


Src Family Kinases Facilitate the Crosstalk between CGRP and Cytokines in Sensitizing Trigeminal Ganglion via Transmitting CGRP Receptor/PKA Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Mouse TG Tissue Culture
2.3. ELISA
2.4. Multiplex Immunoassay
2.5. qPCR
2.6. Western Blot
2.7. Immunohistochemistry
2.8. Statistical Analysis
3. Results
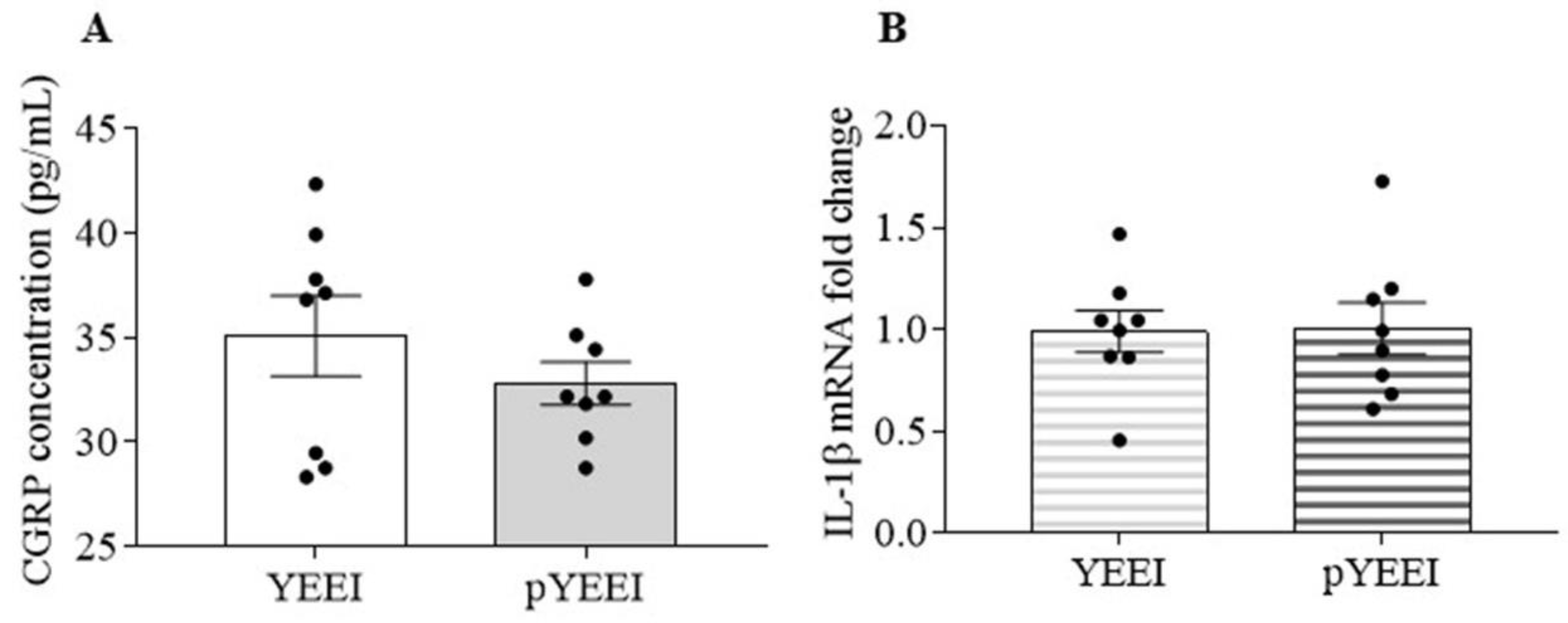
3.1. pYEEI Alone Did Not Increase CGRP Release and IL-1β Gene Expression in the Mouse TG
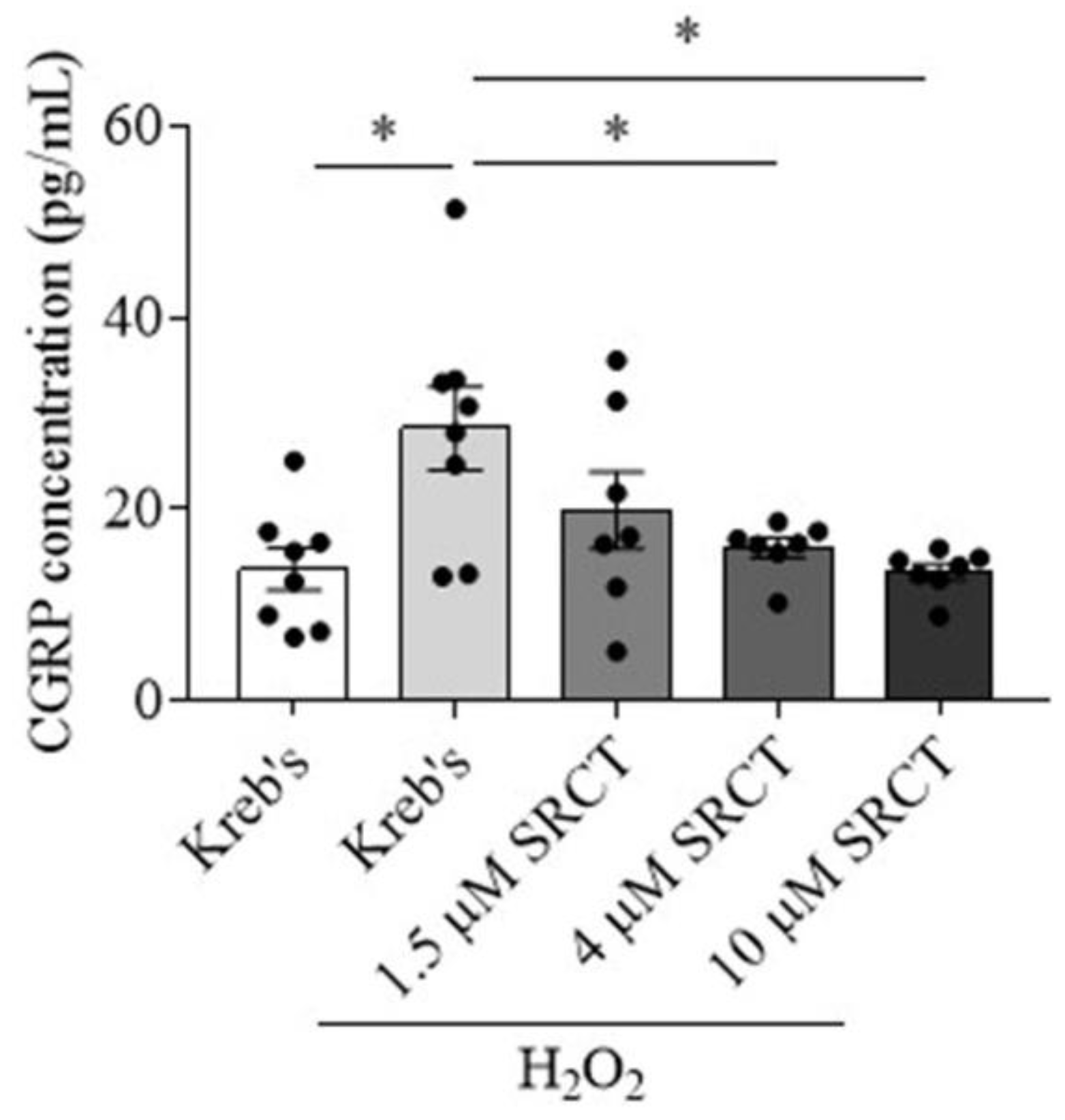
3.2. Saracatinib Reduced CGRP Release Induced by H2O2 in the Mouse TG
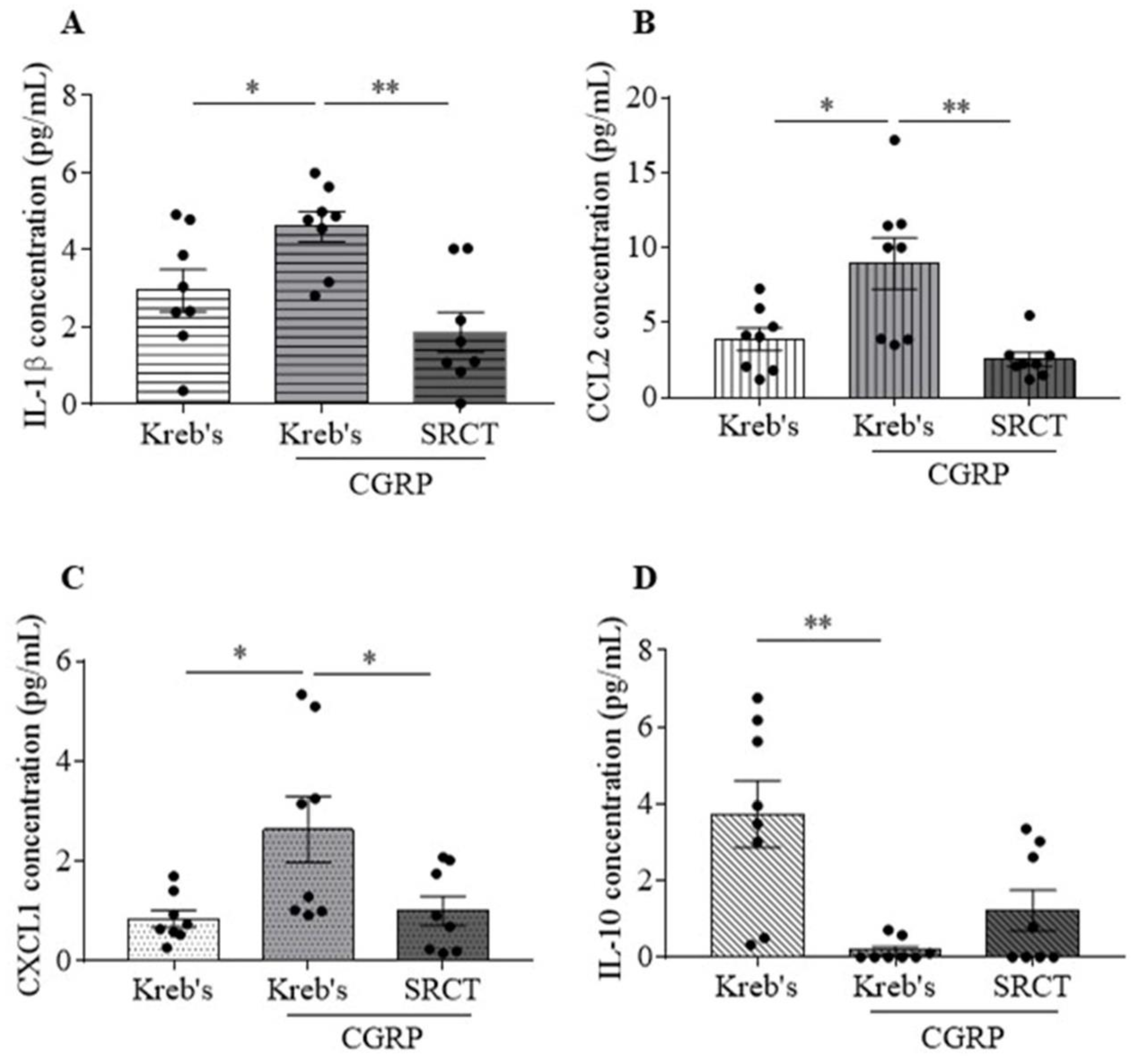
3.3. Saracatinib Reduced IL-1β, CCL2, and CXCL1 Release Induced by CGRP in the Mouse TG
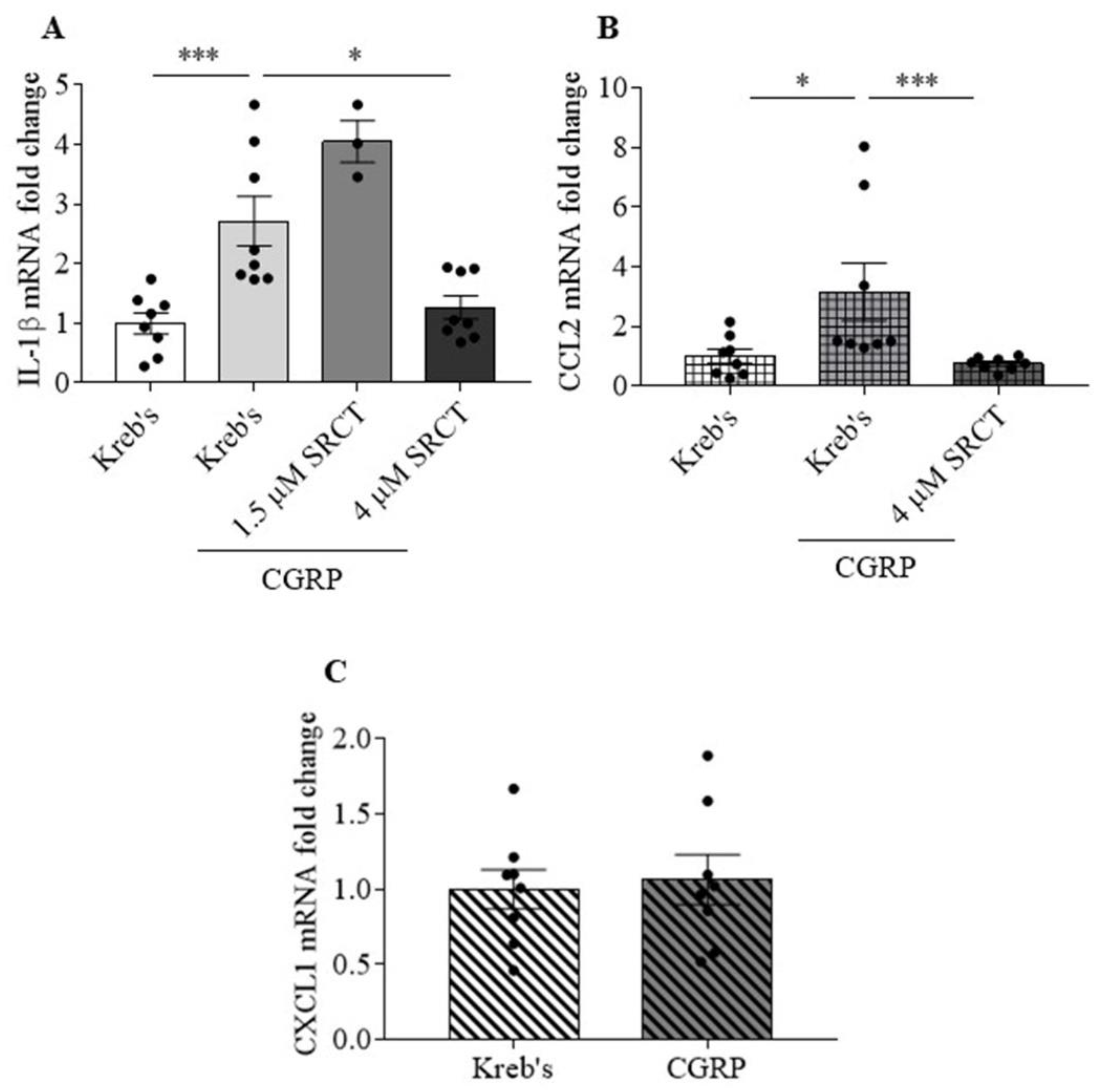
3.4. Saracatinib Reduced IL-1β and CCL2 Gene Expression Induced by CGRP in the Mouse TG
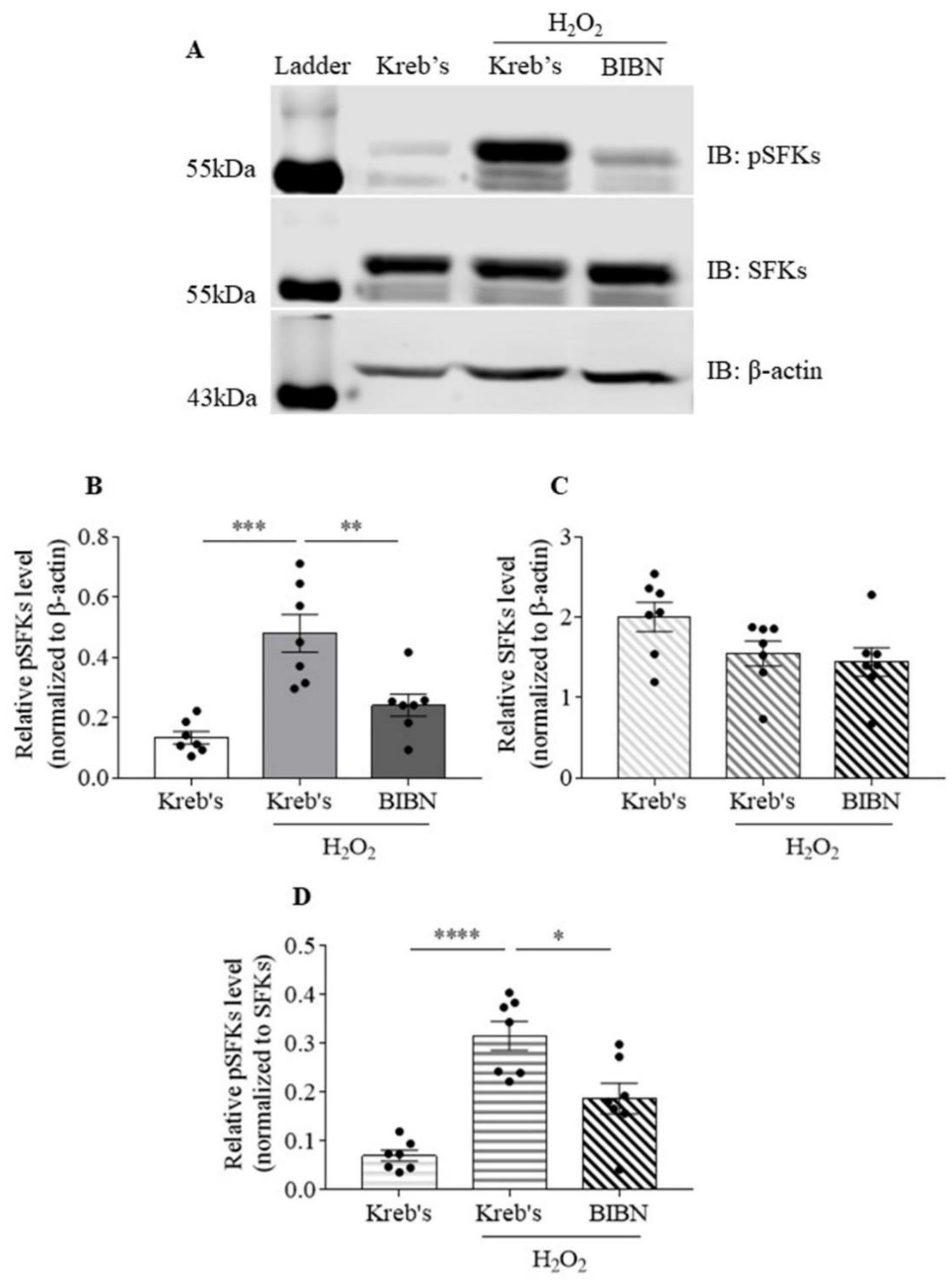
3.5. The Protein Level of Phosphorylated SFKs at Y416 Was Increased by H2O2, Which Was Reduced by BIBN4096 in the Mouse TG
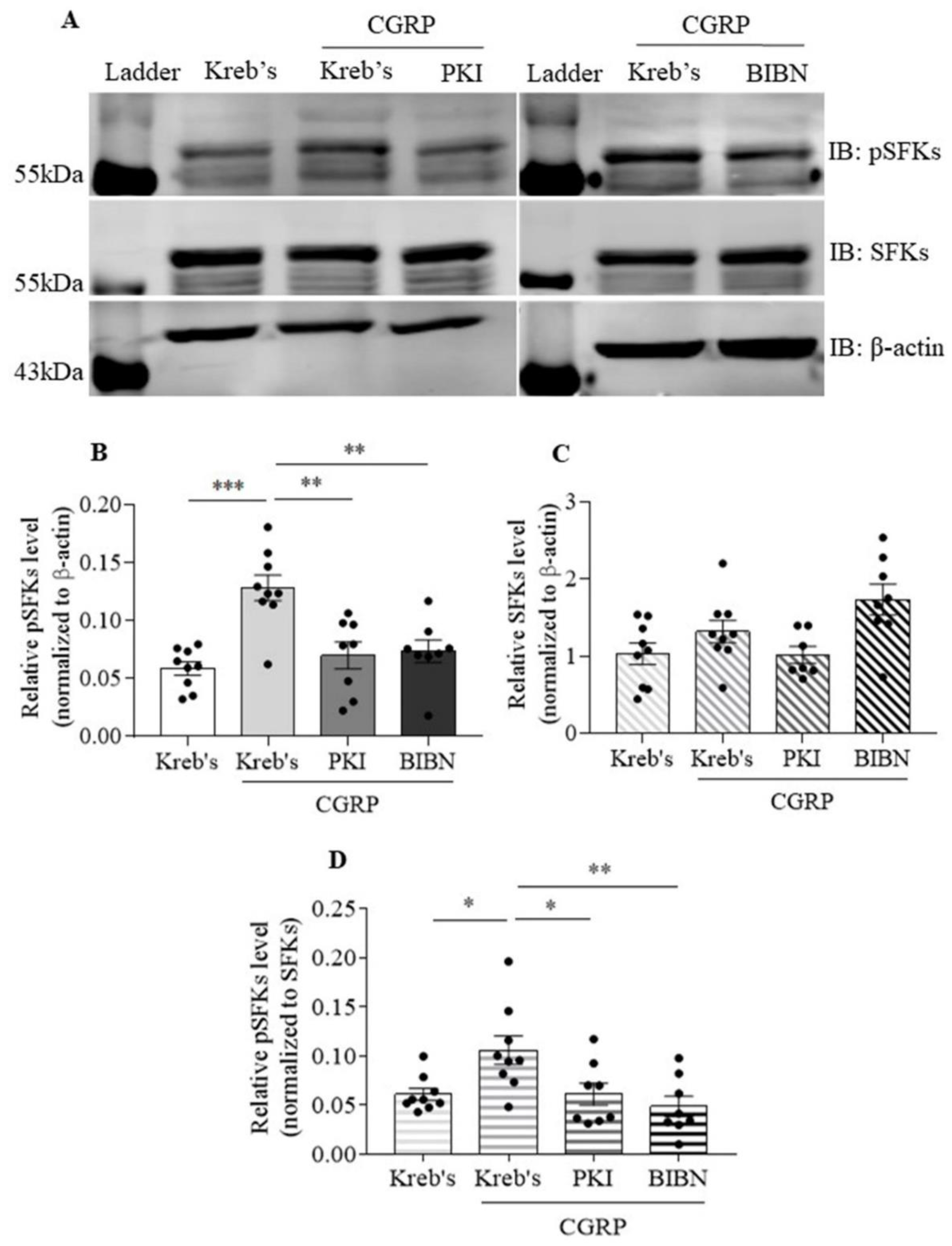
3.6. The Protein Level of Phosphorylated SFKs at Y416 Was Increased by CGRP, Which Was Reduced by Both PKI (14-22) Amide and BIBN4096 in the Mouse TG
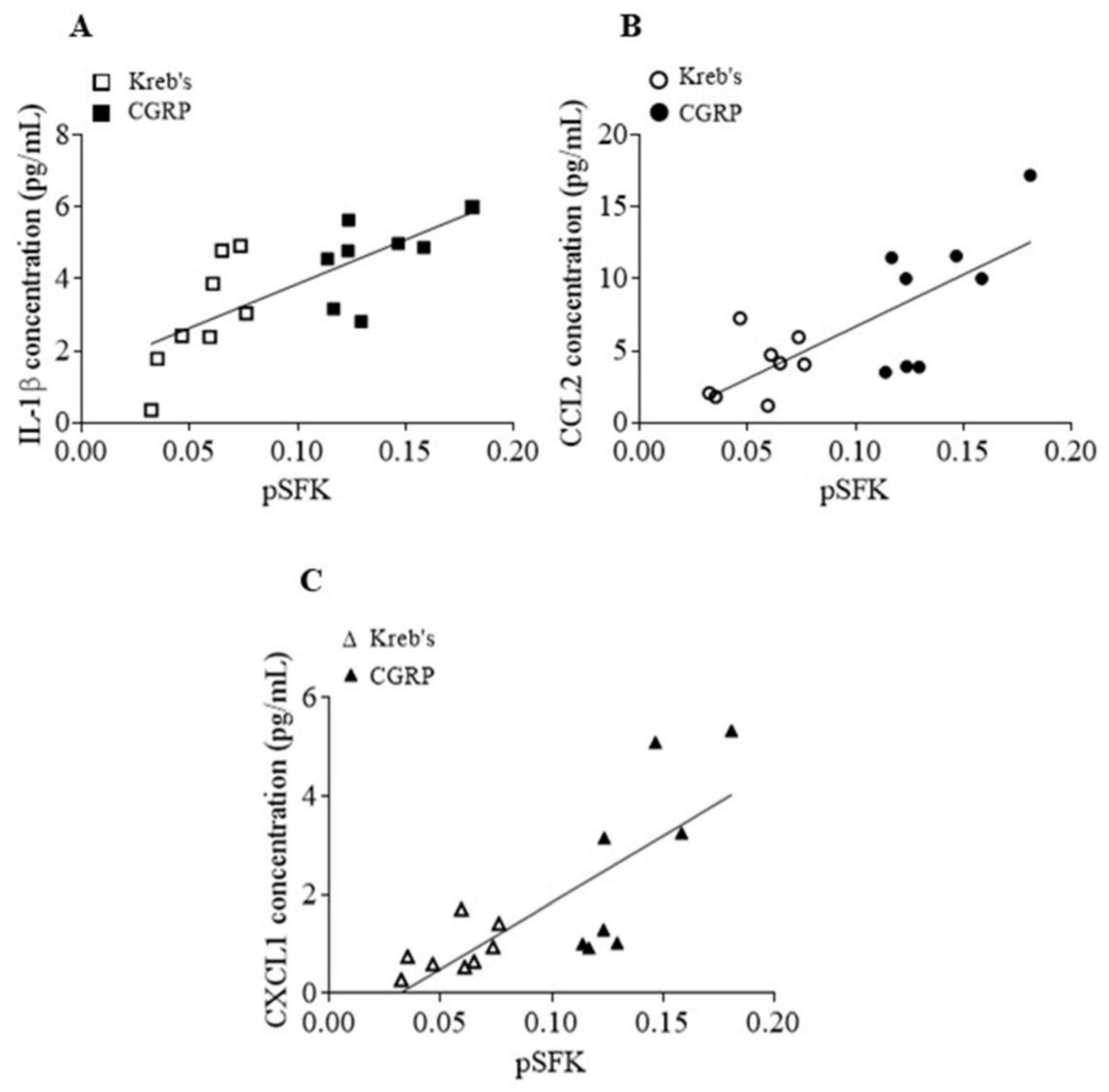
3.7. The Protein Levels of Phosphorylated SFKs at Y416 and Released Cytokines Induced by CGRP Were Positively Correlated in the Mouse TG
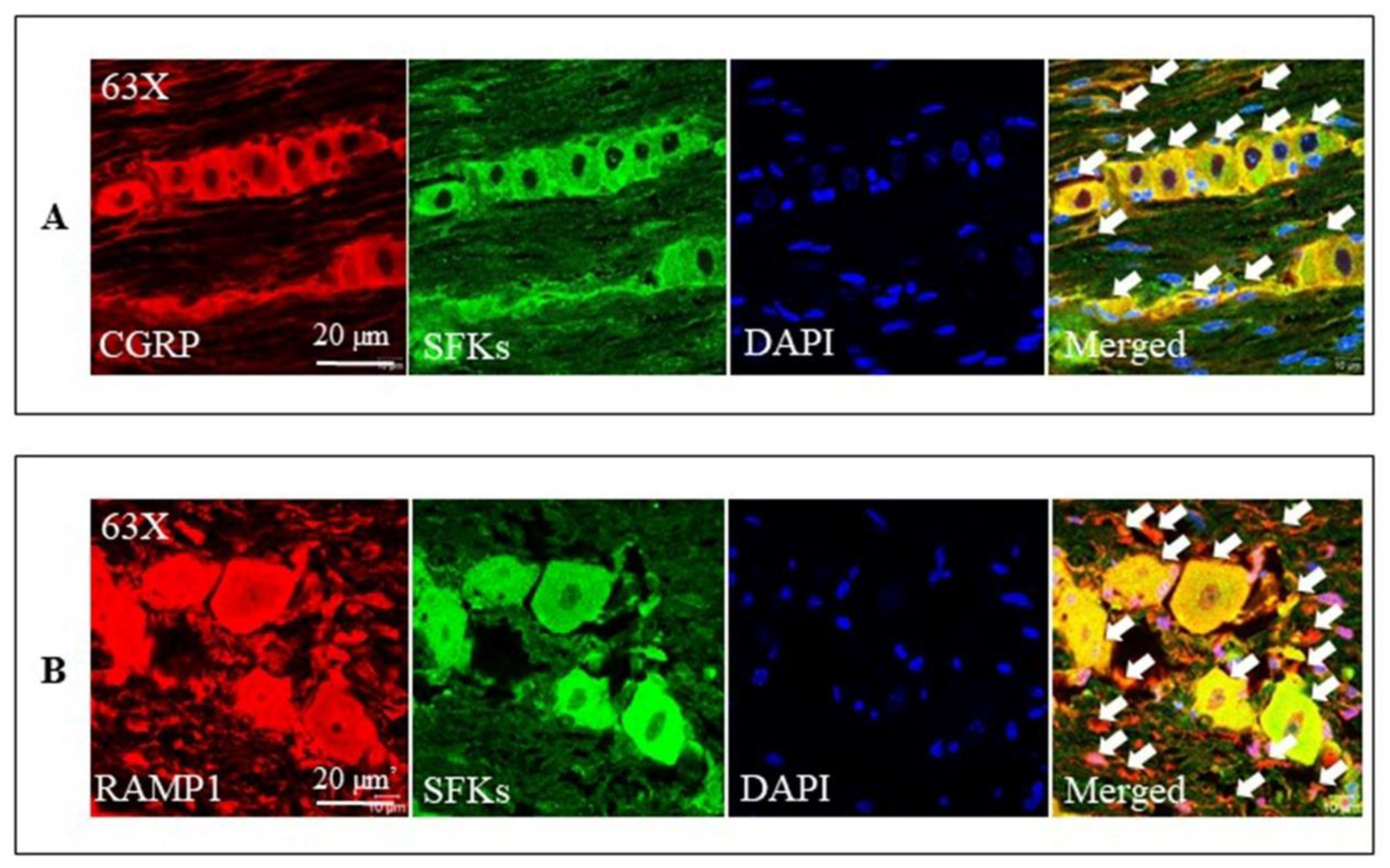
3.8. SFKs Co-Localized with CGRP and RAMP1 in the Mouse TG
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashina, M.; Katsarava, Z.; Do, T.P.; Buse, D.C.; Pozo-Rosich, P.; Özge, A.; Krymchantowski, A.V.; Lebedeva, E.R.; Ravishankar, K.; Yu, S.; et al. Migraine: Epidemiology and systems of care. Lancet 2021, 397, 1485–1495. [Google Scholar] [CrossRef]
- Bernstein, C.; Burstein, R. Sensitization of the Trigeminovascular Pathway: Perspective and Implications to Migraine Pathophysiology. J. Clin. Neurol. 2012, 8, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Noseda, R.; Burstein, R. Migraine pathophysiology: Anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain 2013, 154, S44–S53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashina, M.; Hansen, J.M.; Do, T.P.; Melo-Carrillo, A.; Burstein, R.; Moskowitz, M.A. Migraine and the trigeminovascular system-40 years and counting. Lancet Neurol. 2019, 18, 795–804. [Google Scholar] [CrossRef]
- Ray, B.S.; Wolff, H.G. Experimental studies on headache: Pain-sensitive structures of the head and their significance in heaadache. Arch. Surg. 1940, 41, 813–856. [Google Scholar] [CrossRef]
- Moskowitz, M.A.; Macfarlane, R. Neurovascular and molecular mechanisms in migraine headaches. Cerebrovasc. Brain Metab. Rev. 1993, 5, 159–177. [Google Scholar] [PubMed]
