
Dimethylmyricacene: An In Vitro and In Silico Study of a Semisynthetic Non-Camptothecin Derivative Compound, Targeting Human DNA Topoisomerase 1B
 ,
,  , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein and Reagents
2.2. Cells and Culture Conditions
2.3. Production and Characterization of DM
2.4. Dose Dependent and Time Course Assays
2.5. Cleavage and Religation Kinetics
2.6. Cell Viability Assays
2.7. Molecular Docking
2.8. Classical Molecular Dynamics Simulations
2.9. Trajectory Analysis
2.10. Statistics
3. Results
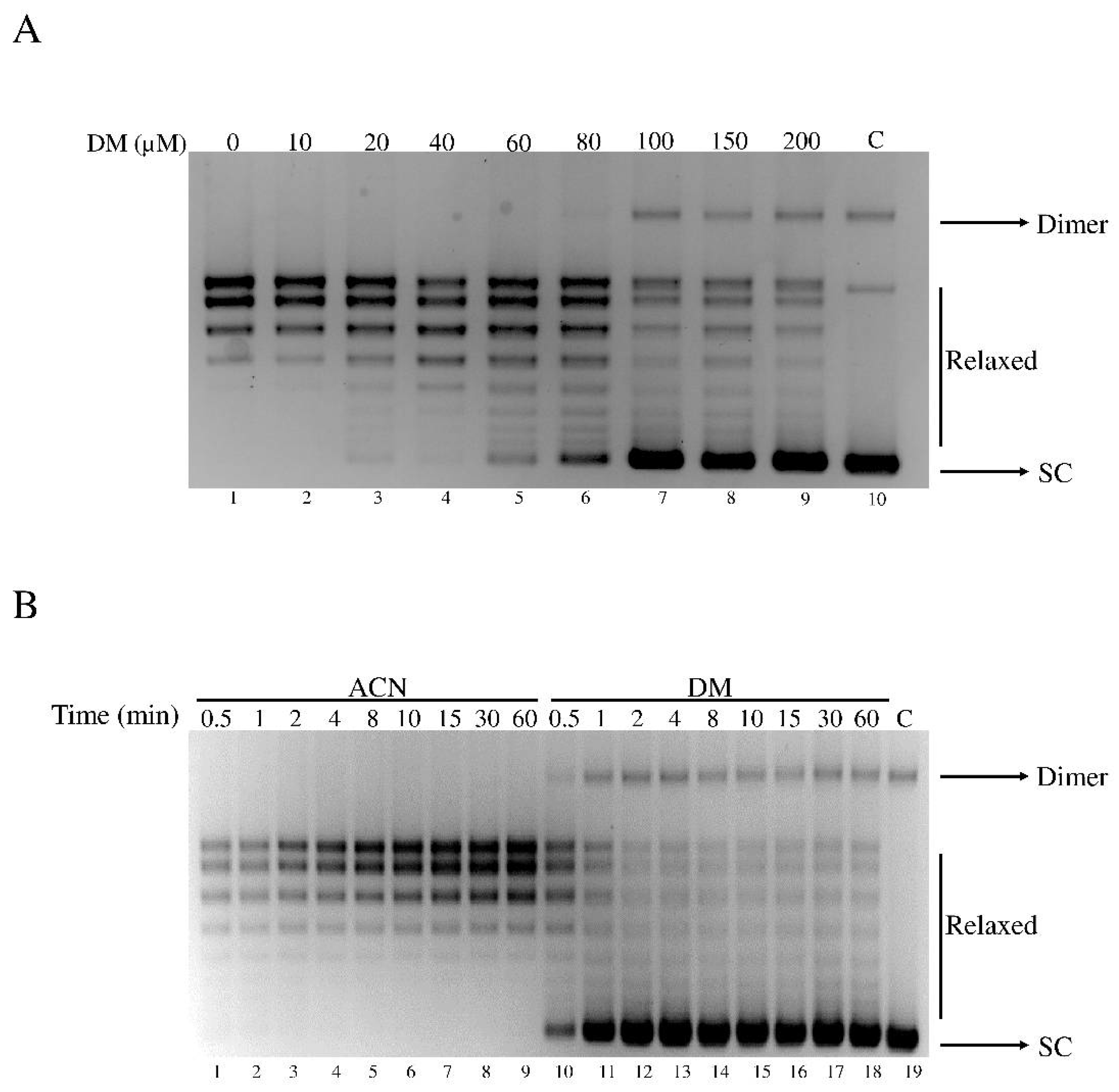
3.1. DM inhibits the Catalytic Activity of hTop1
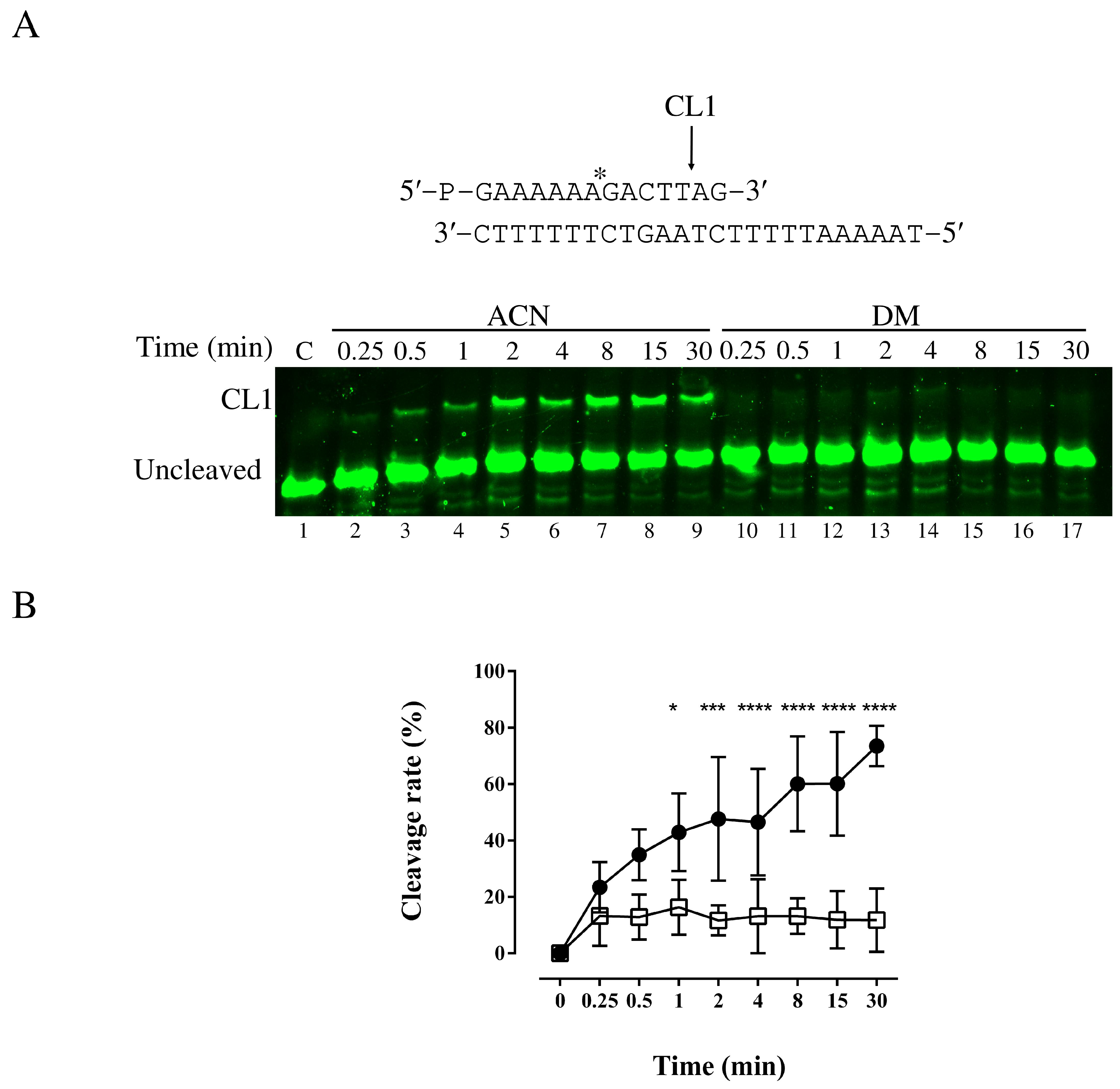
3.2. Cleavage Assays in the Absence and Presence of DM
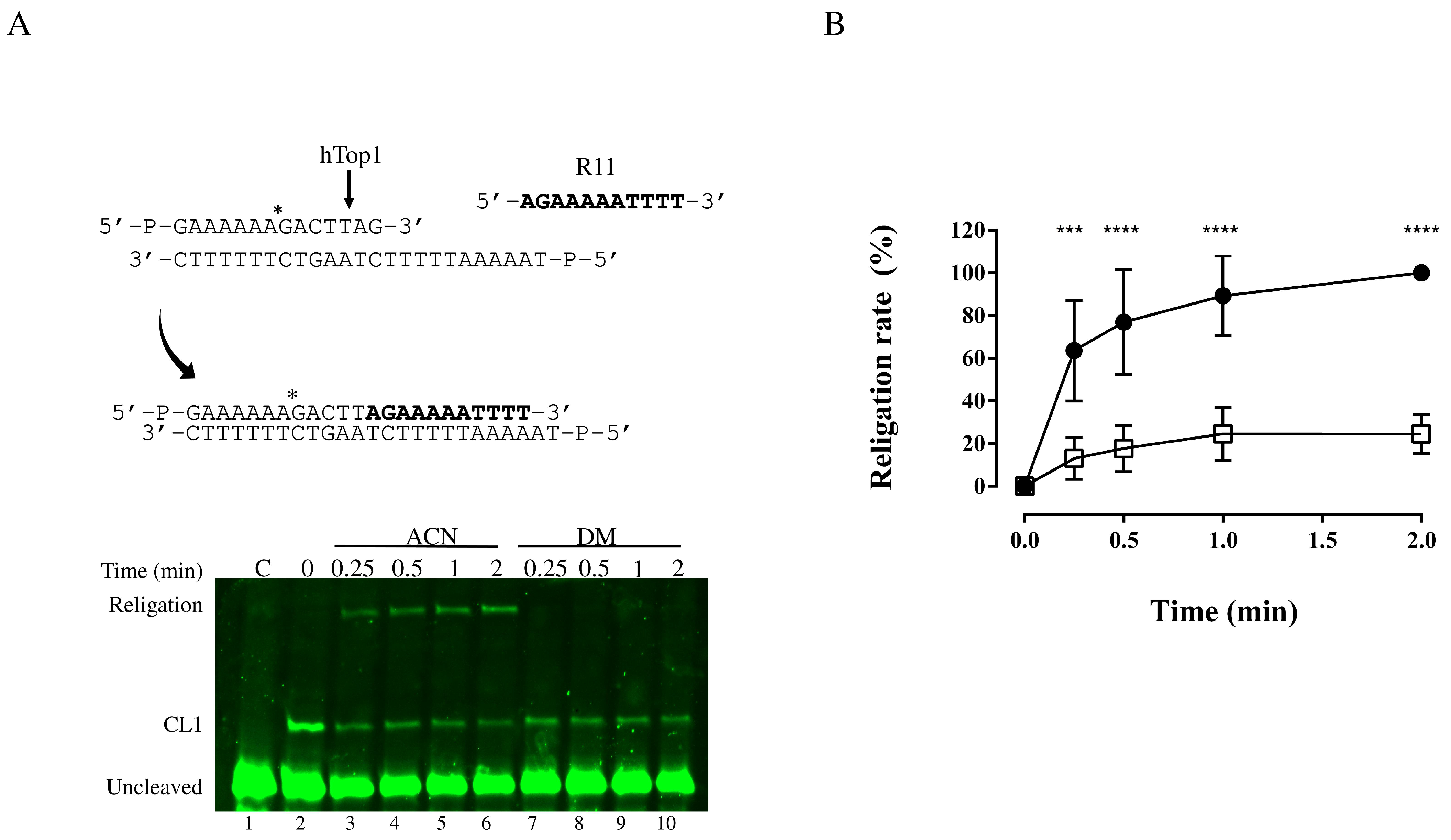
3.3. Religation Kinetics
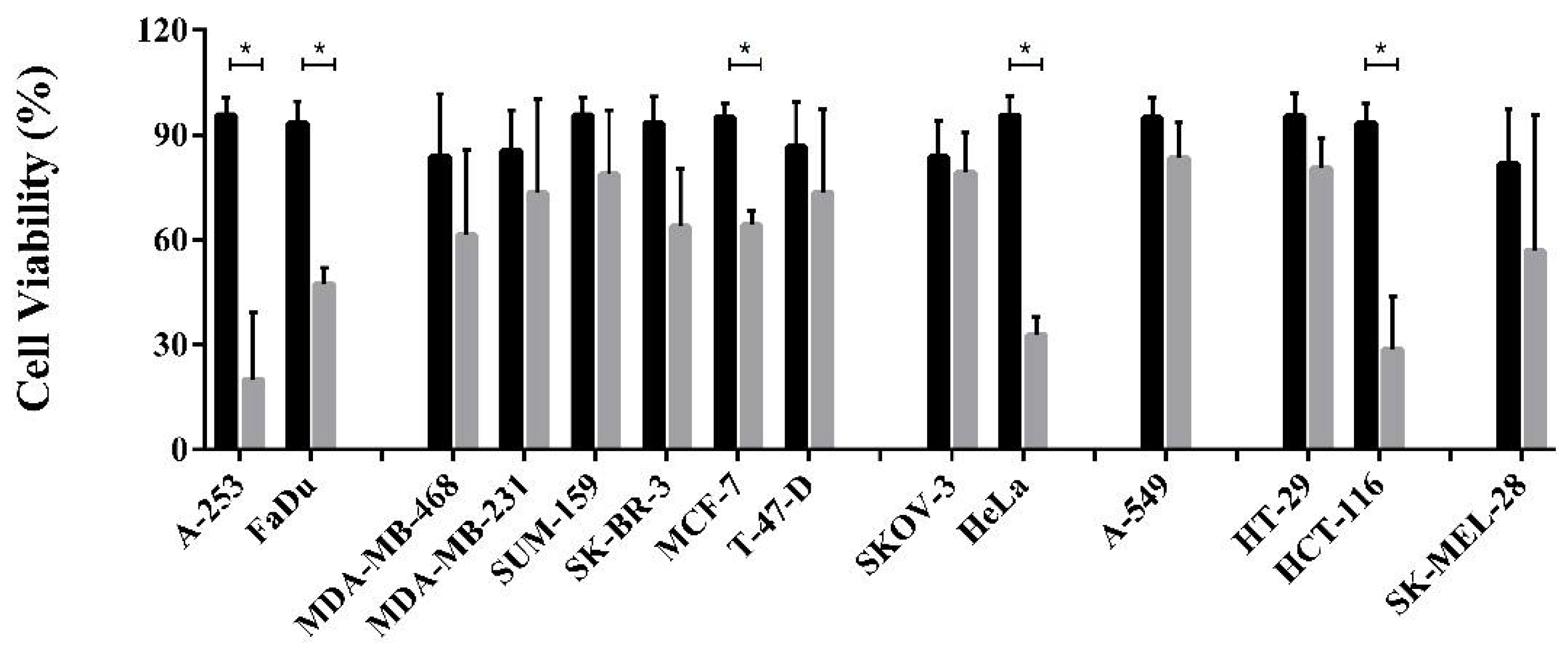
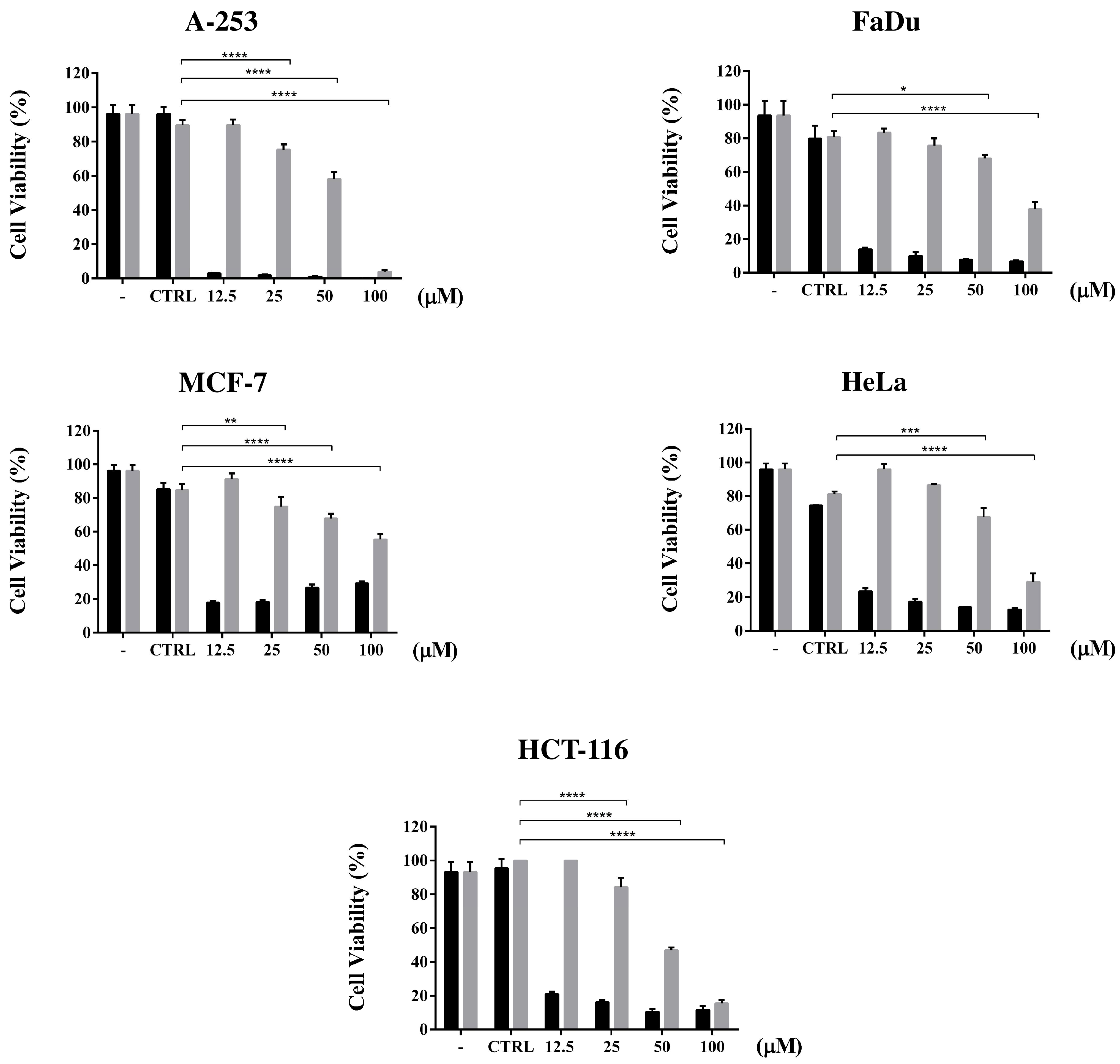
3.4. Cell Viability Assay and Dose Dependent Effect
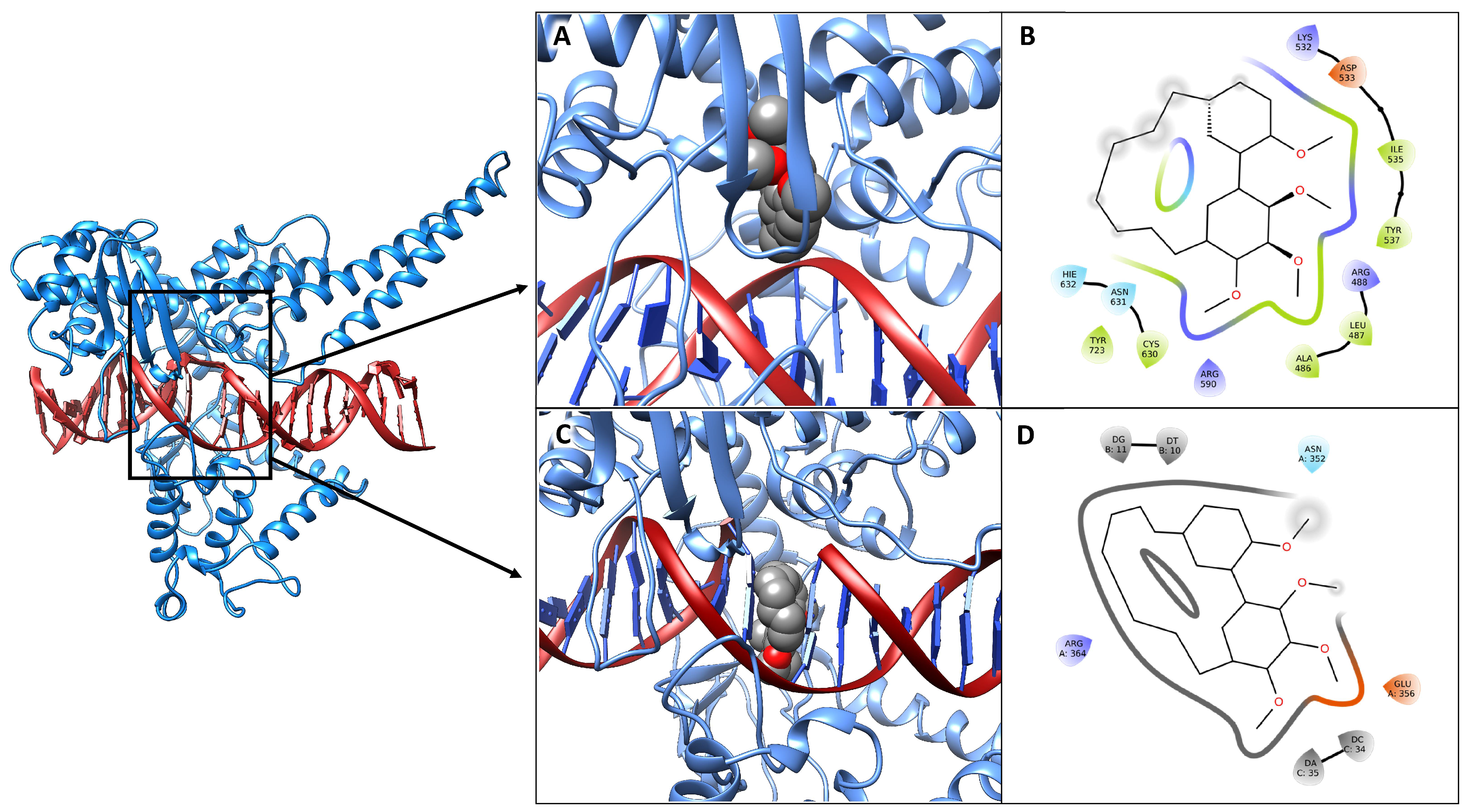
3.5. Molecular Docking Simulations
3.6. MM/GBSA Analysis
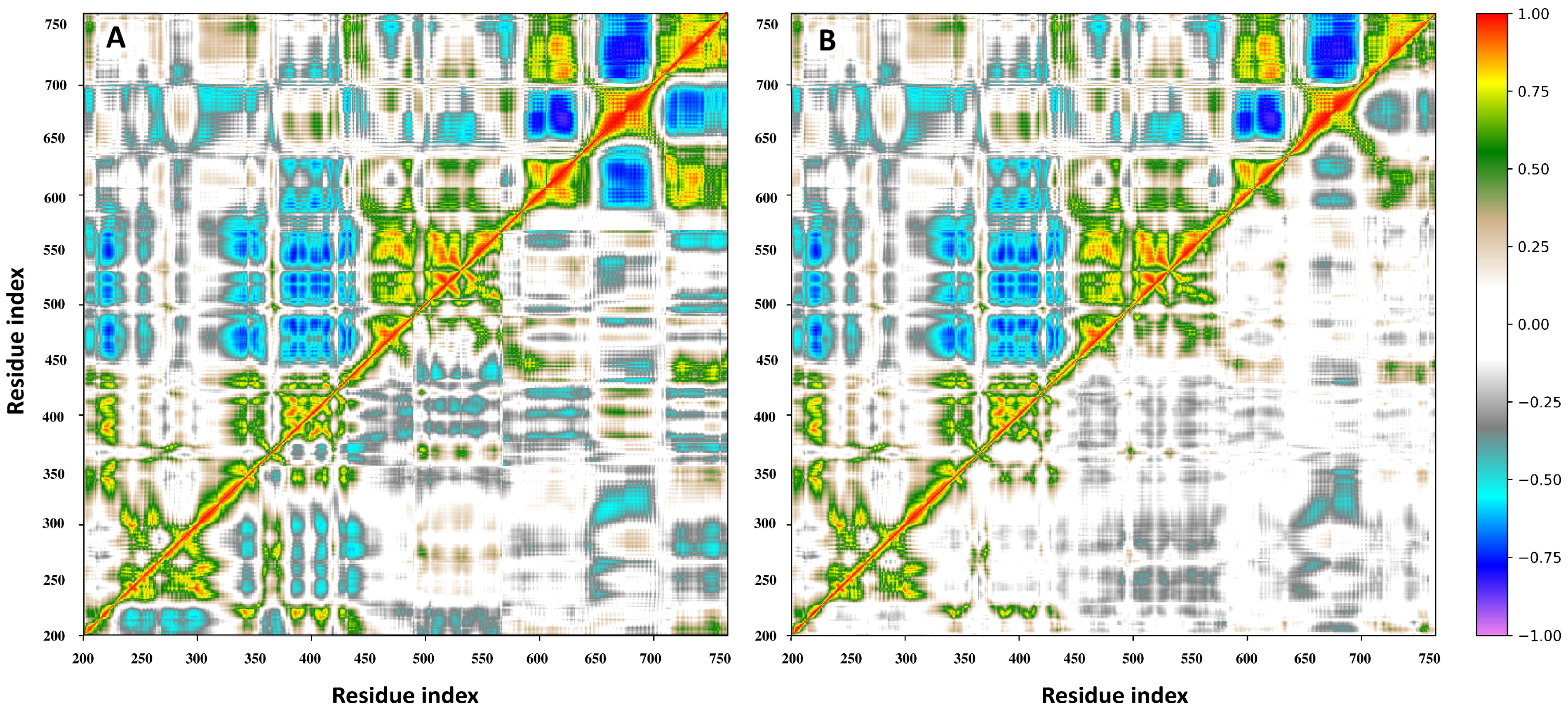
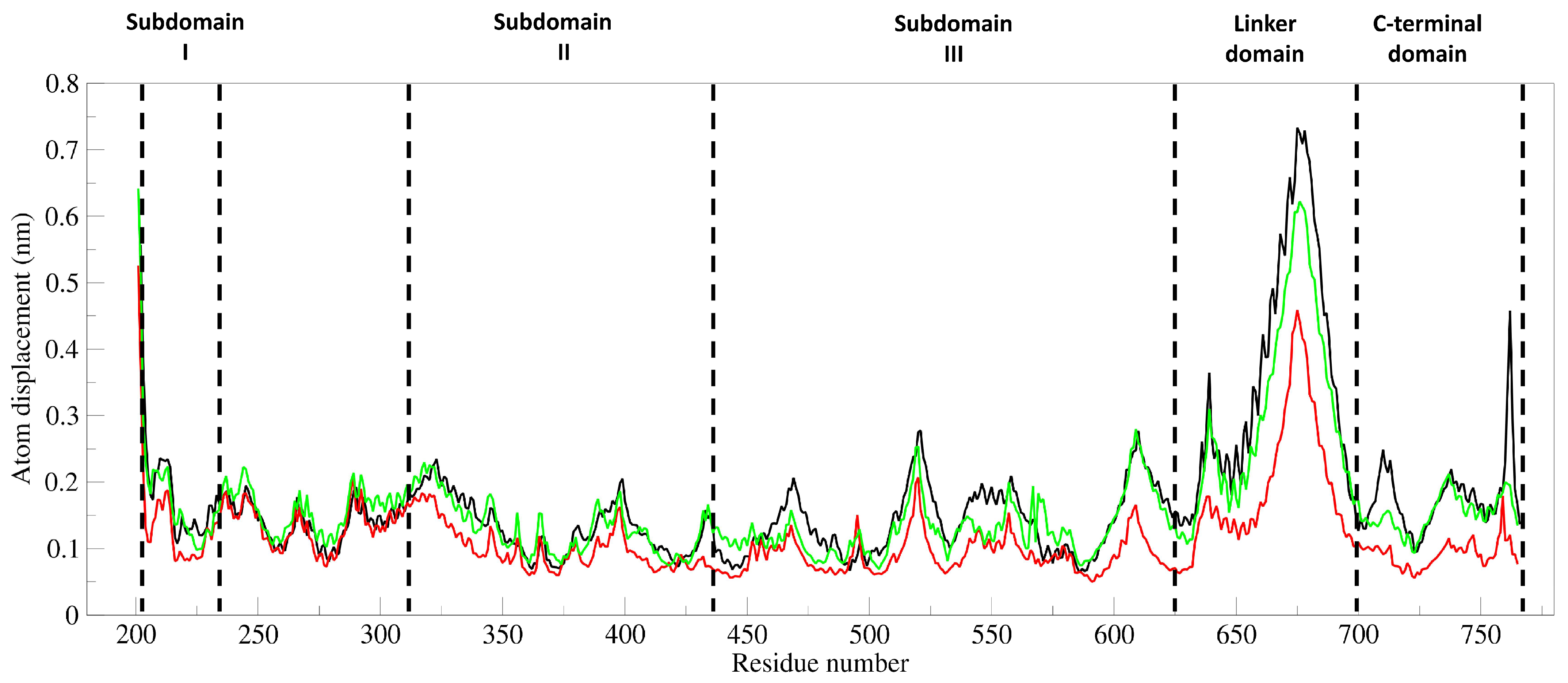
3.7. Principal Component and RMSF Analyses
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Stewart, L.; Ireton, G.C.; Champoux, J.J. The domain organization of human topoisomerase I. J. Biol. Chem. 1996, 271, 7602–7628. [Google Scholar] [CrossRef] [Green Version]
- Soren, B.C.; Dasari, J.B.; Ottaviani, A.; Lacovelli, F.; Fiorani, P. Topoisomerase IB: A relaxing enzyme for stressed DNA. Cancer Drug Resist 2019, 3, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [Green Version]
