
Rifaximin Modifies Gut Microbiota and Attenuates Inflammation in Parkinson’s Disease: Preclinical and Clinical Studies

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Study Participants and Rifaximin Treatment
2.2. Experimental Animals and Rifaximin Treatment
2.3. GM: 16S rRNA Assessment
2.4. Protein Quantification: Western Blot Analysis
2.5. Immunohistochemistry
2.6. Human Plasma Cytokine Quantification
2.7. Clinical Assessments
2.8. Behavioral Assessments
3. Results
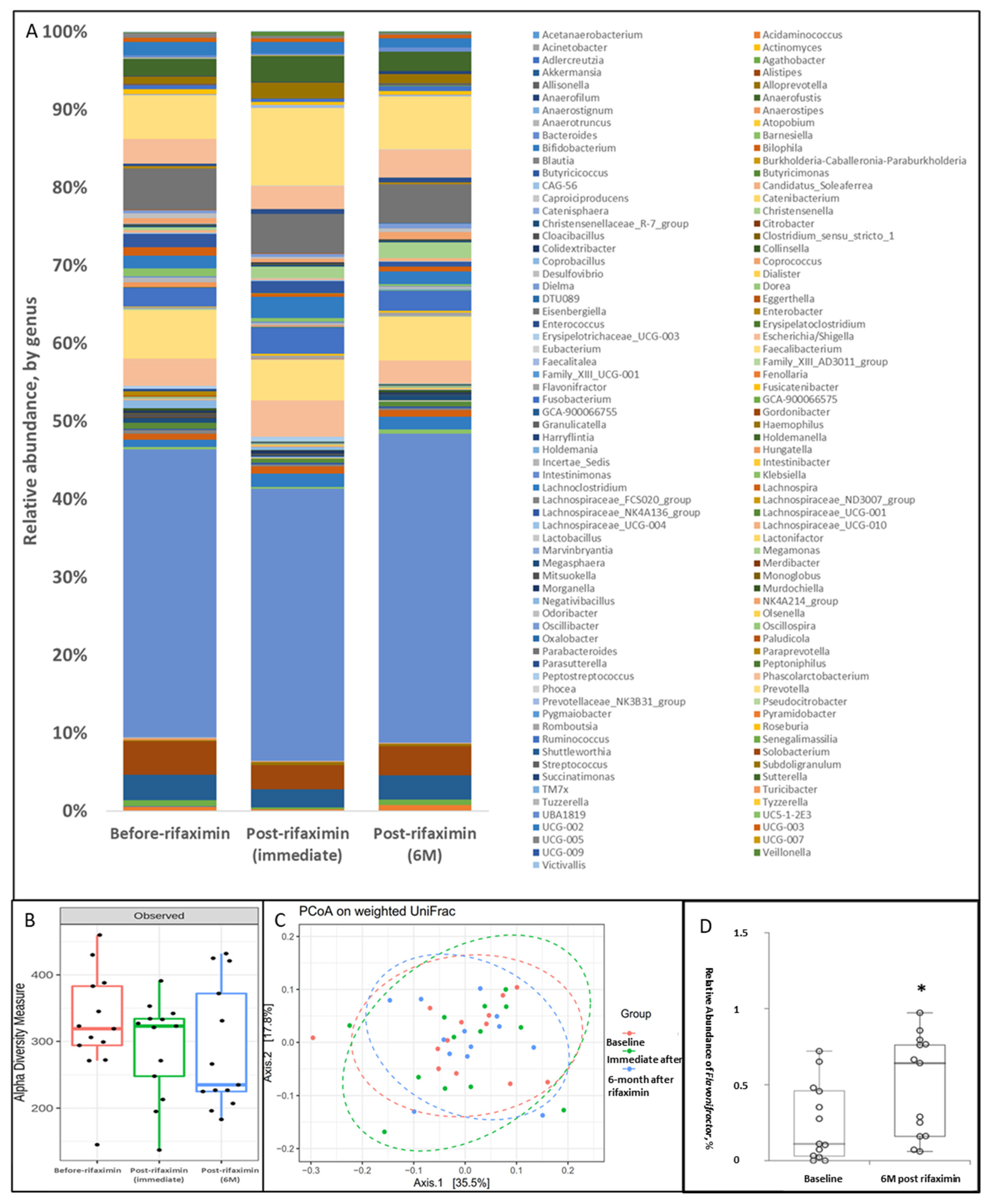
3.1. Rifaximin Treatment Altered the GM of Transgenic PD Mice
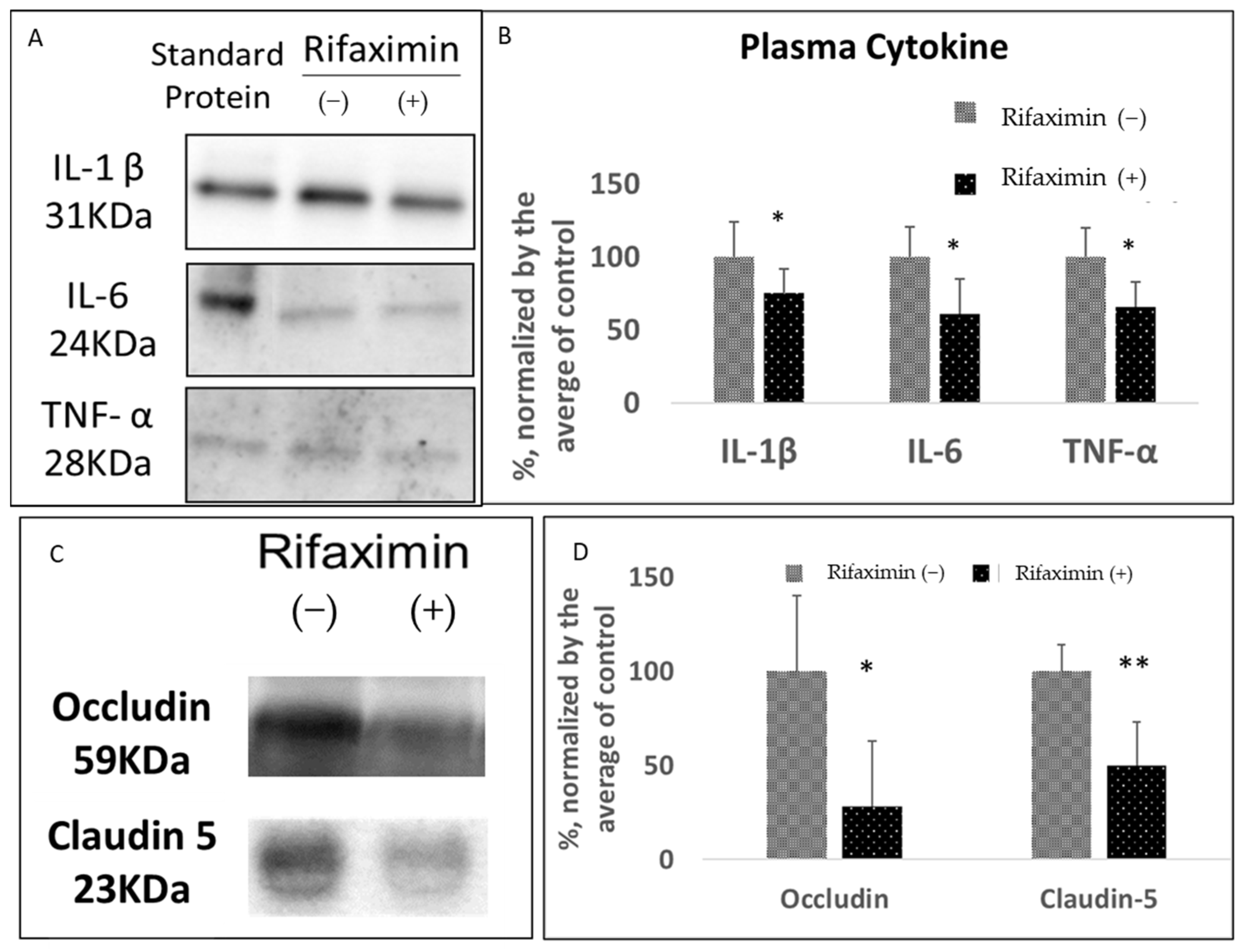
3.2. Rifaximin Treatment Preserved Intestinal Epithelial Integrity and Reduced Systemic Inflammation in Transgenic PD Mice
3.3. Rifaximin Treatment Prevented Motor and Cognitive Dysfunction in Transgenic PD Mice
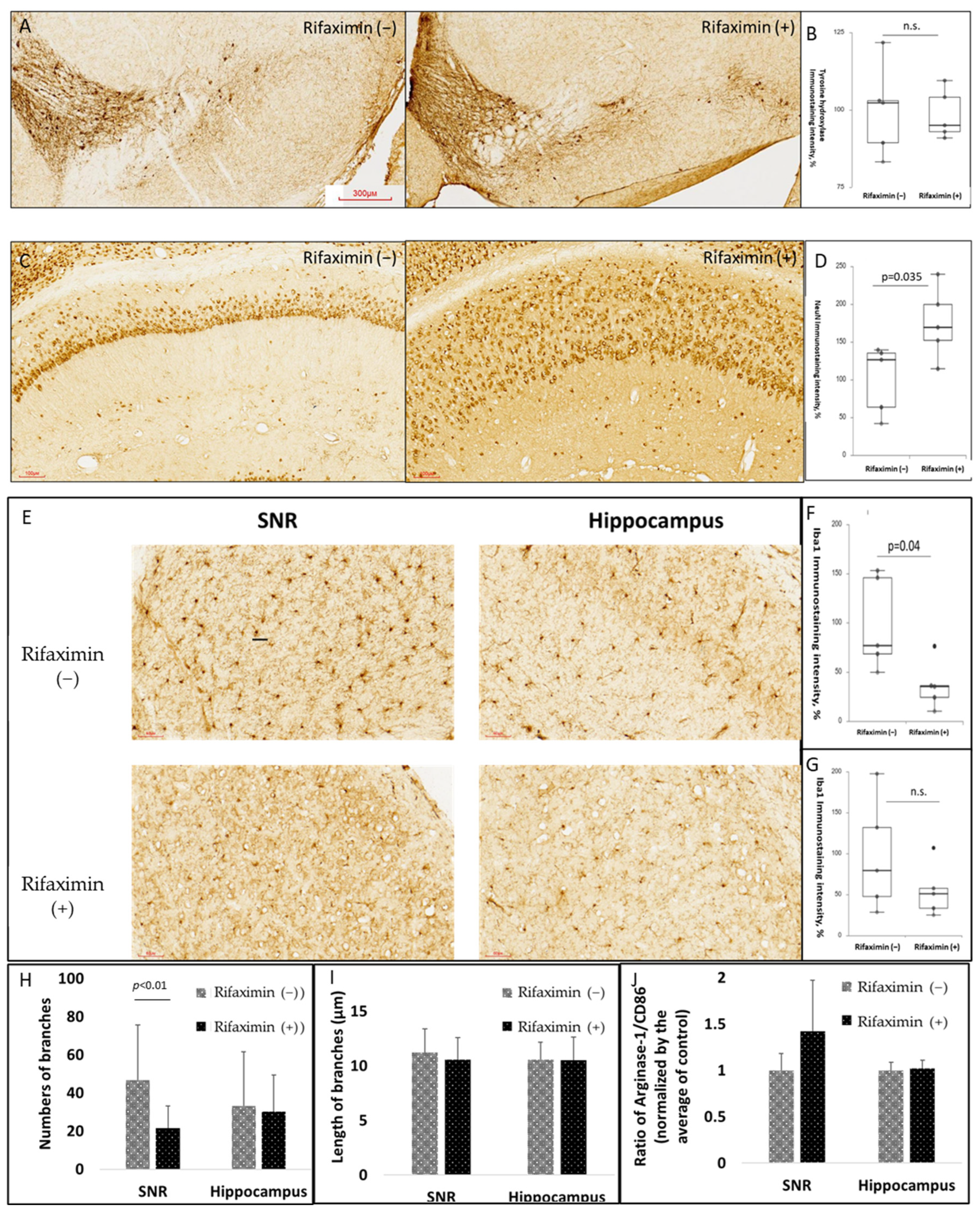
3.4. Rifaximin Treatment Attenuated Neuroinflammation in the Midbrain and Neurodegeneration in the Hippocampus of Transgenic PD Mice
3.5. Effect of Rifaximin Treatment on Patients with PD: Alterations in GM
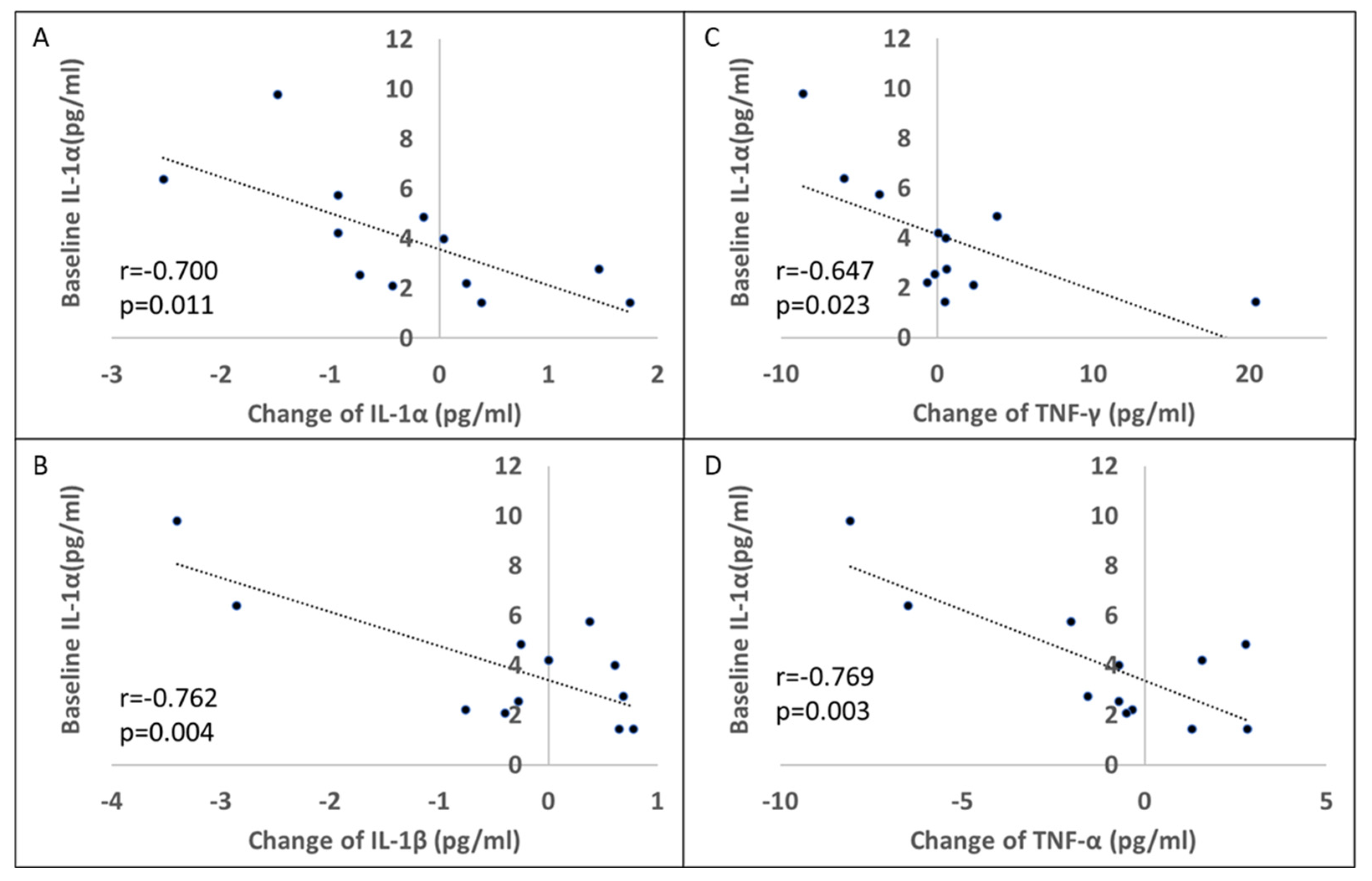
3.6. Effect of Rifaximin Treatment on Patients with PD: Serum Cytokine Profile
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BBB | blood-brain barrier |
| ELISA | enzyme-linked immunosorbent assay |
| GM | gut microbiota |
| IFN | interferon |
| IL | interleukin |
| Iba1 | ionized calcium-binding adapter molecule 1 |
| MMSE | Mini-Mental Status Examination |
| NOR | novel object recognition |
| PD | Parkinson’s disease |
| PCoA | principal coordinate analysis |
| rRNA | ribosomal RNA |
| rpm | revolutions per minute |
| SV | sequence variants |
| TH | tyrosine hydroxylase |
| TNF | tumor necrosis factor |
| UPDRS | Unified Parkinson’s Disease Rating Scale |
References
- de Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e627. [Google Scholar] [CrossRef]
- Shannon, K.M.; Keshavarzian, A.; Dodiya, H.B.; Jakate, S.; Kordower, J.H. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov. Disord. 2012, 27, 716–719. [Google Scholar] [CrossRef]