- Olesen, J.; Burstein R Fau-Ashina, M.; Ashina M Fau-Tfelt-Hansen, P.; Tfelt-Hansen, P. Origin of pain in migraine: Evidence for peripheral sensitisation. Lancet Neurol. 2009, 8, 679–690. [Google Scholar] [CrossRef]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K.; Krause, D.N. CGRP as the target of new migraine therapies—successful translation from bench to clinic. Nat. Rev. Neurol. 2018, 14, 338–350. [Google Scholar] [CrossRef]
- Chatchaisak, D.; Connor, M.; Srikiatkhachorn, A.; Chetsawang, B. The potentiating effect of calcitonin gene-related peptide on transient receptor potential vanilloid-1 activity and the electrophysiological responses of rat trigeminal neurons to nociceptive stimuli. J. Physiol. Sci. 2018, 68, 261–268. [Google Scholar] [CrossRef]
- Edvinsson, J.C.A.; Warfvinge, K.; Krause, D.N.; Blixt, F.W.; Sheykhzade, M.; Edvinsson, L.; Haanes, K.A. C-fibers may modulate adjacent Aδ-fibers through axon-axon CGRP signaling at nodes of Ranvier in the trigeminal system. J. Headache Pain 2019, 20, 105. [Google Scholar] [CrossRef]
- Afroz, S.; Arakaki, R.; Iwasa, T.; Oshima, M.; Hosoki, M.; Inoue, M.; Baba, O.; Okayama, Y.; Matsuka, Y. CGRP Induces Differential Regulation of Cytokines from Satellite Glial Cells in Trigeminal Ganglia and Orofacial Nociception. Int. J. Mol. Sci. 2019, 20, 711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thalakoti, S.; Patil, V.V.; Damodaram, S.; Vause, C.V.; Langford, L.E.; Freeman, S.E.; Durham, P.L. Neuron–Glia Signaling in Trigeminal Ganglion: Implications for Migraine Pathology. Headache 2007, 47, 1008–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vause, C.V.; Durham, P.L. Calcitonin gene-related peptide differentially regulates gene and protein expression in trigeminal glia cells: Findings from array analysis. Neurosci. Lett. 2010, 473, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Afroz, S.; Arakaki, R.; Iwasa, T.; Waskitho, A.; Oshima, M.; Matsuka, Y. Role of CGRP in Neuroimmune Interaction via NF-κB Signaling Genes in Glial Cells of Trigeminal Ganglia. Int. J. Mol. Sci. 2020, 21, 6005. [Google Scholar] [CrossRef]
- Bowen, E.J.; Schmidt, T.W.; Firm, C.S.; Russo, A.F.; Durham, P.L. Tumor necrosis factor-α stimulation of calcitonin gene-related peptide expression and secretion from rat trigeminal ganglion neurons. J. Neurochem. 2006, 96, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capuano, A.; De Corato, A.; Lisi, L.; Tringali, G.; Navarra, P.; Russo, C.D. Proinflammatory-Activated Trigeminal Satellite Cells Promote Neuronal Sensitization: Relevance for Migraine Pathology. Mol. Pain 2009, 5, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messlinger, K.; Russo, A.F. Current understanding of trigeminal ganglion structure and function in headache. Cephalalgia Int. J. Headache 2018, 39, 1661–1674. [Google Scholar] [CrossRef]
- Iyengar, S.; Johnson, K.W.; Ossipov, M.H.; Aurora, S.K. CGRP and the Trigeminal System in Migraine. Headache 2019, 59, 659–681. [Google Scholar] [CrossRef] [Green Version]
