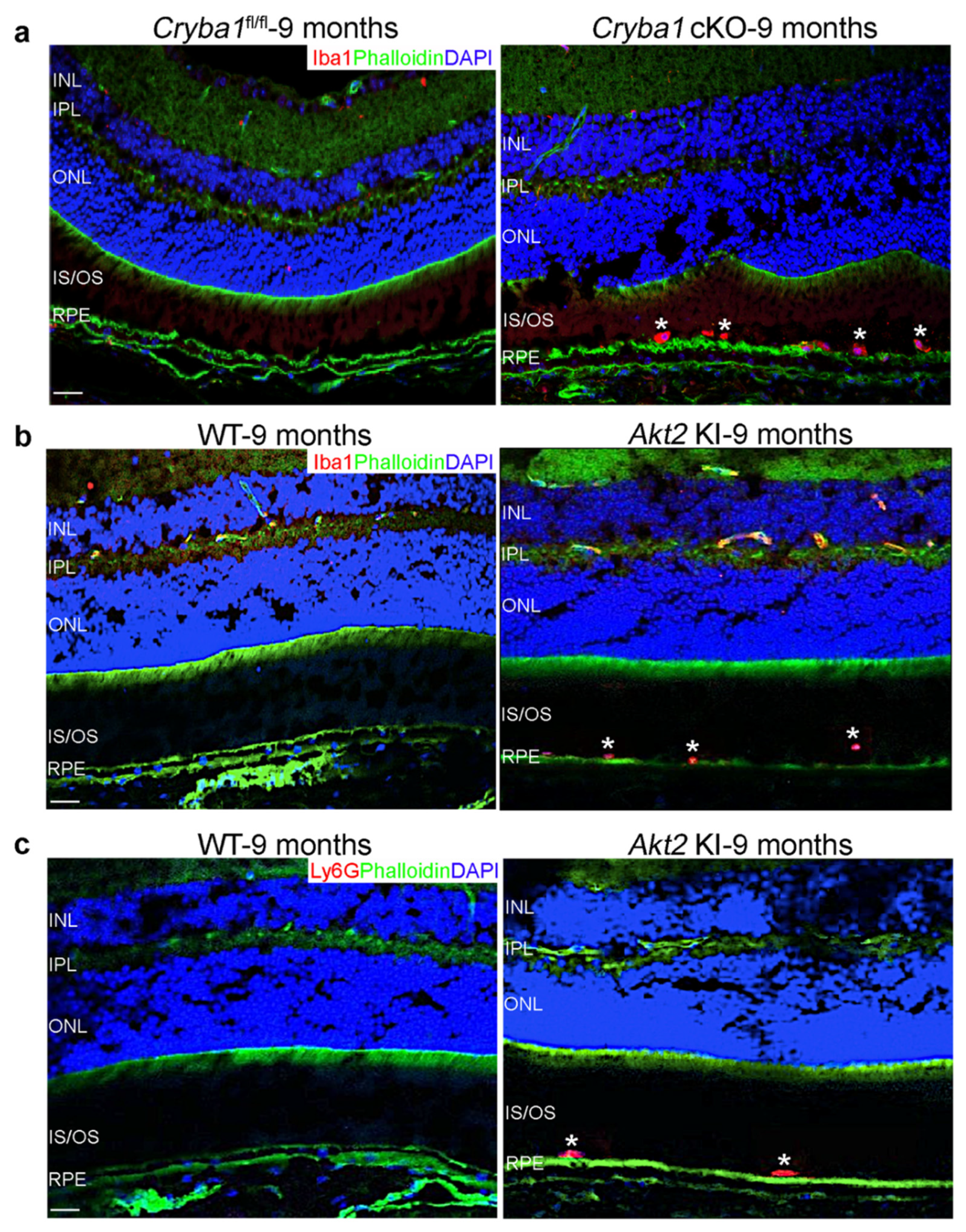
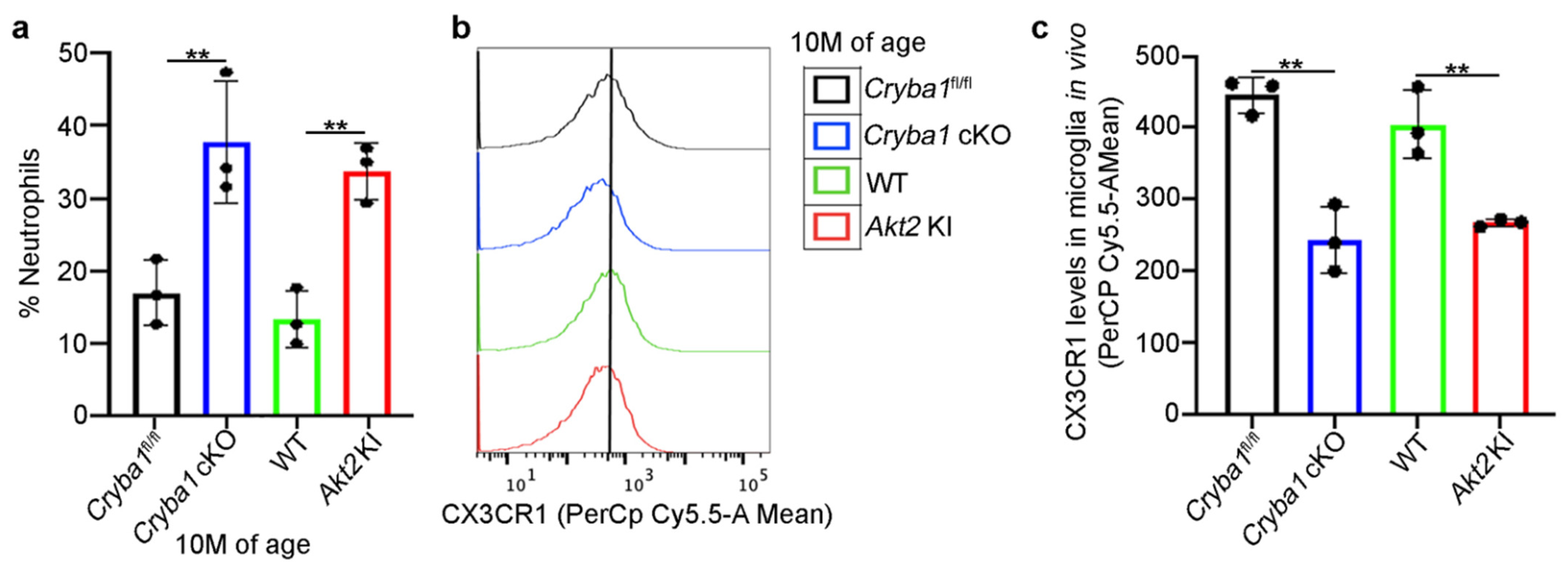
3.1. Microglia and Neutrophils Infiltrate the SRS in a Mouse Model upon Activation of Retinal Inflammation
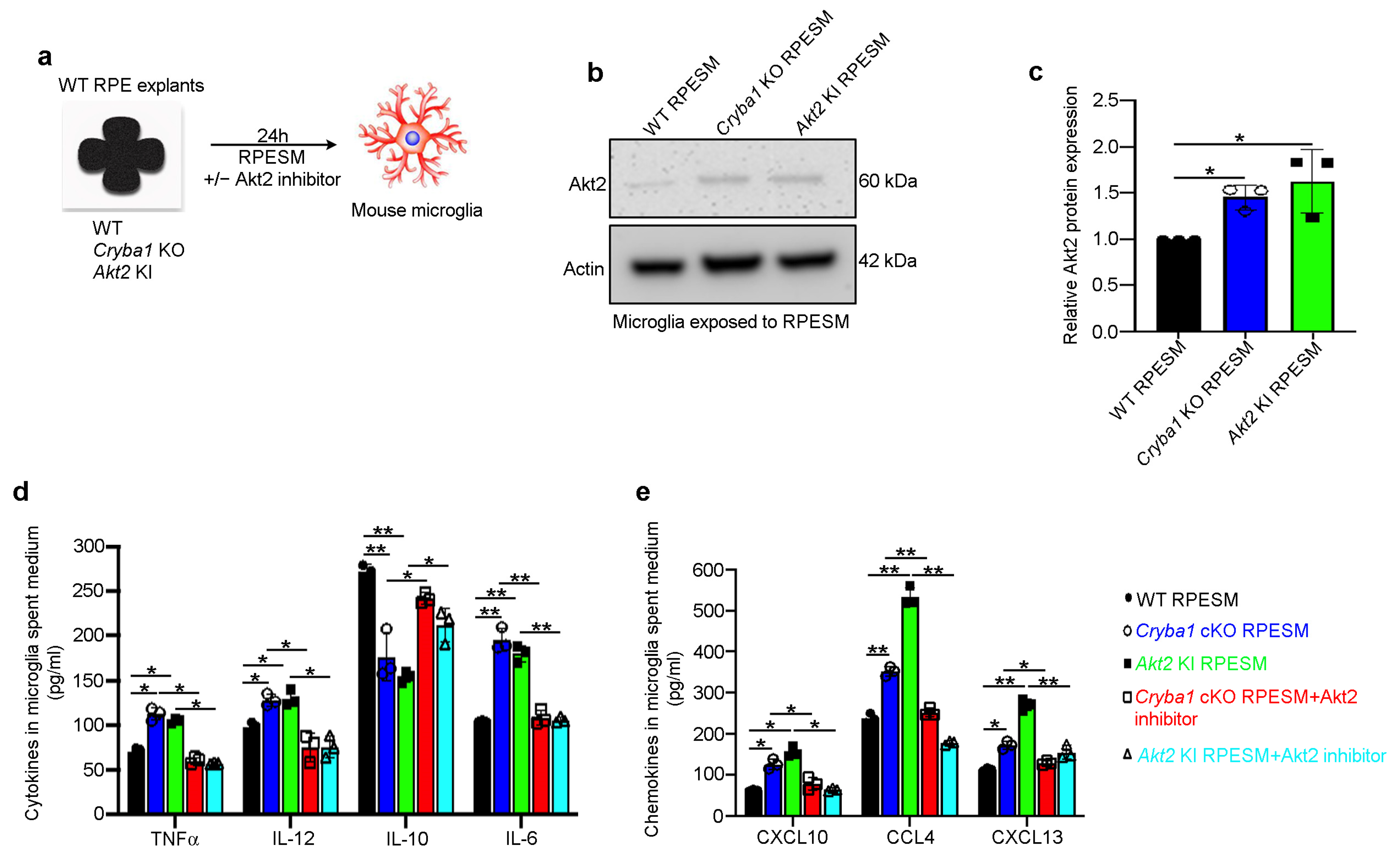
3.2. RPE Drives Microglia Activation through the Akt2 Signaling Pathway
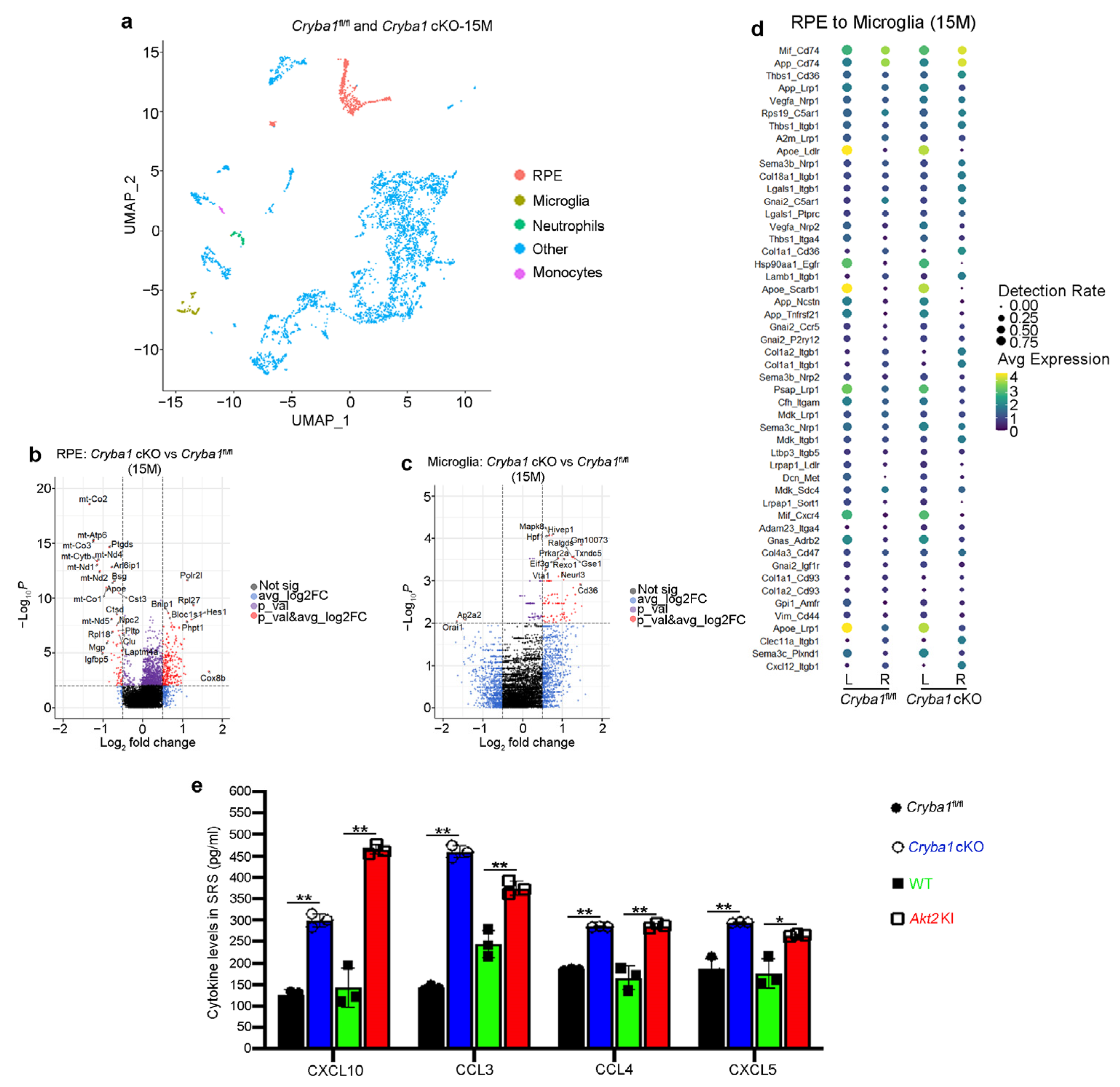
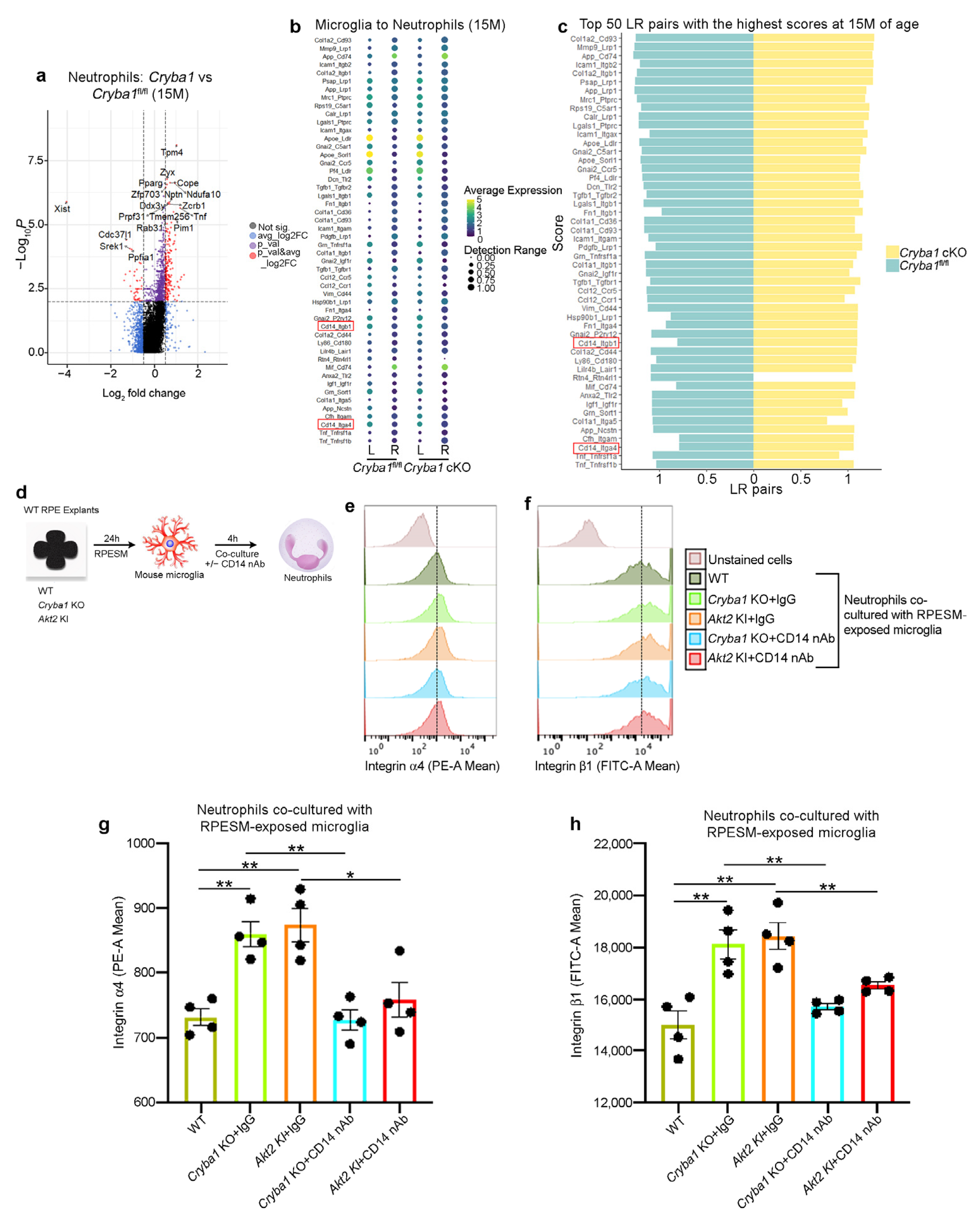
3.3. Microglia and Neutrophils Interact in the SRS of a Mouse Model of Dry AMD
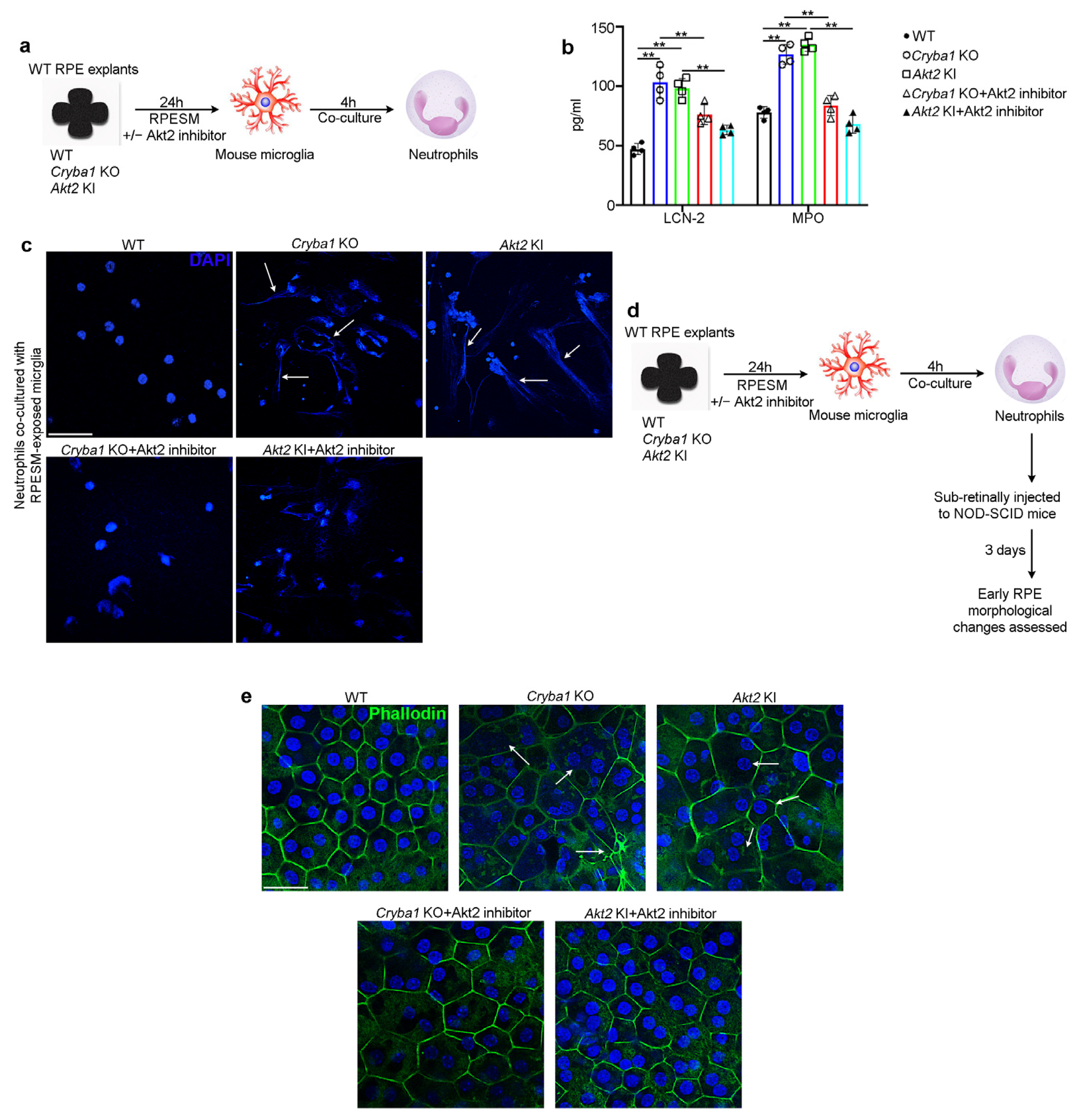
3.4. Activated Microglia Drive Neutrophil Activation and Subsequent Retinal Degeneration
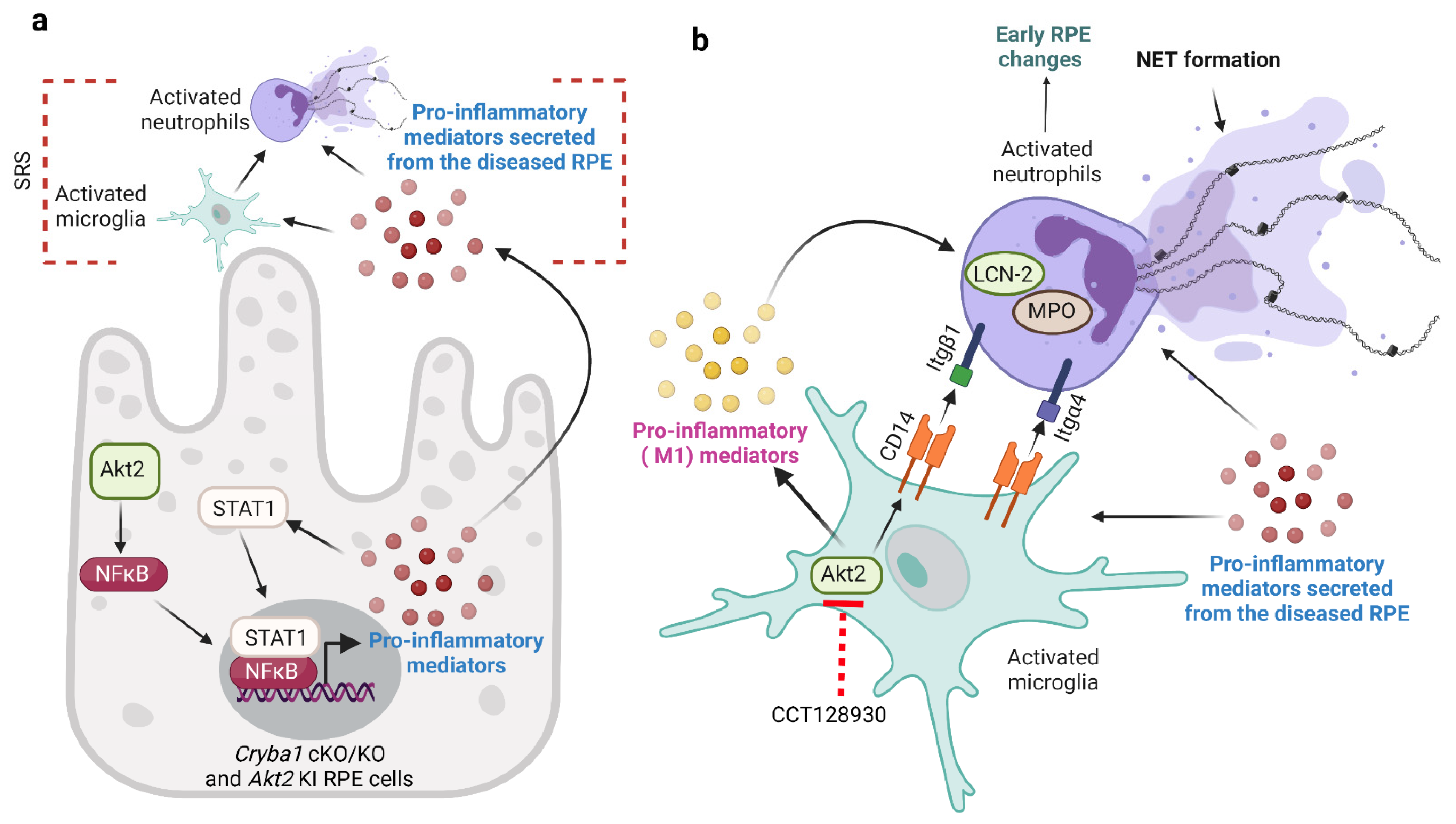
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meizlish, M.L.; Franklin, R.A.; Zhou, X.; Medzhitov, R. Tissue Homeostasis and Inflammation. Annu. Rev. Immunol. 2021, 39, 557–581. [Google Scholar] [CrossRef]
- Handa, J.T.; Bowes Rickman, C.; Dick, A.D.; Gorin, M.B.; Miller, J.W.; Toth, C.A.; Ueffing, M.; Zarbin, M.; Farrer, L.A. A systems biology approach towards understanding and treating non-neovascular age-related macular degeneration. Nat. Commun. 2019, 10, 3347. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Zou, J.; Yoshida, S.; Jiang, B.; Zhou, Y. The Role of Inflammation in Age-Related Macular Degeneration. Int. J. Biol. Sci. 2020, 16, 2989–3001. [Google Scholar] [CrossRef]
- Whitcup, S.M.; Sodhi, A.; Atkinson, J.P.; Holers, V.M.; Sinha, D.; Rohrer, B.; Dick, A.D. The role of the immune response in age-related macular degeneration. Int. J. Inflam. 2013, 2013, 348092. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106-16. [Google Scholar] [CrossRef] [Green Version]