- Shannon, K.M.; Keshavarzian, A.; Mutlu, E.; Dodiya, H.B.; Daian, D.; Jaglin, J.A.; Kordower, J.H. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov. Disord. 2012, 27, 709–715. [Google Scholar] [CrossRef]
- Hilton, D.; Stephens, M.; Kirk, L.; Edwards, P.; Potter, R.; Zajicek, J.; Broughton, E.; Hagan, H.; Carroll, C. Accumulation of α-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol. 2014, 127, 235–241. [Google Scholar] [CrossRef]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease—The gut–brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e1412. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Chen, C.C.; Chiang, H.L.; Liou, J.M.; Chang, C.M.; Lu, T.P.; Chuang, E.Y.; Tai, Y.C.; Cheng, C.; Lin, H.Y.; et al. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflammation 2019, 16, 129. [Google Scholar] [CrossRef]
- Aho, V.T.E.; Pereira, P.A.B.; Voutilainen, S.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 2019, 44, 691–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopfner, F.; Künstner, A.; Müller, S.H.; Künzel, S.; Zeuner, K.E.; Margraf, N.G.; Deuschl, G.; Baines, J.F.; Kuhlenbäumer, G. Gut microbiota in Parkinson disease in a northern German cohort. Brain Res. 2017, 1667, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Savva, G.M.; Bedarf, J.R.; Charles, I.G.; Hildebrand, F.; Narbad, A. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinson’s Dis. 2021, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Fong, W.; Li, Q.; Yu, J. Gut microbiota modulation: A novel strategy for prevention and treatment of colorectal cancer. Oncogene 2020, 39, 4925–4943. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef]
- Gillis, J.C.; Brogden, R.N. Rifaximin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic potential in conditions mediated by gastrointestinal bacteria. Drugs 1995, 49, 467–484. [Google Scholar] [CrossRef]
- Taylor, D.N.; Hamer, D.H.; Shlim, D.R. Medications for the prevention and treatment of travellers’ diarrhea. J. Travel Med. 2017, 24, S17–S22. [Google Scholar] [CrossRef] [Green Version]
