

Advanced Glycation End Products and Inflammation in Type 1 Diabetes Development
 , ,
, ,  , and
, and 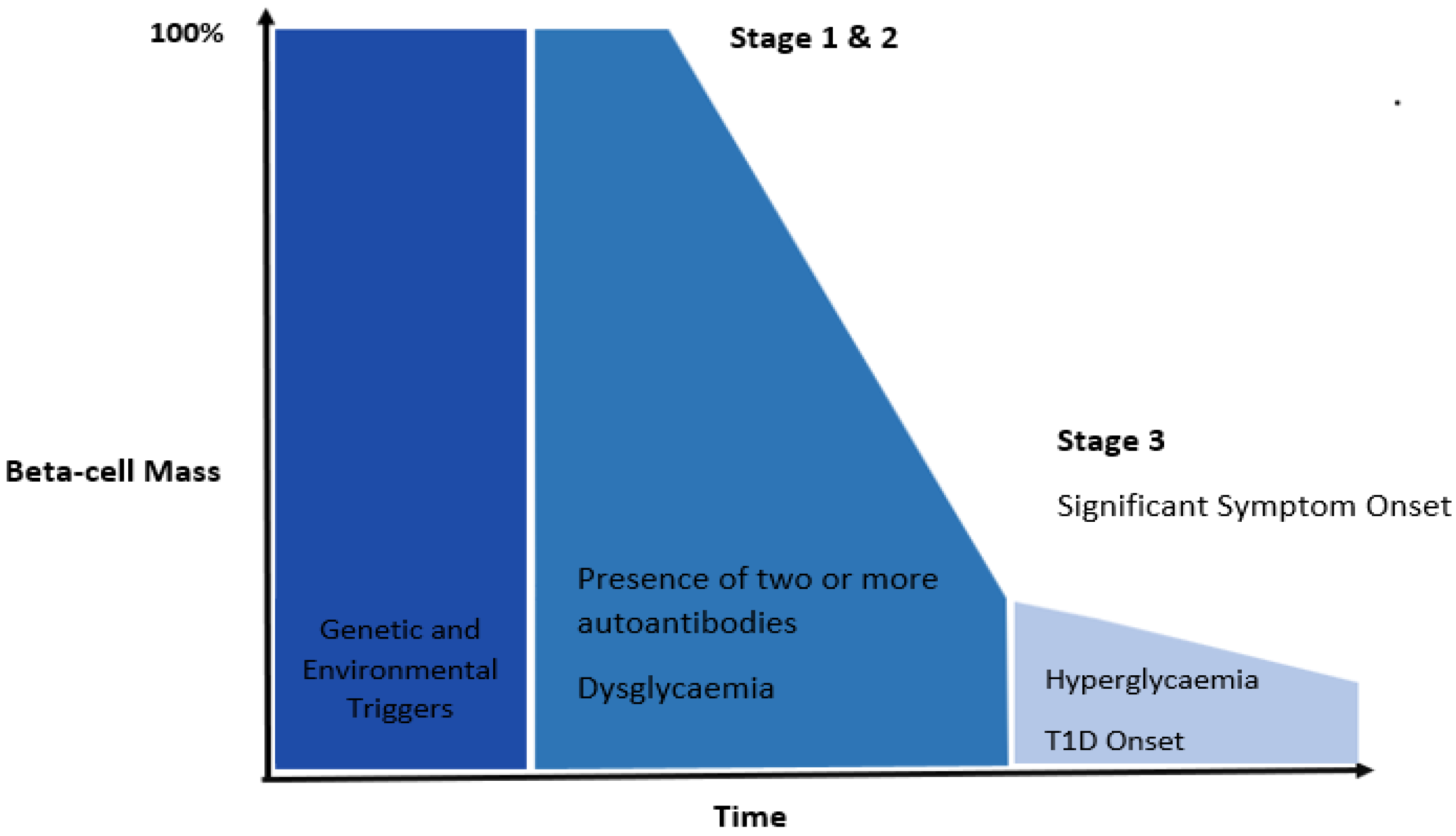{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Main Text
2.1. Pathophysiology of Type 1 Diabetes
2.2. Genetic Susceptibility
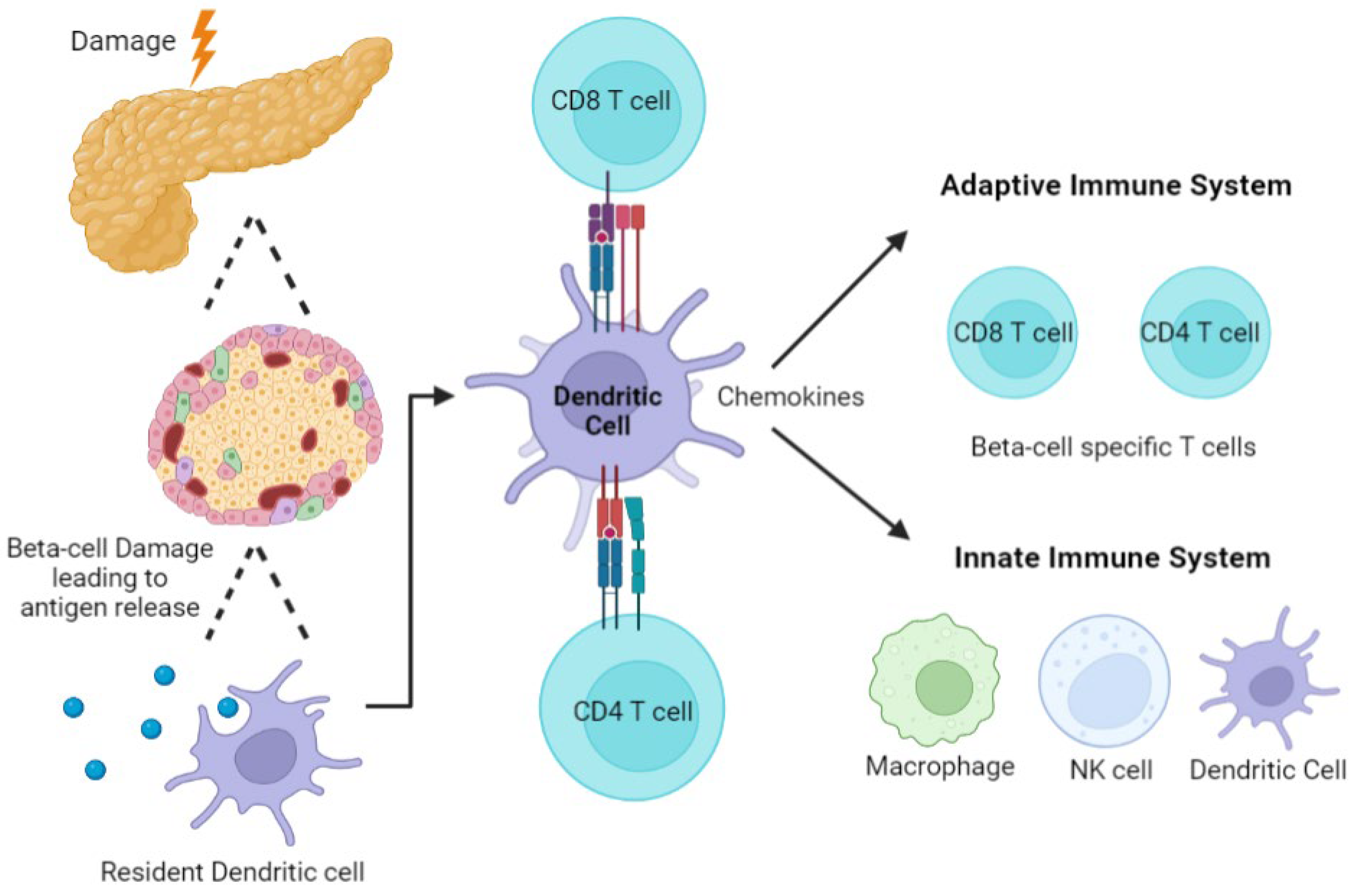
2.3. Autoimmunity and Role of the Immune System

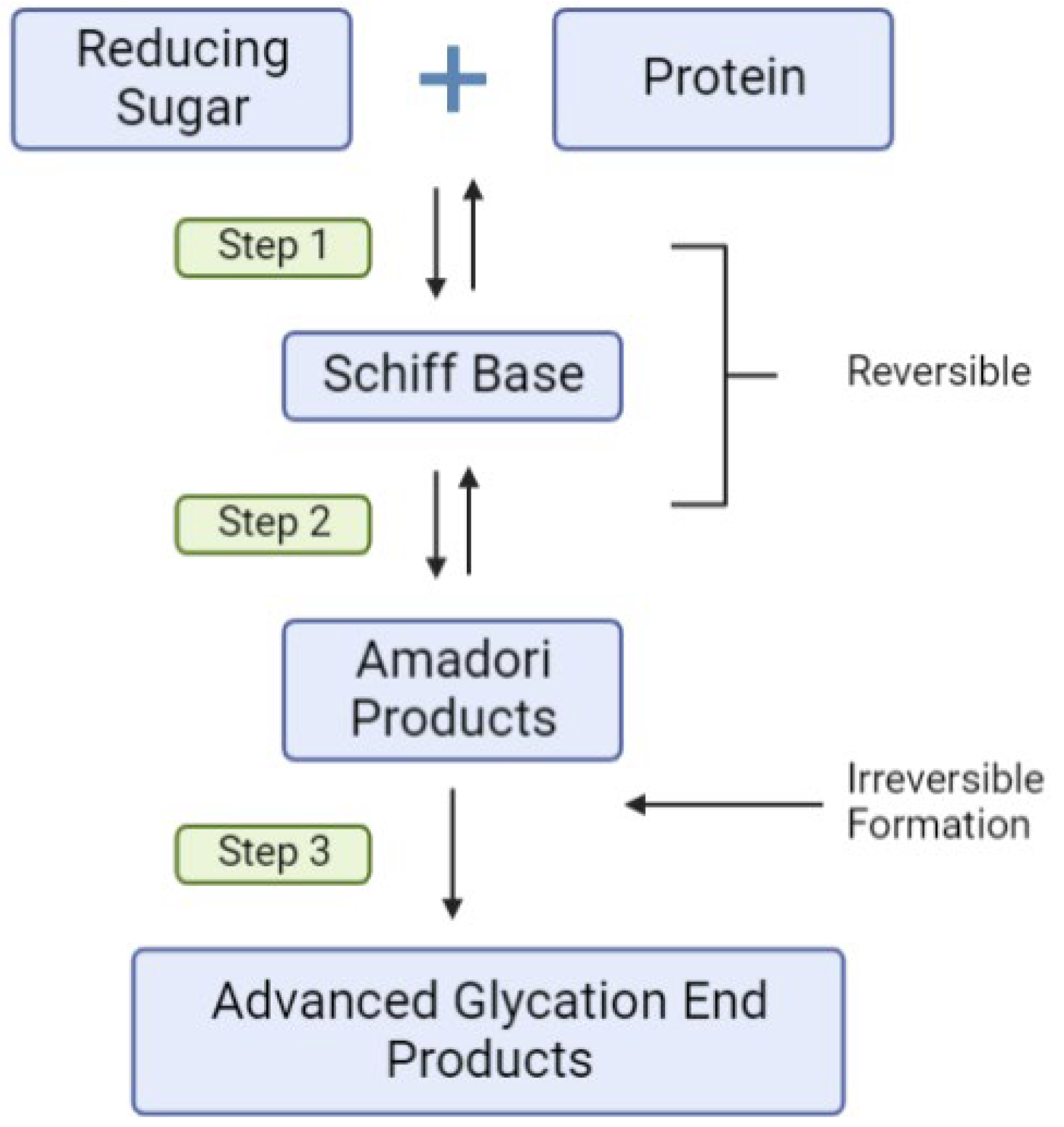
2.4. Environmental Triggers, Dietary AGEs, Inflammation and T1D Risk
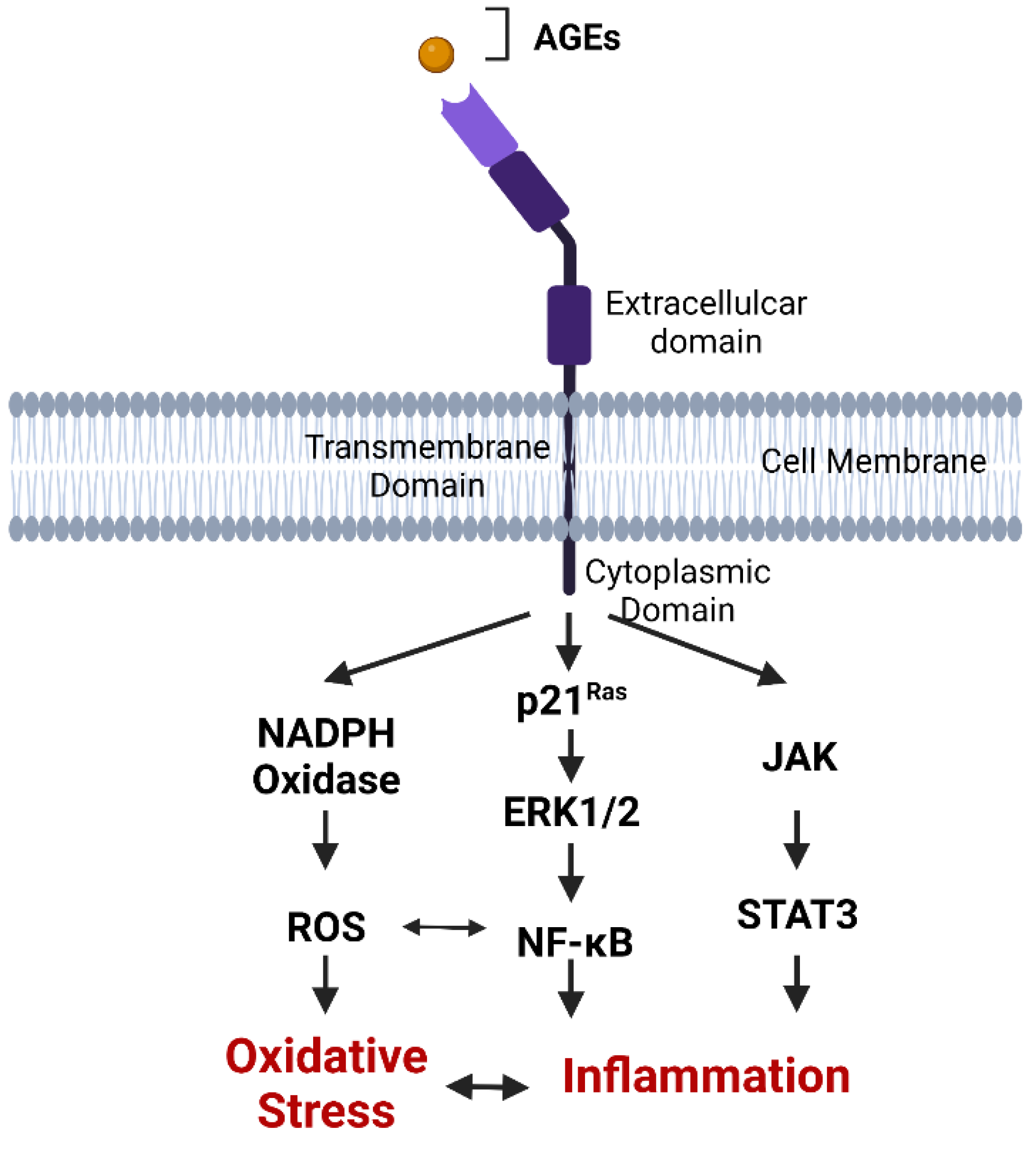
2.5. AGE Binding to RAGE
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mobasseri, M.; Shirmohammadi, M.; Amiri, T.; Vahed, N.; Hosseini Fard, H.; Ghojazadeh, M. Prevalence and incidence of type 1 diabetes in the world: A systematic review and meta-analysis. Health Promot. Perspect. 2020, 10, 98–115. [Google Scholar] [CrossRef]
- Saberzadeh-Ardestani, B.; Karamzadeh, R.; Basiri, M.; Hajizadeh-Saffar, E.; Farhadi, A.; Shapiro, A.M.J.; Tahamtani, Y.; Baharvand, H. Type 1 Diabetes Mellitus: Cellular and Molecular Pathophysiology at a Glance. Cell J. 2018, 20, 294–301. [Google Scholar] [CrossRef]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Leslie, R.D.; Evans-Molina, C.; Freund-Brown, J.; Buzzetti, R.; Dabelea, D.; Gillespie, K.M.; Goland, R.; Jones, A.G.; Kacher, M.; Phillips, L.S.; et al. Adult-Onset Type 1 Diabetes: Current Understanding and Challenges. Diabetes Care 2021, 44, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Moran, A.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Greenbaum, C.J.; Herold, K.C.; Marks, J.B.; Raskin, P.; et al. Interleukin-1 antagonism in type 1 diabetes of recent onset: Two multicentre, randomised, double-blind, placebo-controlled trials. Lancet 2013, 381, 1905–1915. [Google Scholar] [CrossRef] [Green Version]
- Feutren, G.; Papoz, L.; Assan, R.; Vialettes, B.; Karsenty, G.; Vexiau, P.; Du Rostu, H.; Rodier, M.; Sirmai, J.; Lallemand, A.; et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset: Results of a multicentre double-blind trial. Lancet 1986, 2, 119–124. [Google Scholar] [CrossRef]
- Bone, R.N.; Evans-Molina, C. Combination Immunotherapy for Type 1 Diabetes. Curr. Diabetes Rep. 2017, 17, 50. [Google Scholar] [CrossRef]
- Australian National Diabetes Audit. Australian National Diabetes Audit—Australian Quality Clinical Audit 2019 Annual Report; Australian Government Department of Health: Canberra, Australia, 2019.
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Fousteri, G.; Ippolito, E.; Ahmed, R.; Hamad, A.R.A. Beta-cell Specific Autoantibodies: Are they Just an Indicator of Type 1 Diabetes? Curr. Diabetes Rev. 2017, 13, 322–329. [Google Scholar] [CrossRef]
- Ilonen, J.; Lempainen, J.; Hammais, A.; Laine, A.P.; Harkonen, T.; Toppari, J.; Veijola, R.; Knip, M.; Finnish Pediatric Diabetes, R. Primary islet autoantibody at initial seroconversion and autoantibodies at diagnosis of type 1 diabetes as markers of disease heterogeneity. Pediatr. Diabetes 2018, 19, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Miao, D.; Babu, S.; Yu, J.; Barker, J.; Klingensmith, G.; Rewers, M.; Eisenbarth, G.S.; Yu, L. Prevalence of autoantibody-negative diabetes is not rare at all ages and increases with older age and obesity. J. Clin. Endocrinol. Metab. 2007, 92, 88–92. [Google Scholar] [CrossRef]
- Chiou, J.; Geusz, R.J.; Okino, M.-L.; Han, J.Y.; Miller, M.; Melton, R.; Beebe, E.; Benaglio, P.; Huang, S.; Korgaonkar, K.; et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 2021, 594, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Role of immune system in type 1 diabetes mellitus pathogenesis. Int. Immunopharmacol. 2014, 22, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, A. Autoreactive T cells in type 1 diabetes. J. Clin. Investig. 2017, 127, 2881–2891. [Google Scholar] [CrossRef]
- Hyttinen, V.; Kaprio, J.; Kinnunen, L.; Koskenvuo, M.; Tuomilehto, J. Genetic Liability of Type 1 Diabetes and the Onset Age Among 22,650 Young Finnish Twin Pairs: A Nationwide Follow-Up Study. Diabetes 2003, 52, 1052–1055. [Google Scholar] [CrossRef] [Green Version]
- Redondo, M.J.; Jeffrey, J.; Fain, P.R.; Eisenbarth, G.S.; Orban, T. Concordance for Islet Autoimmunity among Monozygotic Twins. N. Engl. J. Med. 2008, 359, 2849–2850. [Google Scholar] [CrossRef]
