
Enhancing an Oxidative “Trojan Horse” Action of Vitamin C with Arsenic Trioxide for Effective Suppression of KRAS-Mutant Cancers: A Promising Path at the Bedside

 , , and
, , and
Abstract
:1. Introduction
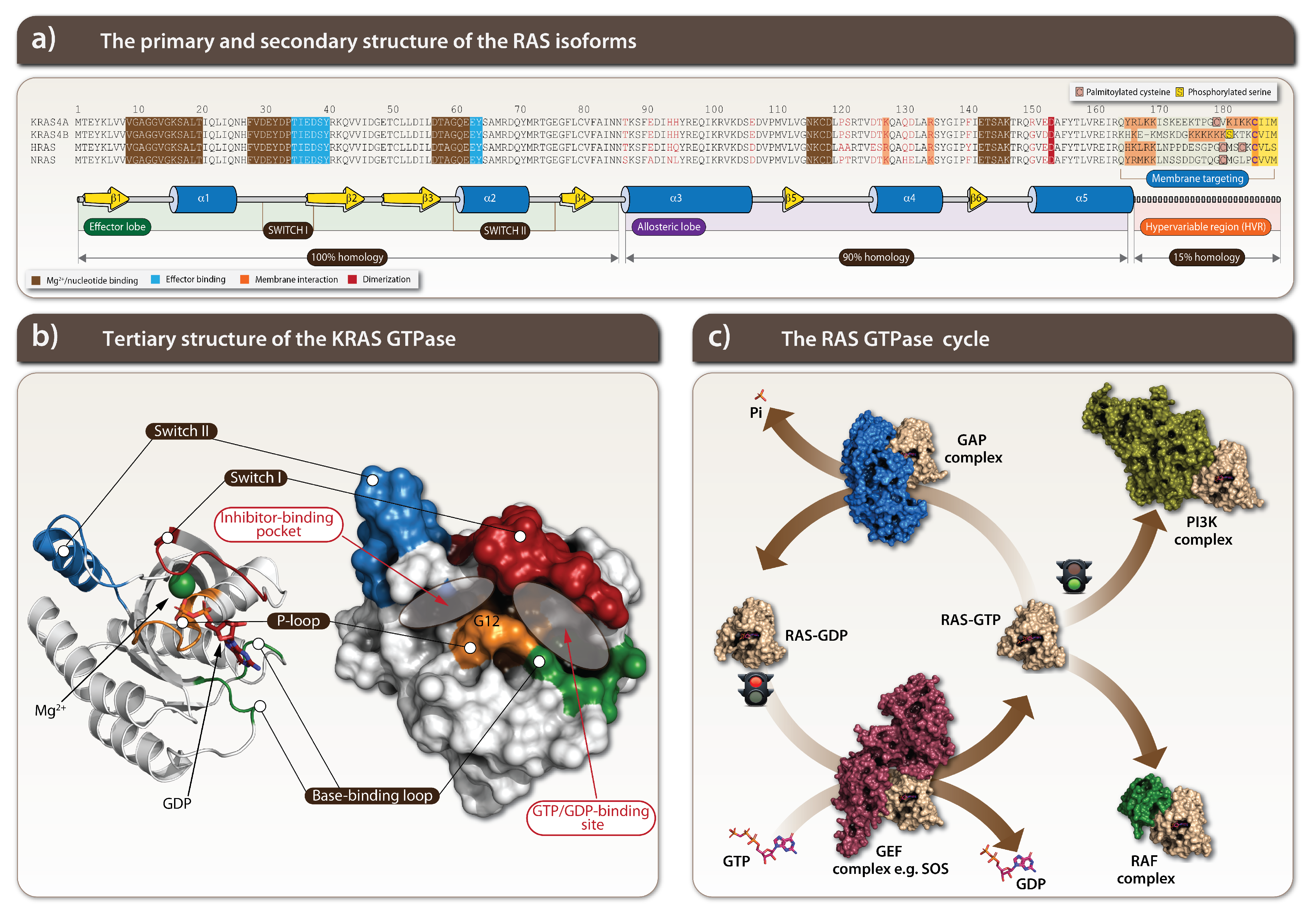
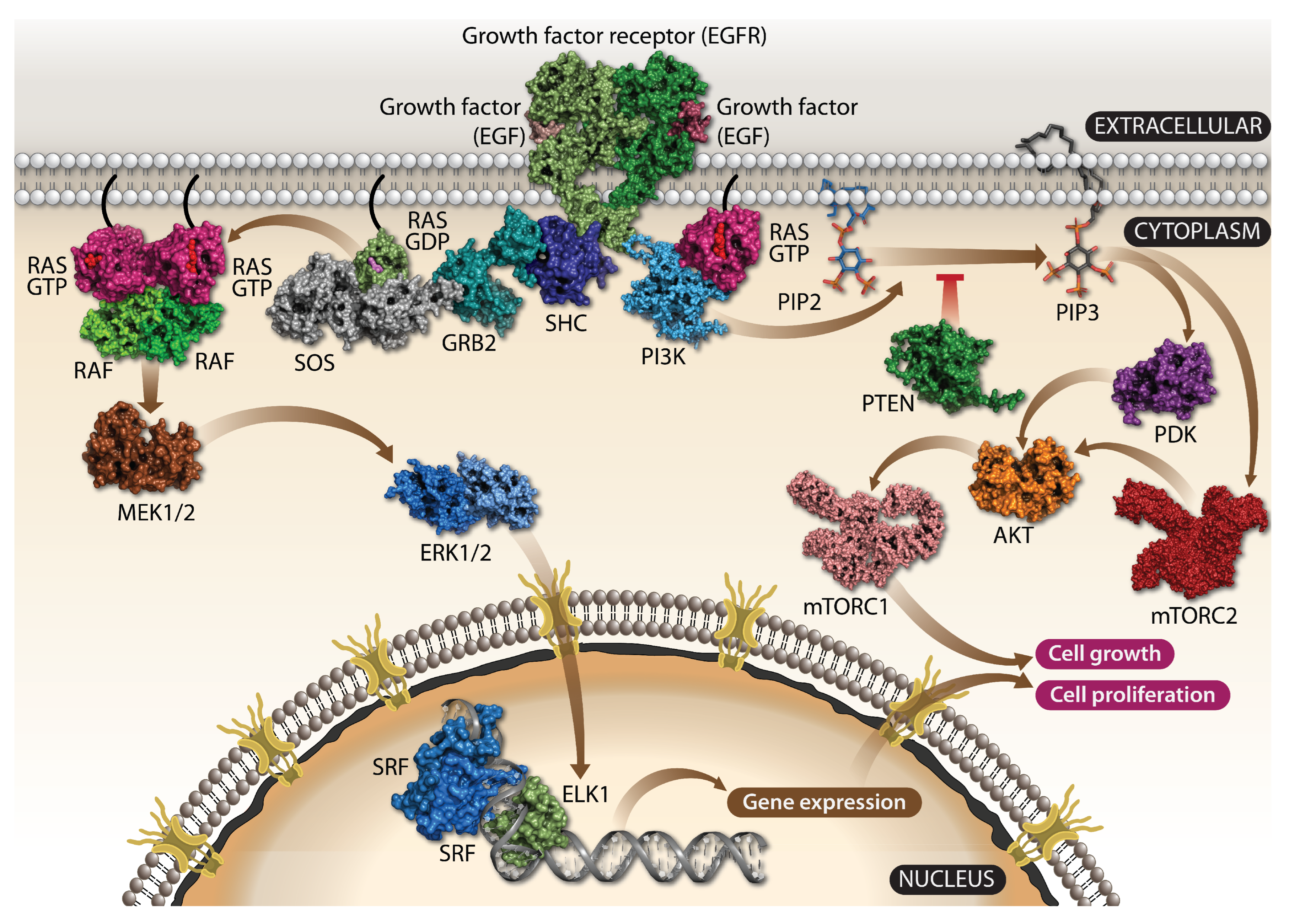
1.1. KRAS Is a Crucial Component of Growth Factor Signaling