- Caraceni, P.; Vargas, V.; Solà, E.; Alessandria, C.; de Wit, K.; Trebicka, J.; Angeli, P.; Mookerjee, R.P.; Durand, F.; Pose, E.; et al. The Use of Rifaximin in Patients With Cirrhosis. Hepatology 2021, 74, 1660–1673. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Scaldaferri, F.; Petito, V.; Paroni Sterbini, F.; Pecere, S.; Lopetuso, L.R.; Palladini, A.; Gerardi, V.; Masucci, L.; Pompili, M.; et al. The Role of Antibiotics in Gut Microbiota Modulation: The Eubiotic Effects of Rifaximin. Dig. Dis. 2016, 34, 269–278. [Google Scholar] [CrossRef]
- Fasano, A.; Bove, F.; Gabrielli, M.; Petracca, M.; Zocco, M.A.; Ragazzoni, E.; Barbaro, F.; Piano, C.; Fortuna, S.; Tortora, A.; et al. The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov. Disord. 2013, 28, 1241–1249. [Google Scholar] [CrossRef]
- Suhocki, P.V.; Ronald, J.S.; Diehl, A.M.E.; Murdoch, D.M.; Doraiswamy, P.M. Probing gut-brain links in Alzheimer’s disease with rifaximin. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2022, 8, e12225. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, M.I.; Galter, D. The MitoPark Mouse—An animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat. Disord. 2009, 15 (Suppl. S3), S185–S188. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Morrison, H. Quantifying Microglia Morphology from Photomicrographs of Immunohistochemistry Prepared Tissue Using ImageJ. J. Vis. Exp. 2018, 136, e57648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obrenovich, M.E.M. Leaky Gut, Leaky Brain? Microorganisms 2018, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Lasek-Bal, A.; Kokot, A.; Gendosz de Carrillo, D.; Student, S.; Pawletko, K.; Krzan, A.; Puz, P.; Bal, W.; Jędrzejowska-Szypułka, H. Plasma Levels of Occludin and Claudin-5 in Acute Stroke Are Correlated with the Type and Location of Stroke but Not with the Neurological State of Patients-Preliminary Data. Brain Sci. 2020, 10, 831. [Google Scholar] [CrossRef]