- Messlinger, K.; Balcziak, L.K.; Russo, A.F. Cross-talk signaling in the trigeminal ganglion: Role of neuropeptides and other mediators. J. Neural Transm. 2020, 127, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Park, K.A.; Fehrenbacher, J.C.; Thompson, E.L.; Duarte, D.B.; Hingtgen, C.M.; Vasko, M.R. Signaling pathways that mediate nerve growth factor-induced increase in expression and release of calcitonin gene-related peptide from sensory neurons. Neuroscience 2010, 171, 910–923. [Google Scholar] [CrossRef]
- Nie, L.; Jiang, L.; Quinn, J.P.; Grubb, B.D.; Wang, M. TRPA1-Mediated Src Family Kinases Activity Facilitates Cortical Spreading Depression Susceptibility and Trigeminovascular System Sensitization. Int. J. Mol. Sci. 2021, 22, 12273. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-J.; Ou, Y.-C.; Chang, C.-Y.; Pan, H.-C.; Lin, S.-Y.; Liao, S.-L.; Raung, S.-L.; Chen, S.-Y.; Chang, C.-J. Src signaling involvement in Japanese encephalitis virus-induced cytokine production in microglia. Neurochem. Int. 2011, 58, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Portugal Camila, C.; Socodato, R.; Canedo, T.; Silva Cátia, M.; Martins, T.; Coreixas Vivian, S.M.; Loiola Erick, C.; Gess, B.; Röhr, D.; Santiago Ana, R.; et al. Caveolin-1–mediated internalization of the vitamin C transporter SVCT2 in microglia triggers an inflammatory phenotype. Sci. Signal. 2017, 10, eaal2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Li, Y.; Zhong, X.; Hu, Y.; Liu, P.; Zhao, Y.; Deng, Z.; Liu, X.; Liu, S.; Zhong, Y. Src-family kinases activation in spinal microglia contributes to central sensitization and chronic pain after lumbar disc herniation. Mol. Pain 2017, 13, 1744806917733637. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Low, C.Y.B.; Wong, S.Y.; Lai, M.K.P.; Tan, M.G.K. Selective induction of alternatively spliced FynT isoform by TNF facilitates persistent inflammatory responses in astrocytes. Sci. Rep. 2017, 7, 43651. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-Y.; Zhou, H.-R.; Wang, S.; Liu, C.-Y.; Qin, G.-C.; Fu, Q.-Q.; Zhou, J.-Y.; Chen, L.-X. NR2B-Tyr phosphorylation regulates synaptic plasticity in central sensitization in a chronic migraine rat model. J. Headache Pain 2018, 19, 102. [Google Scholar] [CrossRef] [Green Version]
- Staehr, C.; Hangaard, L.; Bouzinova, E.V.; Kim, S.; Rajanathan, R.; Boegh Jessen, P.; Luque, N.; Xie, Z.; Lykke-Hartmann, K.; Sandow, S.L.; et al. Smooth muscle Ca2+ sensitization causes hypercontractility of middle cerebral arteries in mice bearing the familial hemiplegic migraine type 2 associated mutation. J. Cereb. Blood Flow Metab. 2018, 39, 1570–1587. [Google Scholar] [CrossRef]
- Nie, L.; Ma, D.; Quinn, J.P.; Wang, M. Src family kinases activity is required for transmitting purinergic P2X7 receptor signaling in cortical spreading depression and neuroinflammation. J. Headache Pain 2021, 22, 146. [Google Scholar] [CrossRef]