- Makino, S.; Kunimoto, K.; Muraoka, Y.; Mizushima, Y.; Katagiri, K.; Tochino, Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu 1980, 29, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.G.; Mathews, C.E.; Driver, J.P. The Role of NOD Mice in Type 1 Diabetes Research: Lessons from the Past and Recommendations for the Future. Front. Endocrinol. 2018, 9, 51. [Google Scholar] [CrossRef]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Hogquist, K.A. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeker, L.T.; Bour-Jordan, H.; Bluestone, J.A. Breakdown in peripheral tolerance in type 1 diabetes in mice and humans. Cold Spring Harb. Perspect. Med. 2012, 2, a007807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdem, N.; Montero, E.; Roep, B.O. Breaking and restoring immune tolerance to pancreatic beta-cells in type 1 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2021, 28, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Rewers, M.; Ludvigsson, J. Environmental risk factors for type 1 diabetes. Lancet 2016, 387, 2340–2348. [Google Scholar] [CrossRef] [Green Version]
- Esposito, S.; Toni, G.; Tascini, G.; Santi, E.; Berioli, M.G.; Principi, N. Environmental Factors Associated with Type 1 Diabetes. Front. Endocrinol. 2019, 10, 592. [Google Scholar] [CrossRef]
- Redondo, M.J.; Steck, A.K.; Pugliese, A. Genetics of type 1 diabetes. Pediatr. Diabetes 2018, 19, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Roep, B.O.; Thomaidou, S.; van Tienhoven, R.; Zaldumbide, A. Type 1 diabetes mellitus as a disease of the beta-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 2021, 17, 150–161. [Google Scholar] [CrossRef]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, A.; et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef] [Green Version]
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Prim. 2017, 3, 17016. [Google Scholar] [CrossRef]
- Claessens, L.A.; Wesselius, J.; van Lummel, M.; Laban, S.; Mulder, F.; Mul, D.; Nikolic, T.; Aanstoot, H.J.; Koeleman, B.P.C.; Roep, B.O. Clinical and genetic correlates of islet-autoimmune signatures in juvenile-onset type 1 diabetes. Diabetologia 2020, 63, 351–361. [Google Scholar] [CrossRef]
- Pundziute-Lyckå, A.; Persson, L.A.; Cedermark, G.; Jansson-Roth, A.; Nilsson, U.; Westin, V.; Dahlquist, G. Diet, growth, and the risk for type 1 diabetes in childhood: A matched case-referent study. Diabetes Care 2004, 27, 2784–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kibirige, M.; Metcalf, B.; Renuka, R.; Wilkin, T.J. Testing the Accelerator Hypothesis: The relationship between body mass and age at diagnosis of type 1 diabetes. Diabetes Care 2003, 26, 2865–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knerr, I.; Wolf, J.; Reinehr, T.; Stachow, R.; Grabert, M.; Schober, E.; Rascher, W.; Holl, R.W. The ‘accelerator hypothesis’: Relationship between weight, height, body mass index and age at diagnosis in a large cohort of 9,248 German and Austrian children with type 1 diabetes mellitus. Diabetologia 2005, 48, 2501–2504. [Google Scholar] [CrossRef] [Green Version]
- Nucci, A.M.; Virtanen, S.M.; Cuthbertson, D.; Ludvigsson, J.; Einberg, U.; Huot, C.; Castano, L.; Aschemeier, B.; Becker, D.J.; Knip, M.; et al. Growth and development of islet autoimmunity and type 1 diabetes in children genetically at risk. Diabetologia 2021, 64, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Ciężki, S.; Kurpiewska, E.; Bossowski, A.; Głowińska-Olszewska, B. Multi-Faceted Influence of Obesity on Type 1 Diabetes in Children—From Disease Pathogenesis to Complications. Front. Endocrinol. 2022, 13, 890833. [Google Scholar] [CrossRef]
- Von Scholten, B.J.; Kreiner, F.F.; Gough, S.C.L.; von Herrath, M. Current and future therapies for type 1 diabetes. Diabetologia 2021, 64, 1037–1048. [Google Scholar] [CrossRef]
- Holmes-Walker, D.J.; Abraham, M.B.; Chee, M.; Jones, T.W. Glycaemic outcomes in Australasian children and adults with type 1 diabetes: Failure to meet targets across the age spectrum. Intern. Med. J. 2022. ahead of print. [Google Scholar] [CrossRef]
- Abdul-Rasoul, M.; Habib, H.; Al-Khouly, M. ‘The honeymoon phase’ in children with type 1 diabetes mellitus: Frequency, duration, and influential factors. Pediatr. Diabetes 2006, 7, 101–107. [Google Scholar] [CrossRef]