- Jiao, X.; He, P.; Li, Y.; Fan, Z.; Si, M.; Xie, Q.; Chang, X.; Huang, D. The Role of Circulating Tight Junction Proteins in Evaluating Blood Brain Barrier Disruption following Intracranial Hemorrhage. Dis. Markers 2015, 2015, 860120. [Google Scholar] [CrossRef] [Green Version]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar]
- Mogilevski, T. The bi-directional role of the gut-brain axis in inflammatory and other gastrointestinal diseases. Curr. Opin. Gastroenterol. 2021, 37, 572–577. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [Green Version]
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The gut microbiome in neurological disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef]
- Cirstea, M.S.; Yu, A.C.; Golz, E.; Sundvick, K.; Kliger, D.; Radisavljevic, N.; Foulger, L.H.; Mackenzie, M.; Huan, T.; Finlay, B.B.; et al. Microbiota Composition and Metabolism Are Associated With Gut Function in Parkinson’s Disease. Mov. Disord. 2020, 35, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of Gut Microbiota in Patients with Parkinson’s Disease. Bull. Exp. Biol. Med. 2017, 162, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Scheperjans, F.; Aho, V.; Pereira, P.A.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- Yu, X.; Jin, Y.; Zhou, W.; Xiao, T.; Wu, Z.; Su, J.; Gao, H.; Shen, P.; Zheng, B.; Luo, Q.; et al. Rifaximin Modulates the Gut Microbiota to Prevent Hepatic Encephalopathy in Liver Cirrhosis Without Impacting the Resistome. Front. Cell. Infect. Microbiol. 2022, 11, 1427. [Google Scholar] [CrossRef]
- Jørgensen, S.F.; Macpherson, M.E.; Bjørnetrø, T.; Holm, K.; Kummen, M.; Rashidi, A.; Michelsen, A.E.; Lekva, T.; Halvorsen, B.; Trøseid, M.; et al. Rifaximin alters gut microbiota profile, but does not affect systemic inflammation—A randomized controlled trial in common variable immunodeficiency. Sci. Rep. 2019, 9, 167. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S.; Heuman, D.M.; Sanyal, A.J.; Hylemon, P.B.; Sterling, R.K.; Stravitz, R.T.; Fuchs, M.; Ridlon, J.M.; Daita, K.; Monteith, P.; et al. Modulation of the metabiome by rifaximin in patients with cirrhosis and minimal hepatic encephalopathy. PLoS ONE 2013, 8, e60042. [Google Scholar] [CrossRef] [Green Version]