- Champoux, J.J. Domains of human topoisomerase I and associated functions. Prog. Nucleic Acid Res. Mol. Biol. 1998, 60, 111–132. [Google Scholar]
- Chillemi, G.; Fiorani, P.; Benedetti, P.; Desideri, A. Protein concerted motions in the DNA–human topoisomerase I complex. Nucleic Acids Res. 2003, 31, 1525–1535. [Google Scholar] [CrossRef]
- Chillemi, G.; Fiorani, P.; Castelli, S.; Bruselles, A.; Benedetti, P.; Desideri, A. Effect on DNA relaxation of the single Thr718Ala mutation in human topoisomerase I: A functional and molecular dynamics study. Nucleic Acids Res. 2005, 33, 3339–3350. [Google Scholar] [CrossRef] [Green Version]
- Krogh, B.O.; Shuman, S. Catalytic mechanism of DNA topoisomerase IB. Mol. Cell 2000, 5, 1035–1041. [Google Scholar] [CrossRef]
- Stewart, L. A Model for the Mechanism of Human Topoisomerase I. Science 1998, 279, 1534–1541. [Google Scholar] [CrossRef]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar]
- Rasheed, Z.A.; Rubin, E.H. Mechanisms of resistance to topoisomerase I-targeting drugs. Oncogene 2003, 22, 7296–7304. [Google Scholar] [CrossRef] [Green Version]
- Mancini, G.; D’Annessa, I.; Coletta, A.; Sanna, N.; Chillemi, G.; Desideri, A. Structural and dynamical effects induced by the anticancer drug topotecan on the human topoisomerase I–DNA complex. PLoS ONE 2010, 5, e10934. [Google Scholar] [CrossRef]