- Jansen, A.; Homo-Delarche, F.; Hooijkaas, H.; Leenen, P.J.; Dardenne, M.; Drexhage, H.A. Immunohistochemical Characterization of Monocytes-Macrophages and Dendritic Cells Involved in the Initiation of the Insulitis and β-Cell Destruction in NOD Mice. Diabetes 1994, 43, 667–675. [Google Scholar] [CrossRef]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.; Atkinson, M.A.; Roep, B.O.; von Herrath, M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef]
- In’t Veld, P.; Lievens, D.; de Grijse, J.; Ling, Z.; van der Auwera, B.; Pipeleers-Marichal, M.; Gorus, F.; Pipeleers, D. Screening for insulitis in adult autoantibody-positive organ donors. Diabetes 2007, 56, 2400–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell-Thompson, M.; Wasserfall, C.; Kaddis, J.; Albanese-O’Neill, A.; Staeva, T.; Nierras, C.; Moraski, J.; Rowe, P.; Gianani, R.; Eisenbarth, G.; et al. Network for Pancreatic Organ Donors with Diabetes (nPOD): Developing a tissue biobank for type 1 diabetes. Diabetes/Metab. Res. Rev. 2012, 28, 608–617. [Google Scholar] [CrossRef]
- In’t Veld, P. Insulitis in human type 1 diabetes: The quest for an elusive lesion. Islets 2011, 3, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Hutchings, P.; Rosen, H.; O’Reilly, L.; Simpson, E.; Gordon, S.; Cooke, A. Transfer of diabetes in mice prevented by blockade of adhesion-promoting receptor on macrophages. Nature 1990, 348, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.S.; Yoon, C.S.; Zbytnuik, L.; van Rooijen, N.; Yoon, J.W. The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J. Exp. Med. 1999, 189, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.J. Big mac attack: Does it play a direct role for monocytes/macrophages in type 1 diabetes? Diabetes 2008, 57, 2922–2923. [Google Scholar] [CrossRef] [Green Version]
- Lehuen, A.; Diana, J.; Zaccone, P.; Cooke, A. Immune cell crosstalk in type 1 diabetes. Nat. Rev. Immunol. 2010, 10, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Probst, H.C.; Lagnel, J.; Kollias, G.; van den Broek, M. Inducible Transgenic Mice Reveal Resting Dendritic Cells as Potent Inducers of CD8+ T Cell Tolerance. Immunity 2003, 18, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Morel, P.A. Dendritic cell subsets in type 1 diabetes: Friend or foe? Front. Immunol. 2013, 4, 415. [Google Scholar] [CrossRef]
- Ganguly, D.; Haak, S.; Sisirak, V.; Reizis, B. The role of dendritic cells in autoimmunity. Nat. Rev. Immunol. 2013, 13, 566–577. [Google Scholar] [CrossRef] [Green Version]
- Creusot, R.J.; Postigo-Fernandez, J.; Teteloshvili, N. Altered Function of Antigen-Presenting Cells in Type 1 Diabetes: A Challenge for Antigen-Specific Immunotherapy? Diabetes 2018, 67, 1481–1494. [Google Scholar] [CrossRef] [Green Version]
- Saxena, V.; Ondr, J.K.; Magnusen, A.F.; Munn, D.H.; Katz, J.D. The Countervailing Actions of Myeloid and Plasmacytoid Dendritic Cells Control Autoimmune Diabetes in the Nonobese Diabetic Mouse. J. Immunol. 2007, 179, 5041–5053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uno, S.; Imagawa, A.; Okita, K.; Sayama, K.; Moriwaki, M.; Iwahashi, H.; Yamagata, K.; Tamura, S.; Matsuzawa, Y.; Hanafusa, T.; et al. Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor-α in patients with recent-onset type 1 diabetes. Diabetologia 2007, 50, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Makala, L.H.; Jin, Y.; Hopkins, D.; Muir, A.; Garge, N.; Podolsky, R.H.; She, J.X. Type 1 diabetes patients have significantly lower frequency of plasmacytoid dendritic cells in the peripheral blood. Clin. Immunol. 2008, 129, 413–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuckovic, S.; Withers, G.; Harris, M.; Khalil, D.; Gardiner, D.; Flesch, I.; Tepes, S.; Greer, R.; Cowley, D.; Cotterill, A.; et al. Decreased blood dendritic cell counts in type 1 diabetic children. Clin. Immunol. 2007, 123, 281–288. [Google Scholar] [CrossRef]
- Wallberg, M.; Cooke, A. Immune mechanisms in type 1 diabetes. Trends Immunol. 2013, 34, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Haase, C.; Skak, K.; Michelsen, B.K.; Markholst, H. Local Activation of Dendritic Cells Leads to Insulitis and Development of Insulin-Dependent Diabetes in Transgenic Mice Expressing CD154 on the Pancreatic β-Cells. Diabetes 2004, 53, 2588–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.M.; Parish, N.M.; Raine, T.; Bland, C.; Sawyer, Y.; de La Pena, H.; Cooke, A. Type 1 diabetes development requires both CD4+ and CD8+ T cells and can be reversed by non-depleting antibodies targeting both T cell populations. Rev. Diabet. Stud. 2009, 6, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.; Kroger, C.J.; Tisch, R.M. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front. Immunol. 2017, 8, 1898. [Google Scholar] [CrossRef]