- Kuti, D.; Winkler, Z.; Horváth, K.; Juhász, B.; Paholcsek, M.; Stágel, A.; Gulyás, G.; Czeglédi, L.; Ferenczi, S.; Kovács, K.J. Gastrointestinal (non-systemic) antibiotic rifaximin differentially affects chronic stress-induced changes in colon microbiome and gut permeability without effect on behavior. Brain Behav. Immun. 2020, 84, 218–228. [Google Scholar] [CrossRef]
- Sinkala, E.; Zyambo, K.; Besa, E.; Kaonga, P.; Nsokolo, B.; Kayamba, V.; Vinikoor, M.; Zulu, R.; Bwalya, M.; Foster, G.R.; et al. Rifaximin Reduces Markers of Inflammation and Bacterial 16S rRNA in Zambian Adults with Hepatosplenic Schistosomiasis: A Randomized Control Trial. Am. J. Trop. Med. Hyg. 2018, 98, 1152–1158. [Google Scholar] [CrossRef] [Green Version]
- Larsen, J.M. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Iljazovic, A.; Roy, U.; Gálvez, E.J.C.; Lesker, T.R.; Zhao, B.; Gronow, A.; Amend, L.; Will, S.E.; Hofmann, J.D.; Pils, M.C.; et al. Perturbation of the gut microbiome by Prevotella spp. enhances host susceptibility to mucosal inflammation. Mucosal Immunol. 2021, 14, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.S.; Griffen, A.L.; Barton, J.A.; Paster, B.J.; Moeschberger, M.L.; Leys, E.J. New bacterial species associated with chronic periodontitis. J. Dent. Res. 2003, 82, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Lucke, K.; Miehlke, S.; Jacobs, E.; Schuppler, M. Prevalence of Bacteroides and Prevotella spp. in ulcerative colitis. J. Med. Microbiol. 2006, 55, 617–624. [Google Scholar] [CrossRef]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.M.; Ke, X.; Hitchcock, D.; Jeanfavre, S.; Avila-Pacheco, J.; Nakata, T.; Arthur, T.D.; Fornelos, N.; Heim, C.; Franzosa, E.A.; et al. Bacteroides-Derived Sphingolipids Are Critical for Maintaining Intestinal Homeostasis and Symbiosis. Cell Host Microbe 2019, 25, 668–680.e667. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Sibai, M.; Altuntaş, E.; Yıldırım, B.; Öztürk, G.; Yıldırım, S.; Demircan, T. Microbiome and Longevity: High Abundance of Longevity-Linked Muribaculaceae in the Gut of the Long-Living Rodent Spalax leucodon. OMICS 2020, 24, 592–601. [Google Scholar] [CrossRef]
- Baldini, F.; Hertel, J.; Sandt, E.; Thinnes, C.C.; Neuberger-Castillo, L.; Pavelka, L.; Betsou, F.; Krüger, R.; Thiele, I.; Aguayo, G.; et al. Parkinson’s disease-associated alterations of the gut microbiome predict disease-relevant changes in metabolic functions. BMC Biology 2020, 18, 62. [Google Scholar] [CrossRef]
- Mao, L.; Zhang, Y.; Tian, J.; Sang, M.; Zhang, G.; Zhou, Y.; Wang, P. Cross-Sectional Study on the Gut Microbiome of Parkinson’s Disease Patients in Central China. Front. Microbiol. 2021, 12, 728479. [Google Scholar] [CrossRef]
- Ferrari, C.C.; Tarelli, R. Parkinson’s disease and systemic inflammation. Parkinsons Dis. 2011, 2011, 436813. [Google Scholar] [CrossRef] [PubMed]
- Brudek, T. Inflammatory Bowel Diseases and Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, S331–S344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weimers, P.; Halfvarson, J.; Sachs, M.C.; Saunders-Pullman, R.; Ludvigsson, J.F.; Peter, I.; Burisch, J.; Olén, O. Inflammatory Bowel Disease and Parkinson’s Disease: A Nationwide Swedish Cohort Study. Inflamm. Bowel Dis. 2018, 25, 111–123. [Google Scholar] [CrossRef]
- Bower, J.H.; Maraganore, D.M.; Peterson, B.J.; Ahlskog, J.E.; Rocca, W.A. Immunologic diseases, anti-inflammatory drugs, and Parkinson disease: A case-control study. Neurology 2006, 67, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Samii, A.; Etminan, M.; Wiens, M.O.; Jafari, S. NSAID use and the risk of Parkinson’s disease: Systematic review and meta-analysis of observational studies. Drugs Aging 2009, 26, 769–779. [Google Scholar] [CrossRef]
- Kinashi, Y.; Hase, K. Partners in Leaky Gut Syndrome: Intestinal Dysbiosis and Autoimmunity. Front. Immunol. 2021, 12, 673708. [Google Scholar] [CrossRef]