- Girmens, J.F.; Sahel, J.A.; Marazova, K. Dry age-related macular degeneration: A currently unmet clinical need. Intractable Rare Dis. Res. 2012, 1, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zibetti, C.; Shang, P.; Sripathi, S.R.; Zhang, P.; Cano, M.; Hoang, T.; Xia, S.; Ji, H.; Merbs, S.L.; et al. ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat. Commun. 2018, 9, 1364. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Luo, C.; Zhao, J.; Devarajan, G.; Xu, H. Immune regulation in the aging retina. Prog. Retin. Eye Res. 2019, 69, 159–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.W.; Hsu, S.; Ng, T.F. The Role of Retinal Pigment Epithelial Cells in Regulation of Macrophages/Microglial Cells in Retinal Immunobiology. Front. Immunol. 2021, 12, 724601. [Google Scholar] [CrossRef]
- Chen, M.; Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J. Leukoc. Biol. 2015, 98, 713–725. [Google Scholar] [CrossRef]
- Yu, C.; Roubeix, C.; Sennlaub, F.; Saban, D.R. Microglia versus Monocytes: Distinct Roles in Degenerative Diseases of the Retina. Trends Neurosci. 2020, 43, 433–449. [Google Scholar] [CrossRef] [PubMed]
- O’Koren, E.G.; Yu, C.; Klingeborn, M.; Wong, A.Y.W.; Prigge, C.L.; Mathew, R.; Kalnitsky, J.; Msallam, R.A.; Silvin, A.; Kay, J.N.; et al. Microglial Function Is Distinct in Different Anatomical Locations during Retinal Homeostasis and Degeneration. Immunity 2019, 50, 723–737.