- Chervonsky, A.V.; Wang, Y.; Wong, F.S.; Visintin, I.; Flavell, R.A.; Janeway, C.A., Jr.; Matis, L.A. The role of Fas in autoimmune diabetes. Cell 1997, 89, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Chatenoud, L.; Thervet, E.; Primo, J.; Bach, J.F. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA 1994, 91, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Calvo, T.; Suwandi, J.S.; Amirian, N.; Zapardiel-Gonzalo, J.; Anquetil, F.; Sabouri, S.; Von Herrath, M.G. Heterogeneity and Lobularity of Pancreatic Pathology in Type 1 Diabetes during the Prediabetic Phase. J. Histochem. Cytochem. 2015, 63, 626–636. [Google Scholar] [CrossRef] [Green Version]
- Itoh, N.; Hanafusa, T.; Miyazaki, A.; Miyagawa, J.; Yamagata, K.; Yamamoto, K.; Waguri, M.; Imagawa, A.; Tamura, S.; Inada, M.; et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J. Clin. Investig. 1993, 92, 2313–2322. [Google Scholar] [CrossRef] [Green Version]
- Assalino, M.; Genevay, M.; Morel, P.; Demuylder-Mischler, S.; Toso, C.; Berney, T. Recurrence of type 1 diabetes after simultaneous pancreas-kidney transplantation in the absence of GAD and IA-2 autoantibodies. Am. J. Transplant. 2012, 12, 492–495. [Google Scholar] [CrossRef]
- Williams, A.J.; Thrower, S.L.; Sequeiros, I.M.; Ward, A.; Bickerton, A.S.; Triay, J.M.; Callaway, M.P.; Dayan, C.M. Pancreatic volume is reduced in adult patients with recently diagnosed type 1 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, E2109–E2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Calvo, T.; Ekwall, O.; Amirian, N.; Zapardiel-Gonzalo, J.; von Herrath, M.G. Increased immune cell infiltration of the exocrine pancreas: A possible contribution to the pathogenesis of type 1 diabetes. Diabetes 2014, 63, 3880–3890. [Google Scholar] [CrossRef] [Green Version]
- Visperas, A.; Vignali, D.A. Are Regulatory T Cells Defective in Type 1 Diabetes and Can We Fix Them? J. Immunol. 2016, 197, 3762–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, C.M.; Peakman, M.; Tree, T.I.M. Regulatory T cell dysfunction in type 1 diabetes: What’s broken and how can we fix it? Diabetologia 2017, 60, 1839–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinberg-Bleyer, Y.; Baeyens, A.; You, S.; Elhage, R.; Fourcade, G.; Gregoire, S.; Cagnard, N.; Carpentier, W.; Tang, Q.; Bluestone, J.; et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J. Exp. Med. 2010, 207, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, C.J.; Ward, N.C.; Pugliese, A.; Malek, T.R. Promoting Immune Regulation in Type 1 Diabetes Using Low-Dose Interleukin-2. Curr. Diabetes Rep. 2016, 16, 46. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Hiam-Galvez, K.J.; Mowery, C.T.; Herold, K.C.; Gitelman, S.E.; Esensten, J.H.; Liu, W.; Lares, A.P.; Leinbach, A.S.; Lee, M.; et al. The effect of low-dose IL-2 and Treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight 2021, 6, e147474. [Google Scholar] [CrossRef] [PubMed]
- Long, S.A.; Rieck, M.; Sanda, S.; Bollyky, J.B.; Samuels, P.L.; Goland, R.; Ahmann, A.; Rabinovitch, A.; Aggarwal, S.; Phippard, D.; et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes 2012, 61, 2340–2348. [Google Scholar] [CrossRef] [Green Version]
- Gardner, G.; Fraker, C.A. Natural Killer Cells as Key Mediators in Type I Diabetes Immunopathology. Front. Immunol. 2021, 12, 722979. [Google Scholar] [CrossRef]
- Stene, L.C.; Oikarinen, S.; Hyoty, H.; Barriga, K.J.; Norris, J.M.; Klingensmith, G.; Hutton, J.C.; Erlich, H.A.; Eisenbarth, G.S.; Rewers, M. Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: The Diabetes and Autoimmunity Study in the Young (DAISY). Diabetes 2010, 59, 3174–3180. [Google Scholar] [CrossRef] [Green Version]
- Lönnrot, M.; Salminen, K.; Knip, M.; Savola, K.; Kulmala, P.; Leinikki, P.; Hyypiä, T.; Akerblom, H.K.; Hyöty, H. Enterovirus RNA in serum is a risk factor for beta-cell autoimmunity and clinical type 1 diabetes: A prospective study. J. Med. Virol. 2000, 61, 214–220. [Google Scholar] [CrossRef]