- Williamson, D.J.; Hargreaves, R.J.; Hill, R.G.; Shepheard, S.L. Sumatriptan Inhibits Neurogenic Vasodilation of Dural Blood Vessels in the Anaesthetized Rat—Intravital Microscope Studies. Cephalalgia Int. J. Headache 1997, 17, 525–531. [Google Scholar] [CrossRef]
- Storer, R.J.; Akerman, S.; Goadsby, P.J. Calcitonin gene-related peptide (CGRP) modulates nociceptive trigeminovascular transmission in the cat. Br. J. Pharmacol. 2004, 142, 1171–1181. [Google Scholar] [CrossRef]
- Coste, J.; Voisin, D.L.; Miraucourt, L.S.; Dallel, R.; Luccarini, P. Dorsal horn NK1-expressing neurons control windup of downstream trigeminal nociceptive neurons. Pain 2008, 137, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Dux, M.; Rosta, J.; Messlinger, K. TRP Channels in the Focus of Trigeminal Nociceptor Sensitization Contributing to Primary Headaches. Int. J. Mol. Sci. 2020, 21, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Steven, E.S.; Manas, C.; Gerald, G.; Tony, P.; Wayne, G.H.; Fred, K.; Tom, B.; Sheldon, R.; Robert, J.L.; et al. SH2 domains recognize specific phosphopeptide sequences. Cell 1993, 72, 767–778. [Google Scholar] [CrossRef]
- Pellicena, P.; Stowell, K.R.; Miller, W.T. Enhanced Phosphorylation of Src Family Kinase Substrates Containing SH2 Domain Binding Sites*. J. Biol. Chem. 1998, 273, 15325–15328. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.-M.; Askalan, R.; Keil Gary, J.; Salter Michael, W. NMDA Channel Regulation by Channel-Associated Protein Tyrosine Kinase Src. Science 1997, 275, 674–678. [Google Scholar] [CrossRef]
- Sun, X.-D.; Wang, A.; Ma, P.; Gong, S.; Tao, J.; Yu, X.-M.; Jiang, X. Regulation of the firing activity by PKA-PKC-Src family kinases in cultured neurons of hypothalamic arcuate nucleus. J. Neurosci. Res. 2020, 98, 384–403. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Brodeur Sr Fau-Gish, G.; Gish G Fau-Songyang, Z.; Songyang Z Fau-Cantley, L.C.; Cantley Lc Fau-Laudano, A.P.; Laudano Ap Fau-Pawson, T.; Pawson, T. Regulation of c-Src tyrosine kinase activity by the Src SH2 domain. Oncogene 1993, 8, 1119–1126. [Google Scholar]
- Xu, W.; Doshi A Fau-Lei, M.; Lei M Fau-Eck, M.J.; Eck Mj Fau-Harrison, S.C.; Harrison, S.C. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 1999, 3, 629–638. [Google Scholar] [CrossRef]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef]
- IHS, H.C.C.O.T. Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition. Cephalalgia Int. J. Headache 2018, 38, 1–211. [Google Scholar]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shatillo, A.; Koroleva, K.; Giniatullina, R.; Naumenko, N.; Slastnikova, A.A.; Aliev, R.R.; Bart, G.; Atalay, M.; Gu, C.; Khazipov, R.; et al. Cortical spreading depression induces oxidative stress in the trigeminal nociceptive system. Neuroscience 2013, 253, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Hennequin, L.F.; Allen, J.; Breed, J.; Curwen, J.; Fennell, M.; Green, T.P.; Lambert-van der Brempt, C.; Morgentin, R.; Norman, R.A.; Olivier, A.; et al. N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5- (tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a Novel, Highly Selective, Orally Available, Dual-Specific c-Src/Abl Kinase Inhibitor. J. Med. Chem. 2006, 49, 6465–6488. [Google Scholar] [CrossRef] [PubMed]