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, O.; Lavalette, S.; Hu, S.J.; Housset, M.; Raoul, W.; Eandi, C.; Sahel, J.A.; Sullivan, P.M.; Guillonneau, X.; Sennlaub, F. APOE Isoforms Control Pathogenic Subretinal Inflammation in Age-Related Macular Degeneration. J. Neurosci. 2015, 35, 13568–13576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calippe, B.; Augustin, S.; Beguier, F.; Charles-Messance, H.; Poupel, L.; Conart, J.B.; Hu, S.J.; Lavalette, S.; Fauvet, A.; Rayes, J.; et al. Complement Factor H Inhibits CD47-Mediated Resolution of Inflammation. Immunity 2017, 46, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combadière, C.; Feumi, C.; Raoul, W.; Keller, N.; Rodéro, M.; Pézard, A.; Lavalette, S.; Houssier, M.; Jonet, L.; Picard, E.; et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J. Clin. Investig. 2007, 117, 2920–2928. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Shang, P.; Yazdankhah, M.; Bhutto, I.; Hose, S.; Montezuma, S.R.; Luo, T.; Chattopadhyay, S.; Qian, J.; Lutty, G.A.; et al. Activating the AKT2-nuclear factor-κB-lipocalin-2 axis elicits an inflammatory response in age-related macular degeneration. J. Pathol. 2017, 241, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Padmanabhan, A.; Vaidya, T.; Watson, A.M.; Bhutto, I.A.; Hose, S.; Shang, P.; Stepicheva, N.; Yazdankhah, M.; Weiss, J.; et al. Neutrophils homing into the retina trigger pathology in early age-related macular degeneration. Commun. Biol. 2019, 2, 348. [Google Scholar] [CrossRef] [Green Version]
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10, 1975. [Google Scholar] [CrossRef] [Green Version]