- Simonen-Tikka, M.L.; Pflueger, M.; Klemola, P.; Savolainen-Kopra, C.; Smura, T.; Hummel, S.; Kaijalainen, S.; Nuutila, K.; Natri, O.; Roivainen, M.; et al. Human enterovirus infections in children at increased risk for type 1 diabetes: The Babydiet study. Diabetologia 2011, 54, 2995–3002. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Rosolowsky, E.; Pacaud, D.; Huang, C.; Lemay, J.A.; Brockman, N.; Rath, M.; Doulla, M. Diabetic ketoacidosis at type 1 diabetes diagnosis in children during the COVID-19 pandemic. Pediatr. Diabetes 2021, 22, 552–557. [Google Scholar] [CrossRef]
- Qeadan, F.; Tingey, B.; Egbert, J.; Pezzolesi, M.G.; Burge, M.R.; Peterson, K.A.; Honda, T. The associations between COVID-19 diagnosis, type 1 diabetes, and the risk of diabetic ketoacidosis: A nationwide cohort from the US using the Cerner Real-World Data. PLoS ONE 2022, 17, e0266809. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Vannucci, S.J.; Yan, S.S.D.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheijen, J.; Clevers, E.; Engelen, L.; Dagnelie, P.C.; Brouns, F.; Stehouwer, C.D.A.; Schalkwijk, C.G. Analysis of advanced glycation endproducts in selected food items by ultra-performance liquid chromatography tandem mass spectrometry: Presentation of a dietary AGE database. Food Chem. 2016, 190, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bugel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. 2013, 60, 10–37. [Google Scholar] [CrossRef]
- Jud, P.; Sourij, H. Therapeutic options to reduce advanced glycation end products in patients with diabetes mellitus: A review. Diabetes Res. Clin. Pract. 2019, 148, 54–63. [Google Scholar] [CrossRef]
- Sergi, D.; Boulestin, H.; Campbell, F.M.; Williams, L.M. The Role of Dietary Advanced Glycation End Products in Metabolic Dysfunction. Mol. Nutr. Food Res. 2021, 65, e1900934. [Google Scholar] [CrossRef]
- Sharma, C.; Kaur, A.; Thind, S.S.; Singh, B.; Raina, S. Advanced glycation End-products (AGEs): An emerging concern for processed food industries. J. Food Sci. Technol. 2015, 52, 7561–7576. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.H.; Lin, X.; Bu, C.; Zhang, X. Role of advanced glycation end products in mobility and considerations in possible dietary and nutritional intervention strategies. Nutr. Metab. 2018, 15, 72. [Google Scholar] [CrossRef] [Green Version]
- Huebschmann, A.G.; Regensteiner, J.G.; Vlassara, H.; Reusch, J.E. Diabetes and advanced glycoxidation end products. Diabetes Care 2006, 29, 1420–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular effects of advanced glycation endproducts: Clinical effects and molecular mechanisms. Mol. Metab. 2014, 3, 94–108. [Google Scholar] [CrossRef]
- Qu, W.; Yuan, X.; Zhao, J.; Zhang, Y.; Hu, J.; Wang, J.; Li, J. Dietary advanced glycation end products modify gut microbial composition and partially increase colon permeability in rats. Mol. Nutr. Food Res. 2017, 61, 1700118. [Google Scholar] [CrossRef]
- Seiquer, I.; Rubio, L.A.; Peinado, M.J.; Delgado-Andrade, C.; Navarro, M.P. Maillard reaction products modulate gut microbiota composition in adolescents. Mol. Nutr. Food Res. 2014, 58, 1552–1560. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, D. Thermal processing of food reduces gut microbiota diversity of the host and triggers adaptation of the microbiota: Evidence from two vertebrates. Microbiome 2018, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Marungruang, N.; Fåk, F.; Tareke, E. Heat-treated high-fat diet modifies gut microbiota and metabolic markers in apoe−/− mice. Nutr. Metab. 2016, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Sun, L.; Zhang, S.; Zhao, X.; Gang, X.; Wang, G. Evaluating the Causal Role of Gut Microbiota in Type 1 Diabetes and Its Possible Pathogenic Mechanisms. Front. Endocrinol. 2020, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Snelson, M.; Coughlan, M.T. Dietary Advanced Glycation End Products: Digestion, Metabolism and Modulation of Gut Microbial Ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Bos, D.C.; de Ranitz-Greven, W.L.; de Valk, H.W. Advanced Glycation End Products, Measured as Skin Autofluorescence and Diabetes Complications: A Systematic Review. Diabetes Technol. Ther. 2011, 13, 773–779. [Google Scholar] [CrossRef]
- Beyan, H.; Riese, H.; Hawa, M.I.; Beretta, G.; Davidson, H.W.; Hutton, J.C.; Burger, H.; Schlosser, M.; Snieder, H.; Boehm, B.O.; et al. Glycotoxin and autoantibodies are additive environmentally determined predictors of type 1 diabetes: A twin and population study. Diabetes 2012, 61, 1192–1198. [Google Scholar] [CrossRef] [Green Version]