- Fury, M.G.; Baxi, S.; Shen, R.; Kelly, K.W.; Lipson, B.L.; Carlson, D.; Stambuk, H.; Haque, S.; Pfister, D.G. Phase II Study of Saracatinib (AZD0530) for Patients with Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma (HNSCC). Anticancer Res. 2011, 31, 249. [Google Scholar] [PubMed]
- Renouf, D.J.; Moore, M.J.; Hedley, D.; Gill, S.; Jonker, D.; Chen, E.; Walde, D.; Goel, R.; Southwood, B.; Gauthier, I.; et al. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Investig. New Drugs 2012, 30, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Fujisaka, Y.; Onozawa, Y.; Kurata, T.; Yasui, H.; Goto, I.; Yamazaki, K.; Machida, N.; Watanabe, J.; Shimada, H.; Shi, X.; et al. First report of the safety, tolerability, and pharmacokinetics of the Src kinase inhibitor saracatinib (AZD0530) in Japanese patients with advanced solid tumours. Invest. New Drugs 2013, 31, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, H.B.; Wagner, A.F.; Bowen, G.S.; Good, S.P.; MacAvoy, M.G.; Strittmatter, K.A.; Kaufman, A.C.; Rosenberg, B.J.; Sekine-Konno, T.; Varma, P.; et al. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- van Dyck, C.H.; Nygaard, H.B.; Chen, K.; Donohue, M.C.; Raman, R.; Rissman, R.A.; Brewer, J.B.; Koeppe, R.A.; Chow, T.W.; Rafii, M.S.; et al. Effect of AZD0530 on Cerebral Metabolic Decline in Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2019, 76, 1219–1229. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, A.C.; Salazar, S.V.; Haas, L.T.; Yang, J.; Kostylev, M.A.; Jeng, A.T.; Robinson, S.A.; Gunther, E.C.; van Dyck, C.H.; Nygaard, H.B.; et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. 2015, 77, 953–971. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-J.; Yu, J.-P.; An, X.; Jia, Z.-W.; Zhang, J.; Su, Y.-X. Attenuation of cartilage pathogenesis in osteoarthritis by blocking the phosphorylation of tyrosine kinase Fyn to β-catenin, AZD0530. Bone 2022, 154, 116259. [Google Scholar] [CrossRef]
- Benemei, S.; Appendino, G.; Geppetti, P. Pleasant natural scent with unpleasant effects: Cluster headache-like attacks triggered by Umbellularia californica. Cephalalgia Int. J. Headache 2009, 30, 744–746. [Google Scholar] [CrossRef] [PubMed]
- Drake, W.M.; Lowe, S.R.; Mirtella, A.; Bartlett, T.J.; Clark, A.J. Desensitisation of calcitonin gene-related peptide responsiveness but not adrenomedullin responsiveness in vascular smooth muscle cells. J. Endocrinol. 2000, 165, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.-Q.; Tu, Y.-J.; Lawand, N.B.; Yan, J.-Y.; Lin, Q.; Willis, W.D. Calcitonin Gene-Related Peptide Receptor Activation Produces PKA- and PKC-Dependent Mechanical Hyperalgesia and Central Sensitization. J. Neurophysiol. 2004, 92, 2859–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.S.; Conner Ac Fau-Poyner, D.R.; Poyner Dr Fau-Hay, D.L.; Hay, D.L. Regulation of signal transduction by calcitonin gene-related peptide receptors. Trends Pharmacol. Sci. 2010, 31, 476–483. [Google Scholar] [CrossRef]