- Valapala, M.; Wilson, C.; Hose, S.; Bhutto, I.A.; Grebe, R.; Dong, A.; Greenbaum, S.; Gu, L.; Sengupta, S.; Cano, M.; et al. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/βA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy 2014, 10, 480–496. [Google Scholar] [CrossRef] [Green Version]
- Valapala, M.; Edwards, M.; Hose, S.; Grebe, R.; Bhutto, I.A.; Cano, M.; Berger, T.; Mak, T.W.; Wawrousek, E.; Handa, J.T.; et al. Increased Lipocalin-2 in the retinal pigment epithelium of Cryba1 cKO mice is associated with a chronic inflammatory response. Aging Cell 2014, 13, 1091–1094. [Google Scholar] [CrossRef]
- Shang, P.; Valapala, M.; Grebe, R.; Hose, S.; Ghosh, S.; Bhutto, I.A.; Handa, J.T.; Lutty, G.A.; Lu, L.; Wan, J.; et al. The amino acid transporter SLC36A4 regulates the amino acid pool in retinal pigmented epithelial cells and mediates the mechanistic target of rapamycin, complex 1 signaling. Aging Cell 2017, 16, 349–359. [Google Scholar] [CrossRef]
- Liu, H.; Stepicheva, N.; Ghosh, S.; Shang, P.; Chowdhury, O.; Daley, R.; Yazdankhah, M.; Gupta, U.; Hose, S.; Valapala, M.; et al. Reducing Akt2 in retinal pigment epithelium cells causes a compensatory increase in Akt1 and attenuates diabetic retinopathy. Nat. Commun. 2022, 13, 6045. [Google Scholar] [CrossRef]
- Gupta, U.; Ghosh, S.; Wallace, C.T.; Shang, P.; Xin, Y.; Nair, A.P.; Yazdankhah, M.; Strizhakova, A.; Ross, M.A.; Liu, H.; et al. Increased LCN2 (lipocalin 2) in the RPE decreases autophagy and activates inflammasome-ferroptosis processes in a mouse model of dry AMD. Autophagy 2022, 26, 1–20. [Google Scholar] [CrossRef]