- Coughlan, M.T.; Yap, F.Y.; Tong, D.C.; Andrikopoulos, S.; Gasser, A.; Thallas-Bonke, V.; Webster, D.E.; Miyazaki, J.; Kay, T.W.; Slattery, R.M.; et al. Advanced glycation end products are direct modulators of beta-cell function. Diabetes 2011, 60, 2523–2532. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhao, C.; Zhang, X.H.; Zheng, F.; Cai, W.; Vlassara, H.; Ma, Z.A. Advanced glycation end products inhibit glucose-stimulated insulin secretion through nitric oxide-dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology 2009, 150, 2569–2576. [Google Scholar] [CrossRef]
- Borg, D.J.; Yap, F.Y.T.; Keshvari, S.; Simmons, D.G.; Gallo, L.A.; Fotheringham, A.K.; Zhuang, A.; Slattery, R.M.; Hasnain, S.Z.; Coughlan, M.T.; et al. Perinatal exposure to high dietary advanced glycation end products in transgenic NOD8.3 mice leads to pancreatic beta cell dysfunction. Islets 2018, 10, 10–24. [Google Scholar] [CrossRef]
- Peppa, M.; He, C.; Hattori, M.; McEvoy, R.; Zheng, F.; Vlassara, H. Fetal or neonatal low-glycotoxin environment prevents autoimmune diabetes in NOD mice. Diabetes 2003, 52, 1441–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mericq, V.; Piccardo, C.; Cai, W.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H.; Uribarri, J. Maternally transmitted and food-derived glycotoxins: A factor preconditioning the young to diabetes? Diabetes Care 2010, 33, 2232–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, J.M.; Soderlund, J.; Yap, F.Y.; Knip, M.; Andrikopoulos, S.; Ilonen, J.; Simell, O.; Veijola, R.; Sourris, K.C.; Coughlan, M.T.; et al. Receptor for advanced glycation end-products (RAGE) provides a link between genetic susceptibility and environmental factors in type 1 diabetes. Diabetologia 2011, 54, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Sourris, K.C.; de Courten, M.P.; Dougherty, S.L.; Chand, V.; Lyons, J.G.; Bertovic, D.; Coughlan, M.T.; Schlaich, M.P.; Soldatos, G.; et al. Advanced glycation end products (AGEs) are cross-sectionally associated with insulin secretion in healthy subjects. Amino Acids 2014, 46, 321–326. [Google Scholar] [CrossRef]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diabetes Rep. 2014, 14, 453. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, H.B.; Smith, R.J. Fatty liver disease in diabetes mellitus. Hepatobiliary Surg. Nutr. 2015, 4, 101–108. [Google Scholar] [CrossRef]
- Barros, B.S.V.; Santos, D.C.; Pizarro, M.H.; del Melo, L.G.N.; Gomes, M.B. Type 1 Diabetes and Non-Alcoholic Fatty Liver Disease: When Should We Be Concerned? A Nationwide Study in Brazil. Nutrients 2017, 9, 878. [Google Scholar] [CrossRef] [Green Version]
- De Courten, B.; de Courten, M.P.; Soldatos, G.; Dougherty, S.L.; Straznicky, N.; Schlaich, M.; Sourris, K.C.; Chand, V.; Scheijen, J.L.; Kingwell, B.A.; et al. Diet low in advanced glycation end products increases insulin sensitivity in healthy overweight individuals: A double-blind, randomized, crossover trial. Am. J. Clin. Nutr. 2016, 103, 1426–1433. [Google Scholar] [CrossRef]
- Vlassara, H.; Cai, W.; Tripp, E.; Pyzik, R.; Yee, K.; Goldberg, L.; Tansman, L.; Chen, X.; Mani, V.; Fayad, Z.A.; et al. Oral AGE restriction ameliorates insulin resistance in obese individuals with the metabolic syndrome: A randomised controlled trial. Diabetologia 2016, 59, 2181–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harcourt, B.E.; Sourris, K.C.; Coughlan, M.T.; Walker, K.Z.; Dougherty, S.L.; Andrikopoulos, S.; Morley, A.L.; Thallas-Bonke, V.; Chand, V.; Penfold, S.A.; et al. Targeted reduction of advanced glycation improves renal function in obesity. Kidney Int. 2011, 80, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.; Herath, C.B.; Jia, Z.; Goodwin, M.; Mak, K.Y.; Watt, M.J.; Forbes, J.M.; Angus, P.W. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J. Hepatol. 2014, 60, 832–838. [Google Scholar] [CrossRef]
- Leung, C.; Herath, C.B.; Jia, Z.; Andrikopoulos, S.; Brown, B.E.; Davies, M.J.; Rivera, L.R.; Furness, J.B.; Forbes, J.M.; Angus, P.W. Dietary advanced glycation end-products aggravate non-alcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 8026–8040. [Google Scholar] [CrossRef] [Green Version]
- Egana-Gorrono, L.; Lopez-Diez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights from Human Subjects and Animal Models. Front. Cardiovasc. Med. 2020, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int. J. Inflam. 2013, 2013, 403460. [Google Scholar] [CrossRef] [Green Version]