- Obara, Y.; Labudda, K.; Dillon, T.J.; Stork, P.J.S. PKA phosphorylation of Src mediates Rap1 activation in NGF and cAMP signaling in PC12 cells. J. Cell Sci. 2004, 117, 6085–6094. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-B.; Yang, X.; Cao, J.; Li, S.; Liu, Y.-N.; Suo, Z.-W.; Cui, H.-B.; Guo, Z.; Hu, X.-D. cAMP-dependent protein kinase activated Fyn in spinal dorsal horn to regulate NMDA receptor function during inflammatory pain. J. Neurochem. 2011, 116, 93–104. [Google Scholar] [CrossRef]
- Tozzi, A.; de Iure, A.; Di Filippo, M.; Costa, C.; Caproni, S.; Pisani, A.; Bonsi, P.; Picconi, B.; Cupini, L.M.; Materazzi, S.; et al. Critical role of calcitonin gene-related peptide receptors in cortical spreading depression. Proc. Natl. Acad. Sci. USA 2012, 109, 18985–18990. [Google Scholar] [CrossRef] [Green Version]
- Swierczewski, B.E.; Davies, S.J. Developmental regulation of protein kinase A expression and activity in Schistosoma mansoni. Int. J. Parasitol. 2010, 40, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Héroux, M.; Hogue, M.; Lemieux, S.; Bouvier, M. Functional Calcitonin Gene-related Peptide Receptors Are Formed by the Asymmetric Assembly of a Calcitonin Receptor-like Receptor Homo-oligomer and a Monomer of Receptor Activity-modifying Protein-1*. J. Biol. Chem. 2007, 282, 31610–31620. [Google Scholar] [CrossRef] [Green Version]
- Edvinsson, L.; Grell, A.-S.; Warfvinge, K. Expression of the CGRP Family of Neuropeptides and their Receptors in the Trigeminal Ganglion. J. Mol. Neurosci. 2020, 70, 930–944. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.L.; Lopes, A.H.; Guimarães, R.M.; Cunha, T.M. CXCL1/CXCR2 signaling in pathological pain: Role in peripheral and central sensitization. Neurobiol. Dis. 2017, 105, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Khaiboullina, S.F.; Mendelevich, E.G.; Shigapova, L.H.; Shagimardanova, E.; Gazizova, G.; Nikitin, A.; Martynova, E.; Davidyuk, Y.N.; Bogdanov, E.I.; Gusev, O.; et al. Cerebellar Atrophy and Changes in Cytokines Associated with the CACNA1A R583Q Mutation in a Russian Familial Hemiplegic Migraine Type 1 Family. Front. Cell Neurosci. 2017, 11, 263. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.-B.; Xian, H.; Wu, W.-B.; Ma, S.-Y.; Liu, Y.-K.; Liang, Y.-T.; Guo, H.; Kang, J.-J.; Liu, Y.-Y.; Zhang, H.; et al. CCL2 facilitates spinal synaptic transmission and pain via interaction with presynaptic CCR2 in spinal nociceptor terminals. Mol. Brain 2020, 13, 161. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, T.; Qin, T.; Lopes de Morais, A.; Sugimoto, K.; Chung, J.Y.; Morsett, L.; Mulder, I.; Fischer, P.; Suzuki, T.; Anzabi, M.; et al. Non-invasively triggered spreading depolarizations induce a rapid pro-inflammatory response in cerebral cortex. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2020, 40, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, G.; Suzuki, S.; Morishita, N.; Takeshita, M.; Kanou, K.; Takamatsu, T.; Suzuki, S.; Morichi, S.; Watanabe, Y.; Ishida, Y.; et al. Role of Neuroinflammation and Blood-Brain Barrier Permutability on Migraine. Int. J. Mol. Sci. 2021, 22, 8929. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, J.K.; Rühle, V.; Ceppa, E.P.; Neuhuber, W.L.; Bunnett, N.W.; Grady, E.F.; Messlinger, K. Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: Differences between peripheral and central CGRP receptor distribution. J. Comp. Neurol. 2008, 507, 1277–1299. [Google Scholar] [CrossRef]
- Eftekhari, S.; Salvatore, C.A.; Calamari, A.; Kane, S.A.; Tajti, J.; Edvinsson, L. Differential distribution of calcitonin gene-related peptide and its receptor components in the human trigeminal ganglion. Neuroscience 2010, 169, 683–696. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Liu, H.; Warfvinge, K.; Shi, L.; Dovlatyan, M.; Xu, C.; Edvinsson, L. Immunohistochemical localization of the calcitonin gene-related peptide binding site in the primate trigeminovascular system using functional antagonist antibodies. Neuroscience 2016, 328, 165–183. [Google Scholar] [CrossRef]
- Takeda, M.; Tanimoto, T.; Kadoi, J.; Nasu, M.; Takahashi, M.; Kitagawa, J.; Matsumoto, S. Enhanced excitability of nociceptive trigeminal ganglion neurons by satellite glial cytokine following peripheral inflammation. Pain 2007, 129, 155–166. [Google Scholar] [CrossRef]
- Cao, D.-L.; Zhang, Z.-J.; Xie, R.-G.; Jiang, B.-C.; Ji, R.-R.; Gao, Y.-J. Chemokine CXCL1 enhances inflammatory pain and increases NMDA receptor activity and COX-2 expression in spinal cord neurons via activation of CXCR2. Exp. Neurol. 2014, 261, 328–336. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Park, C.-K.; Xie, R.-G.; Berta, T.; Nedergaard, M.; Ji, R.-R. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain 2014, 137, 2193–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dansereau, M.-A.; Midavaine, É.; Bégin-Lavallée, V.; Belkouch, M.; Beaudet, N.; Longpré, J.-M.; Mélik-Parsadaniantz, S.; Sarret, P. Mechanistic insights into the role of the chemokine CCL2/CCR2 axis in dorsal root ganglia to peripheral inflammation and pain hypersensitivity. J. Neuroinflamm. 2021, 18, 79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Saccani, S.; Shin, H.; Nikolajczyk, B.S. Dynamic Protein Associations Define Two Phases of IL-1β Transcriptional Activation. J. Immunol. 2008, 181, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panicker, N.; Sarkar, S.; Harischandra, D.S.; Neal, M.; Kam, T.