- Neumann, J.; Sauerzweig, S.; Rönicke, R.; Gunzer, F.; Dinkel, K.; Ullrich, O.; Gunzer, M.; Reymann, K.G. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: A new mechanism of CNS immune privilege. J. Neurosci. 2008, 28, 5965–5975. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Lyu, P.; Jiang, J.; Zhou, F.; Wang, J.; Blackshaw, S.; Qian, J. LRLoop: A method to predict feedback loops in cell-cell communication. Bioinformatics 2022, in press. [Google Scholar] [CrossRef]
- Nie, Y.; Sun, L.; Wu, Y.; Yang, Y.; Wang, J.; He, H.; Hu, Y.; Chang, Y.; Liang, Q.; Zhu, J.; et al. AKT2 Regulates Pulmonary Inflammation and Fibrosis via Modulating Macrophage Activation. J. Immunol. 2017, 198, 4470–4480. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Gordillo, K.; Shah, R.; Arellanes-Robledo, J.; Cheng, Y.; Ibrahim, J.; Tuma, P.L. Akt1 and Akt2 Isoforms Play Distinct Roles in Regulating the Development of Inflammation and Fibrosis Associated with Alcoholic Liver Disease. Cells 2019, 8, 1337. [Google Scholar] [CrossRef] [Green Version]
- Bhutto, I.A.; McLeod, D.S.; Jing, T.; Sunness, J.S.; Seddon, J.M.; Lutty, G.A. Increased choroidal mast cells and their degranulation in age-related macular degeneration. Br. J. Ophthalmol. 2016, 100, 720–726. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Yu, W.; Chen, S.; Gao, Z.; Xiao, B. Microglia Polarization and Endoplasmic Reticulum Stress in Chronic Social Defeat Stress Induced Depression Mouse. Neurochem. Res. 2018, 43, 985–994. [Google Scholar] [CrossRef]