- Pommier, Y.; Pourquier, P.; Urasaki, Y.; Wu, J.; Laco, G.S. Topoisomerase I inhibitors: Selectivity and cellular resistance. Drug Resist. Updat. 1999, 2, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Sooryakumar, D.; Dexheimer, T.S.; Teicher, B.A.; Pommier, Y. Molecular and Cellular Pharmacology of the Novel Noncamptothecin Topoisomerase I Inhibitor Genz-644282. Mol. Cancer Ther. 2011, 10, 1490–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foto, E.; Özen, Ç.; Zilifdar, F.; Tekiner-Gülbaş, B.; Yıldız, İ.; Akı-Yalçın, E.; Diril, N.; Yalçın, İ. Benzoxazines as new human topoisomerase I inhibitors and potential poisons. DARU J. Pharm. Sci. 2020, 28, 65–73. [Google Scholar] [CrossRef]
- Carballeira, N.M.; Montano, N.; Amador, L.A.; Rodríguez, A.D.; Golovko, M.Y.; Golovko, S.A.; Reguera, R.M.; Álvarez-Velilla, R.; Balaña-Fouce, R. Novel Very Long-Chain α-Methoxylated Δ5,9 Fatty Acids from the Sponge Asteropus Niger Are Effective Inhibitors of Topoisomerases IB. Lipids 2016, 51, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Castelli, S.; Vieira, S.; D’Annessa, I.; Katkar, P.; Musso, L.; Dallavalle, S.; Desideri, A. A derivative of the natural compound kakuol affects DNA relaxation of topoisomerase IB inhibiting the cleavage reaction. Arch. Biochem. Biophys. 2013, 530, 7–12. [Google Scholar] [CrossRef]
- Vieira, S.; Castelli, S.; Falconi, M.; Takarada, J.; Fiorillo, G.; Buzzetti, F.; Lombardi, P.; Desideri, A. Role of 13-(di)phenylalkyl berberine derivatives in the modulation of the activity of human topoisomerase IB. Int. J. Biol. Macromol. 2015, 77, 68–75. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [Green Version]