-I.; Jin, H.; Saminathan, H.; Langley, M.; Charli, A.; Samidurai, M.; et al. Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J. Exp. Med. 2019, 216, 1411–1430. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veerasubramanian, P.K.; Shao, H.; Meli, V.S.; Phan, T.A.Q.; Luu, T.U.; Liu, W.F.; Downing, T.L. A Src-H3 acetylation signaling axis integrates macrophage mechanosensation with inflammatory response. Biomaterials 2021, 279, 121236. [Google Scholar] [CrossRef]
- Qin, X.; Wan, Y.; Wang, X. CCL2 and CXCL1 trigger calcitonin gene-related peptide release by exciting primary nociceptive neurons. J. Neurosci. Res. 2005, 82, 51–62. [Google Scholar] [CrossRef]
- Viviani, B.; Bartesaghi, S.; Gardoni, F.; Vezzani, A.; Behrens, M.M.; Bartfai, T.; Binaglia, M.; Corsini, E.; Di Luca, M.; Galli, C.L.; et al. Interleukin-1β Enhances NMDA Receptor-Mediated Intracellular Calcium Increase through Activation of the Src Family of Kinases. J. Neurosci. 2003, 23, 8692. [Google Scholar] [CrossRef] [Green Version]
- Davis, N.C.; Mann, E.; Behrens, M.M.; Gaidarova, S.; Rebek, M.; Rebek, J.; Bartfai, T. MyD88-dependent and -independent signaling by IL-1 in neurons probed by bifunctional Toll/IL-1 receptor domain/BB-loop mimetics. Proc. Natl. Acad. Sci. USA 2006, 103, 2953–2958. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Smith, D.E.; Ibáñez-Sandoval, O.; Sims, J.E.; Friedman, W.J. Neuron-Specific Effects of Interleukin-1β Are Mediated by a Novel Isoform of the IL-1 Receptor Accessory Protein. J. Neurosci. 2011, 31, 18048. [Google Scholar] [CrossRef] [Green Version]
- Mon, N.N.; Senga, T.; Ito, S. Interleukin-1β activates focal adhesion kinase and Src to induce matrix metalloproteinase-9 production and invasion of MCF-7 breast cancer cells. Oncol. Lett. 2017, 13, 955–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, F.; Wang, Y.; Jiang, L.; Ma, D.; Quinn, J.P.; Wang, M. Sarcoma family kinase activity is required for cortical spreading depression. Cephalalgia Int. J. Headache 2018, 38, 1748–1758. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nie, L.; Sun, K.; Gong, Z.; Li, H.; Quinn, J.P.; Wang, M. Src Family Kinases Facilitate the Crosstalk between CGRP and Cytokines in Sensitizing Trigeminal Ganglion via Transmitting CGRP Receptor/PKA Pathway. Cells 2022, 11, 3498. https://doi.org/10.3390/cells11213498
Nie L, Sun K, Gong Z, Li H, Quinn JP, Wang M. Src Family Kinases Facilitate the Crosstalk between CGRP and Cytokines in Sensitizing Trigeminal Ganglion via Transmitting CGRP Receptor/PKA Pathway. Cells. 2022; 11(21):3498. https://doi.org/10.3390/cells11213498
Chicago/Turabian StyleNie, Lingdi, Kai Sun, Ziyang Gong, Haoyang Li, John P. Quinn, and Minyan Wang. 2022. "Src Family Kinases Facilitate the Crosstalk between CGRP and Cytokines in Sensitizing Trigeminal Ganglion via Transmitting CGRP Receptor/PKA Pathway" Cells 11, no. 21: 3498. https://doi.org/10.3390/cells11213498