- Chan, L.; Chung, C.-C.; Chen, J.-H.; Yu, R.-C.; Hong, C.-T. Cytokine Profile in Plasma Extracellular Vesicles of Parkinson’s Disease and the Association with Cognitive Function. Cells 2021, 10, 604. [Google Scholar] [CrossRef]
- Qin, X.Y.; Zhang, S.P.; Cao, C.; Loh, Y.P.; Cheng, Y. Aberrations in Peripheral Inflammatory Cytokine Levels in Parkinson Disease: A Systematic Review and Meta-analysis. JAMA Neurol. 2016, 73, 1316–1324. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef]
- Leclercq, S.; Mian, F.M.; Stanisz, A.M.; Bindels, L.B.; Cambier, E.; Ben-Amram, H.; Koren, O.; Forsythe, P.; Bienenstock, J. Low-dose penicillin in early life induces long-term changes in murine gut microbiota, brain cytokines and behavior. Nat. Commun. 2017, 8, 15062. [Google Scholar] [CrossRef] [Green Version]
- Logsdon, A.F.; Erickson, M.A.; Rhea, E.M.; Salameh, T.S.; Banks, W.A. Gut reactions: How the blood-brain barrier connects the microbiome and the brain. Exp. Biol. Med. 2018, 243, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; Oeda, T.; Yamamoto, K.; Tomita, S.; Kohsaka, M.; Park, K.; Sugiyama, H.; Sawada, H. Baseline Plasma C-Reactive Protein Concentrations and Motor Prognosis in Parkinson Disease. PLoS ONE 2015, 10, e0136722. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ruiz, C.; Williams-Gray, C.H.; Yarnall, A.J.; Boucher, J.J.; Lawson, R.A.; Wijeyekoon, R.S.; Barker, R.A.; Kolenda, C.; Parker, C.; Burn, D.J.; et al. Senescence and Inflammatory Markers for Predicting Clinical Progression in Parkinson’s Disease: The ICICLE-PD Study. J. Parkinsons Dis. 2020, 10, 193–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, H.L.; DuPont, H.L. Rifaximin: A unique gastrointestinal-selective antibiotic for enteric diseases. Curr. Opin. Gastroenterol. 2010, 26, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, N.M.; Mullen, K.D.; Sanyal, A.; Poordad, F.; Neff, G.; Leevy, C.B.; Sigal, S.; Sheikh, M.Y.; Beavers, K.; Frederick, T.; et al. Rifaximin treatment in hepatic encephalopathy. N. Engl. J. Med. 2010, 362, 1071–1081. [Google Scholar] [CrossRef] [Green Version]