- León, I.E.; Cadavid-Vargas, J.F.; Tiscornia, I.; Porro, V.; Castelli, S.; Katkar, P.; Desideri, A.; Bollati-Fogolin, M.; Etcheverry, S.B. Oxidovanadium(IV) complexes with chrysin and silibinin: Anticancer activity and mechanisms of action in a human colon adenocarcinoma model. J. Biol. Inorg. Chem. 2015, 20, 1175–1191. [Google Scholar] [CrossRef]
- Vutey, V.; Castelli, S.; D’Annessa, I.; Sâmia, L.B.P.; Souza-Fagundes, E.M.; Beraldo, H.; Desideri, A. Human topoisomerase IB is a target of a thiosemicarbazone copper(II) complex. Arch. Biochem. Biophys. 2016, 606, 34–40. [Google Scholar] [CrossRef]
- Yu, Q.; Chen, Y.; Yang, H.; Zhang, H.L.; Agama, K.; Pommier, Y.; An, L.K. The antitumor activity of CYB-L10, a human topoisomerase IB catalytic inhibitor. J. Enzyme Inhib. Med. Chem. 2019, 34, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.-T.; Liu, L.; Shao, C.-L.; Yu, R.-L.; Chen, F.-L.; Yue, S.-J.; Wang, M.; Guo, Z.-L.; Fan, Y.-C.; Guan, H.-S.; et al. Discovery of DNA Topoisomerase I Inhibitors with Low-Cytotoxicity Based on Virtual Screening from Natural Products. Mar. Drugs 2017, 15, 217. [Google Scholar] [CrossRef] [PubMed]
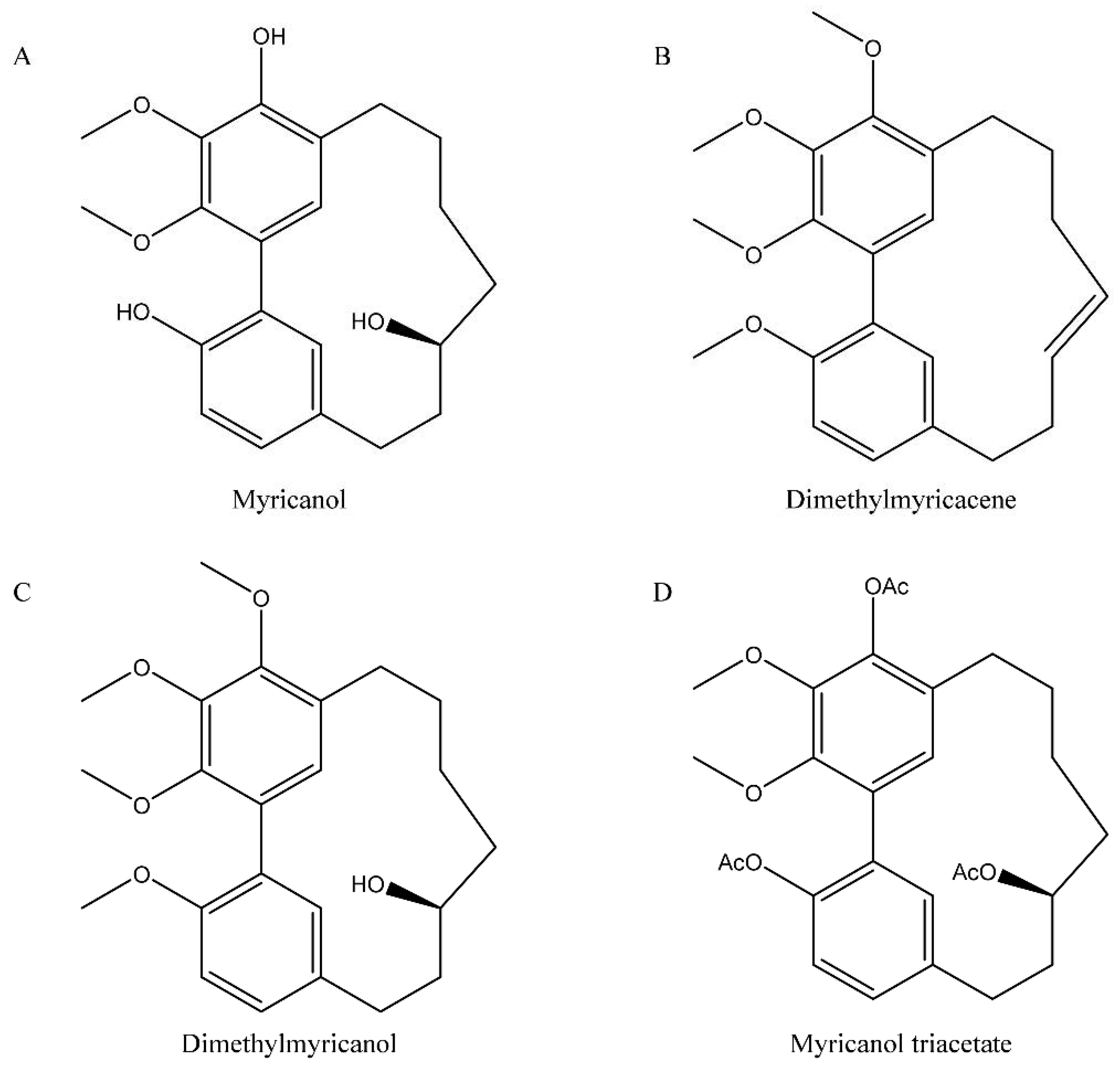