- Goh, S.Y.; Cooper, M.E. Clinical review: The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Rai, V.; Singer, D.; Chabierski, S.; Xie, J.; Reverdatto, S.; Burz, D.S.; Schmidt, A.M.; Hoffmann, R.; Shekhtman, A. Advanced glycation end product recognition by the receptor for AGEs. Structure 2011, 19, 722–732. [Google Scholar] [CrossRef] [Green Version]
- Ohgami, N.; Nagai, R.; Ikemoto, M.; Arai, H.; Miyazaki, A.; Hakamata, H.; Horiuchi, S.; Nakayama, H. CD36, serves as a receptor for advanced glycation endproducts (AGE). J. Diabetes Complicat. 2002, 16, 56–59. [Google Scholar] [CrossRef]
- Horiuchi, S.; Sakamoto, Y.; Sakai, M. Scavenger receptors for oxidized and glycated proteins. Amino Acids 2003, 25, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, N.; Nagai, R.; Miyazaki, A.; Ikemoto, M.; Arai, H.; Horiuchi, S.; Nakayama, H. Scavenger Receptor Class B Type I-mediated Reverse Cholesterol Transport Is Inhibited by Advanced Glycation End Products*. J. Biol. Chem. 2001, 276, 13348–13355. [Google Scholar] [CrossRef] [Green Version]
- Ohgami, N.; Nagai, R.; Ikemoto, M.; Arai, H.; Kuniyasu, A.; Horiuchi, S.; Nakayama, H. CD36, a Member of the Class B Scavenger Receptor Family, as a Receptor for Advanced Glycation End Products *. J. Biol. Chem. 2001, 276, 3195–3202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchałowicz, K.; Rać, M.E. The Multifunctionality of CD36 in Diabetes Mellitus and Its Complications—Update in Pathogenesis, Treatment and Monitoring. Cells 2020, 9, 1877. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Cui, W.; Silverstein, R.L. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J. Exp. Med. 2022, 219, e20211314. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012.e5. [Google Scholar] [CrossRef]
- Vlassara, H.; Li, Y.M.; Imani, F.; Wojciechowicz, D.; Yang, Z.; Liu, F.T.; Cerami, A. Identification of galectin-3 as a high-affinity binding protein for advanced glycation end products (AGE): A new member of the AGE-receptor complex. Mol. Med. 1995, 1, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, T.; Zhou, Z.; Xiao, Y. Emerging roles of Galectin-3 in diabetes and diabetes complications: A snapshot. Rev. Endocr. Metab. Disord. 2022, 23, 569–577. [Google Scholar] [CrossRef]
- Karlsen, A.E.; Størling, Z.M.; Sparre, T.; Larsen, M.R.; Mahmood, A.; Størling, J.; Roepstorff, P.; Wrzesinski, K.; Larsen, P.M.; Fey, S.; et al. Immune-mediated β-cell destruction in vitro and in vivo—A pivotal role for galectin-3. Biochem. Biophys. Res. Commun. 2006, 344, 406–415. [Google Scholar] [CrossRef]
- Saksida, T.; Nikolic, I.; Vujicic, M.; Nilsson, U.J.; Leffler, H.; Lukic, M.L.; Stojanovic, I.; Stosic-Grujicic, S. Galectin-3 deficiency protects pancreatic islet cells from cytokine-triggered apoptosis in vitro. J. Cell. Physiol. 2013, 228, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Mensah-Brown, E.P.K.; Al Rabesi, Z.; Shahin, A.; Al Shamsi, M.; Arsenijevic, N.; Hsu, D.K.; Liu, F.T.; Lukic, M.L. Targeted disruption of the galectin-3 gene results in decreased susceptibility to multiple low dose streptozotocin-induced diabetes in mice. Clin. Immunol. 2009, 130, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhang, Y.; Huang, Y.; Deng, H. Pathophysiology of RAGE in inflammatory diseases. Front. Immunol. 2022, 13, 931473. [Google Scholar] [CrossRef] [PubMed]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef] [Green Version]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef]
- Ebert, H.; Lacruz, M.E.; Kluttig, A.; Simm, A.; Greiser, K.H.; Tiller, D.; Kartschmit, N.; Mikolajczyk, R. Advanced glycation end products and their ratio to soluble receptor are associated with limitations in physical functioning only in women: Results from the CARLA cohort. BMC Geriatr. 2019, 19, 299. [Google Scholar] [CrossRef]
- Le Bagge, S.; Fotheringham, A.K.; Leung, S.S.; Forbes, J.M. Targeting the receptor for advanced glycation end products (RAGE) in type 1 diabetes. Med. Res. Rev. 2020, 40, 1200–1219. [Google Scholar] [CrossRef]
- Salonen, K.M.; Ryhanen, S.J.; Forbes, J.M.; Harkonen, T.; Ilonen, J.; Simell, O.; Veijola, R.; Groop, P.H.; Knip, M. A drop in the circulating concentrations of soluble receptor for advanced glycation end products is associated with seroconversion to autoantibody positivity but not with subsequent progression to clinical disease in children en route to type 1 diabetes. Diabetes Metab. Res. Rev. 2017, 33, e2872. [Google Scholar] [CrossRef]