- Ellwanger, D.C.; Wang, S.; Brioschi, S.; Shao, Z.; Green, L.; Case, R.; Yoo, D.; Weishuhn, D.; Rathanaswami, P.; Bradley, J.; et al. Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2017742118. [Google Scholar] [CrossRef]
- Liu, F.; Ding, X.; Yang, Y.; Li, J.; Tang, M.; Yuan, M.; Hu, A.; Zhan, Z.; Li, Z.; Lu, L. Aqueous humor cytokine profiling in patients with wet AMD. Mol. Vis. 2016, 22, 352–361. [Google Scholar]
- Spindler, J.; Zandi, S.; Pfister, I.B.; Gerhardt, C.; Garweg, J.G. Cytokine profiles in the aqueous humor and serum of patients with dry and treated wet age-related macular degeneration. PLoS ONE 2018, 13, e0203337. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Liu, Y.; Li, Y.; Chen, H.; Liu, X.; Chen, X.; Yue, J.; Zhou, X.; Yang, J. Sulfasalazine alters microglia phenotype by competing endogenous RNA effect of miR-136-5p and long non-coding RNA HOTAIR in cuprizone-induced demyelination. Biochem. Pharmacol. 2018, 155, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef] [PubMed]
- Laffer, B.; Bauer, D.; Wasmuth, S.; Busch, M.; Jalilvand, T.V.; Thanos, S.; Meyer, Z.; Hörste, G.; Loser, K.; Langmann, T.; et al. Loss of IL-10 Promotes Differentiation of Microglia to a M1 Phenotype. Front. Cell Neurosci. 2019, 13, 430. [Google Scholar] [CrossRef]
- Park, J.; Baik, S.H.; Mook-Jung, I.; Irimia, D.; Cho, H. Mimicry of Central-Peripheral Immunity in Alzheimer’s Disease and Discovery of Neurodegenerative Roles in Neutrophil. Front. Immunol. 2019, 10, 2231. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.F. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimers Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, T.A.; Zorzetto-Fernandes, A.L.V.; Cecílio, N.T.; Sardinha-Silva, A.; Fernandes, F.F.; Roque-Barreira, M.C. CD14 is critical for TLR2-mediated M1 macrophage activation triggered by N-glycan recognition. Sci. Rep. 2017, 7, 7083. [Google Scholar] [CrossRef] [Green Version]
- Humphries, J.D.; Humphries, M.J. CD14 is a ligand for the integrin alpha4beta1. FEBS Lett. 2007, 581, 757–763. [Google Scholar] [CrossRef]
- Kirveskari, J.; Bono, P.; Granfors, K.; Leirisalo-Repo, M.; Jalkanen, S.; Salmi, M. Expression of alpha4-integrins on human neutrophils. J. Leukoc. Biol. 2000, 68, 243–250. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Raoul, W.; Feumi, C.; Keller, N.; Lavalette, S.; Houssier, M.; Behar-Cohen, F.; Combadière, C.; Sennlaub, F. Lipid-bloated subretinal microglial cells are at the origin of drusen appearance in CX3CR1-deficient mice. Ophthalmic Res. 2008, 40, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Krogh Nielsen, M.; Hector, S.M.; Allen, K.; Subhi, Y.; Sørensen, T.L. Altered activation state of circulating neutrophils in patients with neovascular age-related macular degeneration. Immun. Ageing 2017, 14, 18. [Google Scholar] [CrossRef]
- Hijioka, M.; Futokoro, R.; Ohto-Nakanishi, T.; Nakanishi, H.; Katsuki, H.; Kitamura, Y. Microglia-released leukotriene B4 promotes neutrophil infiltration and microglial activation following intracerebral hemorrhage. Int. Immunopharmacol. 2020, 85, 106678. [Google Scholar] [CrossRef]