- Jones, J.R.; Lebar, M.D.; Jinwal, U.K.; Abisambra, J.F.; Koren III, J.; Blair, L.; O’Leary, J.C.; Davey, Z.; Trotter, J.; Johnson, A.G.; et al. The Diarylheptanoid (+)-aR,11S-Myricanol and Two Flavones from Bayberry. J. Nat. Prod. 2011, 74, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.D.; Calcul, L.; Smith, C.; Jinwal, U.K.; Fontaine, S.N.; Darling, A.; Seeley, K.; Wojtas, L.; Narayan, M.; Gestwicki, J.E.; et al. Synthesis, stereochemical analysis, and derivatization of myricanol provide new probes that promote autophagic Tau clearance. ACS Chem. Biol. 2015, 10, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Dai, G.; Tong, Y.; Chen, X.; Ren, Z.; Ying, X.; Yang, F.; Chai, K. Myricanol induces apoptotic cell death and anti-tumor activity in non-small cell lung carcinoma in vivo. Int. J. Mol. Sci. 2015, 16, 2717–2731. [Google Scholar] [CrossRef]
- Zuccaro, L.; Tesauro, C.; Cerroni, B.; Ottaviani, A.; Knudsen, B.R.; Balasubramanian, K.; Desideri, A. Rolling circle amplification-based detection of human topoisomerase I activity on magnetic beads. Anal. Biochem. 2014, 451, 42–44. [Google Scholar] [CrossRef]
- Dickey, C.; Jinwal, U.; Calcul, L.; Baker, B.J.; Lebar, M. Myricanol Derivatives and Uses Thereof for Treatment of Neurodegenerative Diseases. U.S. Patent 9,598,338, 21 March 2017. [Google Scholar]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Ottaviani, A.; Welsch, J.; Agama, K.; Pommier, Y.; Desideri, A.; Baker, B.J.; Fiorani, P. From Antarctica to cancer research: A novel human DNA topoisomerase 1B inhibitor from Antarctic sponge Dendrilla antarctica. J. Enzyme Inhib. Med. Chem. 2022, 37, 1404–1410. [Google Scholar] [CrossRef]