- Amboni, M.; Barone, P.; Hausdorff, J.M. Cognitive contributions to gait and falls: Evidence and implications. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 1520–1533. [Google Scholar] [CrossRef]
- Howard, C.E.; Chen, C.L.; Tabachnik, T.; Hormigo, R.; Ramdya, P.; Mann, R.S. Serotonergic Modulation of Walking in Drosophila. Curr. Biol. 2019, 29, 4218–4230.e4218. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n = 13 | |
|---|---|
| Female | 6 |
| Age (year-old) | 61.59 ± 5.34 |
| Disease duration (years) | 1.77 ± 1.74 |
| UPDRS part III | |
| Baseline | 13.69 ± 8.75 |
| 6-month post rifaximin | 12.31 ± 9.21 |
| MMSE | 27.92 ± 2.33 |
| Baseline | 6-Month Post-Rifaximin | p Value | |
|---|---|---|---|
| Interleukin-1α | 4.83 ± 3.94 | 4.25 ± 2.70 | 0.21 |
| Interleukin-1β | 17.26 ± 2.88 | 16.88 ± 1.96 | 0.32 |
| Interleukin-6 | 4.42 ± 1.24 | 4.52 ± 1.77 | 0.80 |
| Interleukin-10 | 15.88 ± 8.60 | 26.82 ± 25.32 | 0.06 |
| Interferon-γ | 7.73 ± 4.41 | 8.41 ± 5.39 | 0.72 |
| Tumor necrosis factor-α | 13.96 ± 8.59 | 13.13 ± 7.47 | 0.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, C.-T.; Chan, L.; Chen, K.-Y.; Lee, H.-H.; Huang, L.-K.; Yang, Y.-C.S.H.; Liu, Y.-R.; Hu, C.-J. Rifaximin Modifies Gut Microbiota and Attenuates Inflammation in Parkinson’s Disease: Preclinical and Clinical Studies. Cells 2022, 11, 3468. https://doi.org/10.3390/cells11213468
Hong C-T, Chan L, Chen K-Y, Lee H-H, Huang L-K, Yang Y-CSH, Liu Y-R, Hu C-J. Rifaximin Modifies Gut Microbiota and Attenuates Inflammation in Parkinson’s Disease: Preclinical and Clinical Studies. Cells. 2022; 11(21):3468. https://doi.org/10.3390/cells11213468
Chicago/Turabian StyleHong, Chien-Tai, Lung Chan, Kai-Yun Chen, Hsun-Hua Lee, Li-Kai Huang, Yu-Chen S. H. Yang, Yun-Ru Liu, and Chaur-Jong Hu. 2022. "Rifaximin Modifies Gut Microbiota and Attenuates Inflammation in Parkinson’s Disease: Preclinical and Clinical Studies" Cells 11, no. 21: 3468. https://doi.org/10.3390/cells11213468