- Kim, Y.R.; Kim, Y.M.; Lee, J.; Park, J.; Lee, J.E.; Hyun, Y.M. Neutrophils Return to Bloodstream Through the Brain Blood Vessel After Crosstalk with Microglia During LPS-Induced Neuroinflammation. Front. Cell Dev. Biol. 2020, 8, 613733. [Google Scholar] [CrossRef]
- Berchtold, D.; Priller, J.; Meisel, C.; Meisel, A. Interaction of microglia with infiltrating immune cells in the different phases of stroke. Brain Pathol. 2020, 30, 1208–1218. [Google Scholar] [CrossRef]
- Arranz, A.; Doxaki, C.; Vergadi, E.; Martinez de la Torre, Y.; Vaporidi, K.; Lagoudaki, E.D.; Ieronymaki, E.; Androulidaki, A.; Venihaki, M.; Margioris, A.N.; et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. USA 2012, 109, 9517–9522. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyce, M.; Xin, Y.; Chowdhury, O.; Shang, P.; Liu, H.; Koontz, V.; Strizhakova, A.; Nemani, M.; Hose, S.; Zigler, J.S., Jr.; et al. Microglia–Neutrophil Interactions Drive Dry AMD-like Pathology in a Mouse Model. Cells 2022, 11, 3535. https://doi.org/10.3390/cells11223535
Boyce M, Xin Y, Chowdhury O, Shang P, Liu H, Koontz V, Strizhakova A, Nemani M, Hose S, Zigler JS Jr., et al. Microglia–Neutrophil Interactions Drive Dry AMD-like Pathology in a Mouse Model. Cells. 2022; 11(22):3535. https://doi.org/10.3390/cells11223535
Chicago/Turabian StyleBoyce, Maeve, Ying Xin, Olivia Chowdhury, Peng Shang, Haitao Liu, Victoria Koontz, Anastasia Strizhakova, Mihir Nemani, Stacey Hose, J. Samuel Zigler, Jr., and et al. 2022. "Microglia–Neutrophil Interactions Drive Dry AMD-like Pathology in a Mouse Model" Cells 11, no. 22: 3535. https://doi.org/10.3390/cells11223535