- Soren, B.C.; Dasari, J.B.; Ottaviani, A.; Messina, B.; Andreotti, G.; Romeo, A.; Iacovelli, F.; Falconi, M.; Desideri, A.; Fiorani, P. In vitro and in silico characterization of an antimalarial compound with antitumor activity targeting human DNA topoisomerase IB. Int. J. Mol. Sci. 2021, 22, 7455. [Google Scholar] [CrossRef]
- Lue, N.; Sharma, A.; Mondragón, A.; Wang, J.C. A 26 kDa yeast DNA topoisomerase I fragment: Crystallographic structure and mechanistic implications. Structure 1995, 3, 1315–1322. [Google Scholar] [CrossRef] [Green Version]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J. Med. Chem. 2005, 48, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2015, 13, 55–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Goga, N.; Rzepiela, A.J.; De Vries, A.H.; Marrink, S.J.; Berendsen, H.J.C. Efficient algorithms for langevin and DPD dynamics. J. Chem. Theory Comput. 2012, 8, 3637–3649. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.Y.; Gong, X.G. A new constant-pressure molecular dynamics method for finite systems. J. Phys. Condens. Matter 2002, 14, L487–L493. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Da Case, D.A.; Betz, R.M.R.; Cerutti, D.D.S.; Te, C.I.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. Amber 2016. Univ. California San Fr. 2016. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Genet. 