- Salonen, K.M.; Ryhanen, S.J.; Forbes, J.M.; Harkonen, T.; Ilonen, J.; Laine, A.P.; Groop, P.H.; Knip, M.; Finnish Pediatric Diabetes, R. Circulating concentrations of soluble receptor for AGE are associated with age and AGER gene polymorphisms in children with newly diagnosed type 1 diabetes. Diabetes Care 2014, 37, 1975–1981. [Google Scholar] [CrossRef]
- Salonen, K.M.; Ryhanen, S.J.; Forbes, J.M.; Borg, D.J.; Harkonen, T.; Ilonen, J.; Simell, O.; Veijola, R.; Groop, P.H.; Knip, M. Decrease in circulating concentrations of soluble receptors for advanced glycation end products at the time of seroconversion to autoantibody positivity in children with prediabetes. Diabetes Care 2015, 38, 665–670. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.C.; Woodward, M.; Neal, B.; Li, Q.; Pickering, R.; Marre, M.; Williams, B.; Perkovic, V.; Cooper, M.E.; Zoungas, S.; et al. Relationship between levels of advanced glycation end products and their soluble receptor and adverse outcomes in adults with type 2 diabetes. Diabetes Care 2015, 38, 1891–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, B.; Desai, D.D.; Downie, M.P.; Chen, Y.; Yan, S.F.; Herold, K.; Schmidt, A.M.; Clynes, R. Receptor for advanced glycation end products expression on T cells contributes to antigen-specific cellular expansion in vivo. J. Immunol. 2007, 179, 8051–8058. [Google Scholar] [CrossRef] [Green Version]
- Leung, S.S.; Borg, D.J.; McCarthy, D.A.; Boursalian, T.E.; Cracraft, J.; Zhuang, A.; Fotheringham, A.K.; Flemming, N.; Watkins, T.; Miles, J.J.; et al. Soluble RAGE Prevents Type 1 Diabetes Expanding Functional Regulatory T Cells. Diabetes 2022, 71, 1994–2008. [Google Scholar] [CrossRef]
- Chen, Y.; Akirav, E.M.; Chen, W.; Henegariu, O.; Moser, B.; Desai, D.; Shen, J.M.; Webster, J.C.; Andrews, R.C.; Mjalli, A.M.; et al. RAGE ligation affects T cell activation and controls T cell differentiation. J. Immunol. 2008, 181, 4272–4278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.Q.; Gong, Z.J.; Xu, S.Q.; Li, X.; Wang, L.K.; Wu, S.M.; Wu, J.H.; Yang, H.F. Advanced glycation end products promote differentiation of CD4+ T helper cells toward pro-inflammatory response. J. Huazhong Univ. Sci. Technol. Med. Sci. 2014, 34, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Akirav, E.M.; Preston-Hurlburt, P.; Garyu, J.; Henegariu, O.; Clynes, R.; Schmidt, A.M.; Herold, K.C. RAGE expression in human T cells: A link between environmental factors and adaptive immune responses. PLoS ONE 2012, 7, e34698. [Google Scholar] [CrossRef] [PubMed]
- Durning, S.P.; Preston-Hurlburt, P.; Clark, P.R.; Xu, D.; Herold, K.C.; Type 1 Diabetes TrialNet Study, G. The Receptor for Advanced Glycation Endproducts Drives T Cell Survival and Inflammation in Type 1 Diabetes Mellitus. J. Immunol. 2016, 197, 3076–3085. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, L.; Wang, F.; Zou, Y.; Li, J.; Luo, J.; Khan, F.; Sun, F.; Li, Y.; Liu, J.; et al. Extracellular HMGB1 exacerbates autoimmune progression and recurrence of type 1 diabetes by impairing regulatory T cell stability. Diabetologia 2020, 63, 987–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Zhong, J.; Wei, W.; Wang, Y.; Huang, Y.; Yang, P.; Purohit, S.; Dong, Z.; Wang, M.H.; She, J.X.; et al. Extracellular high-mobility group box 1 acts as an innate immune mediator to enhance autoimmune progression and diabetes onset in NOD mice. Diabetes 2008, 57, 2118–2127. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, C.; Whiddett, R.O.; Buckle, I.; Chen, C.; Forbes, J.M.; Fotheringham, A.K. Advanced Glycation End Products and Inflammation in Type 1 Diabetes Development. Cells 2022, 11, 3503. https://doi.org/10.3390/cells11213503
Du C, Whiddett RO, Buckle I, Chen C, Forbes JM, Fotheringham AK. Advanced Glycation End Products and Inflammation in Type 1 Diabetes Development. Cells. 2022; 11(21):3503. https://doi.org/10.3390/cells11213503
Chicago/Turabian StyleDu, Chenping, Rani O. Whiddett, Irina Buckle, Chen Chen, Josephine M. Forbes, and Amelia K. Fotheringham. 2022. "Advanced Glycation End Products and Inflammation in Type 1 Diabetes Development" Cells 11, no. 21: 3503. https://doi.org/10.3390/cells11213503