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Iannone, F.; Ambrosino, F.; Bracco, G.; De Rosa, M.; Funel, A.; Guarnieri, G.; Migliori, S.; Palombi, F.; Ponti, G.; Santomauro, G.; et al. CRESCO ENEA HPC clusters: A working example of a multifabric GPFS Spectrum Scale layout. In Proceedings of the 2019 International Conference on High Performance Computing and Simulation, HPCS 2019, Dublin, Ireland, 15–19 July 2019; Institute of Electrical and Electronics Engineers Inc.: Piscataway, NJ, USA, 2019; pp. 1051–1052. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Tesauro, C.; Fiorani, P.; D’Annessa, I.; Chillemi, G.; Turchi, G.; Desideri, A. Erybraedin C, a natural compound from the plant Bituminaria bituminosa, inhibits both the cleavage and religation activities of human topoisomerase I. Biochem. J. 2010, 425, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Fiorani, P.; Tesauro, C.; Mancini, G.; Chillemi, G.; D’Annessa, I.; Graziani, G.; Tentori, L.; Muzi, A.; Desideri, A. Evidence of the crucial role of the linker domain on the catalytic activity of human topoisomerase I by experimental and simulative characterization of the Lys681Ala mutant. Nucleic Acids Res. 2009, 37, 6849–6858. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Site | vdW * (kcal/mol) | Electrostatic (kcal/mol) | Interaction Energy (kcal/mol) |
|---|---|---|---|
| Catalytic site | −42.7 | −17.9 | −32.6 |
| DNA-intercalated | −42.3 | −9.5 | −22.7 |
| Catalytic Site 1 | DNA-Intercalated 1 | ||
|---|---|---|---|
| Residue/Nucleotide | Binding Energy (kcal/mol) | Residue/Nucleotide | Binding Energy (kcal/mol) |
| Ala486 | −2.35 | Ala351 | −1.15 |
| Leu487 | −3.60 | Asn352 | −3.49 |
| Arg488 | −11.93 | Glu356 | −1.72 |
| Lys532 | −1.89 | Arg364 | −0.92 |
| Asp533 | −0.93 | Lys374 | −0.50 |
| Tyr537 | −3.10 | Trp416 | −0.71 |
| Arg590 | −2.94 | Lys425 | −3.41 |
| Cys630 | −2.14 | Tyr426 | −0.47 |
| Asn631 | −5.84 | Met428 | −0.69 |
| His632 | −1.82 | dT10 | −1.96 |
| Tyr723 | −1.25 | dG12 | −0.30 |
| dG 11 | −1.92 | dC34 | −7.30 |
| dA35 | −6.13 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottaviani, A.; Iacovelli, F.; Welsch, J.; Morozzo della Rocca, B.; Desideri, A.; Falconi, M.; Calcul, L.; Baker, B.J.; Fiorani, P. Dimethylmyricacene: An In Vitro and In Silico Study of a Semisynthetic Non-Camptothecin Derivative Compound, Targeting Human DNA Topoisomerase 1B. Cells 2022, 11, 3486. https://doi.org/10.3390/cells11213486
Ottaviani A, Iacovelli F, Welsch J, Morozzo della Rocca B, Desideri A, Falconi M, Calcul L, Baker BJ, Fiorani P. Dimethylmyricacene: An In Vitro and In Silico Study of a Semisynthetic Non-Camptothecin Derivative Compound, Targeting Human DNA Topoisomerase 1B. Cells. 2022; 11(21):3486. https://doi.org/10.3390/cells11213486
Chicago/Turabian StyleOttaviani, Alessio, Federico Iacovelli, Joshua Welsch, Blasco Morozzo della Rocca, Alessandro Desideri, Mattia Falconi, Laurent Calcul, Bill J. Baker, and Paola Fiorani. 2022. "Dimethylmyricacene: An In Vitro and In Silico Study of a Semisynthetic Non-Camptothecin Derivative Compound, Targeting Human DNA Topoisomerase 1B" Cells 11, no. 21: 3486. https://doi.org/10.3390/cells11213486