1.2. KRAS Is a Potent Oncogenic Driver
1.3. Challenges of KRAS Targeting
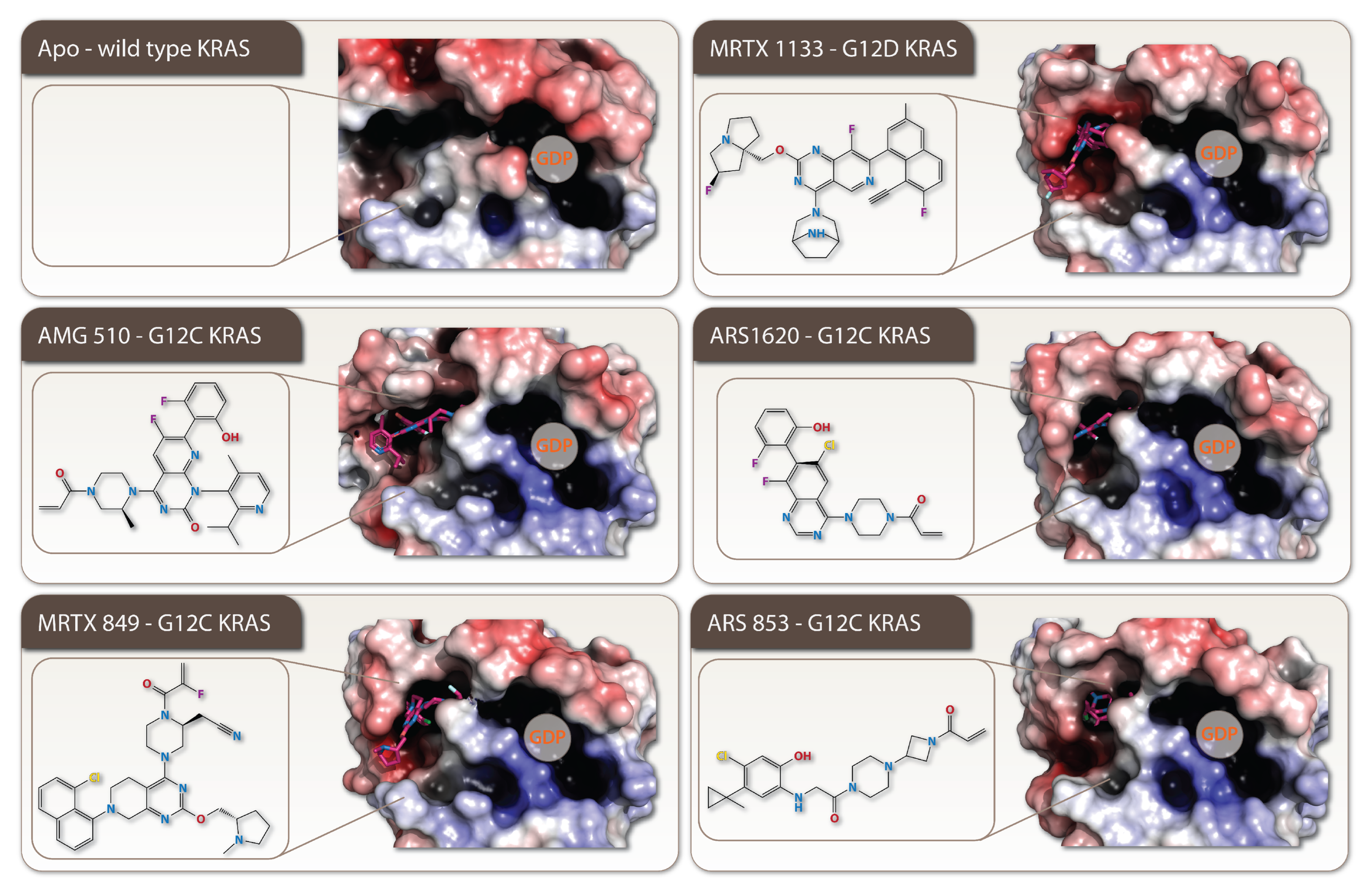
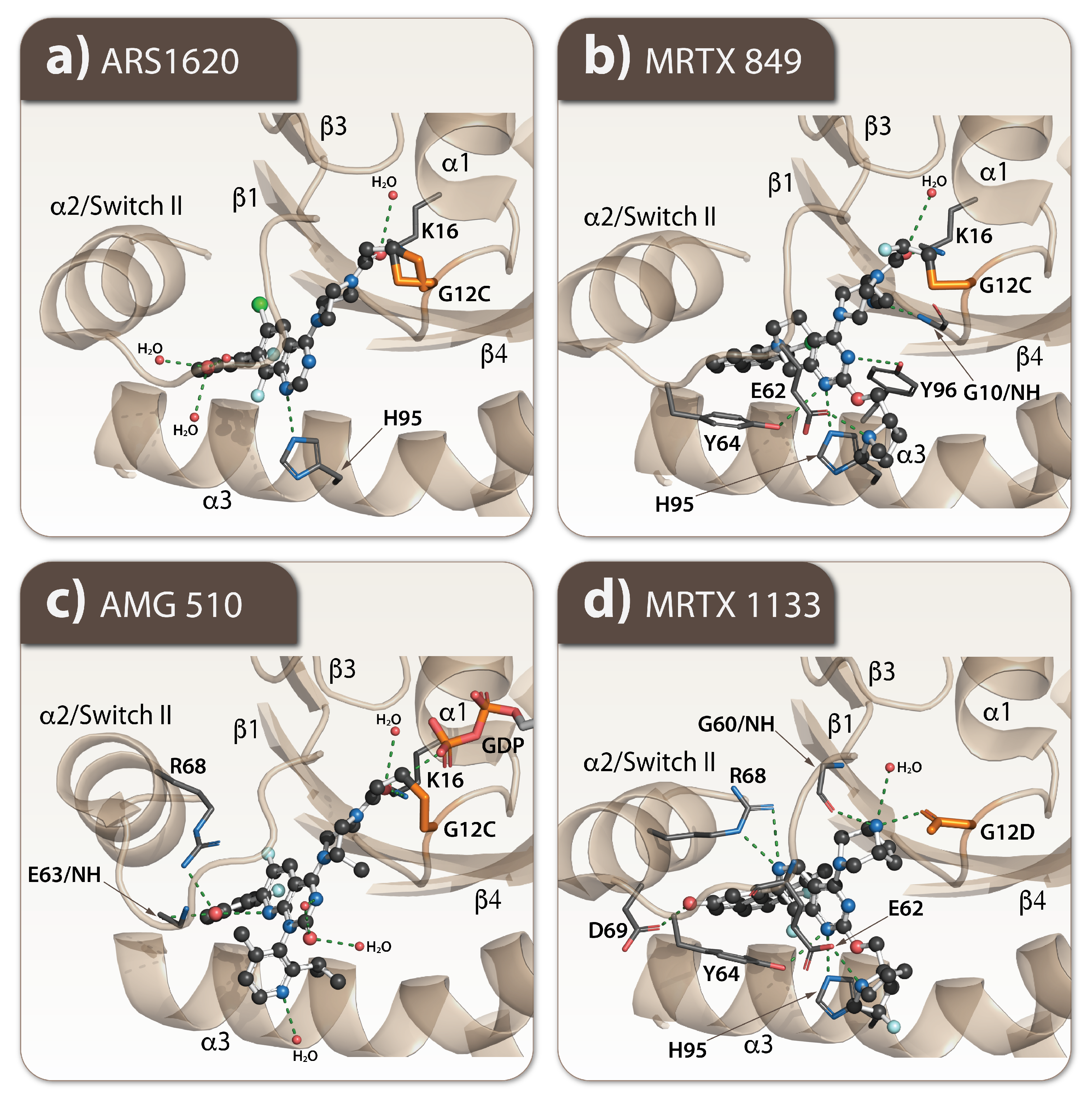
2. Targeting of the Specific KRAS-Mutants
2.1. Every Common Specific KRAS-Mutant: A Case for the Drug Development
2.2. How Resistance Gets Evolved to the Specific KRAS G12C Mutant Inhibitors
3. Deregulation of Metabolism in Cancer
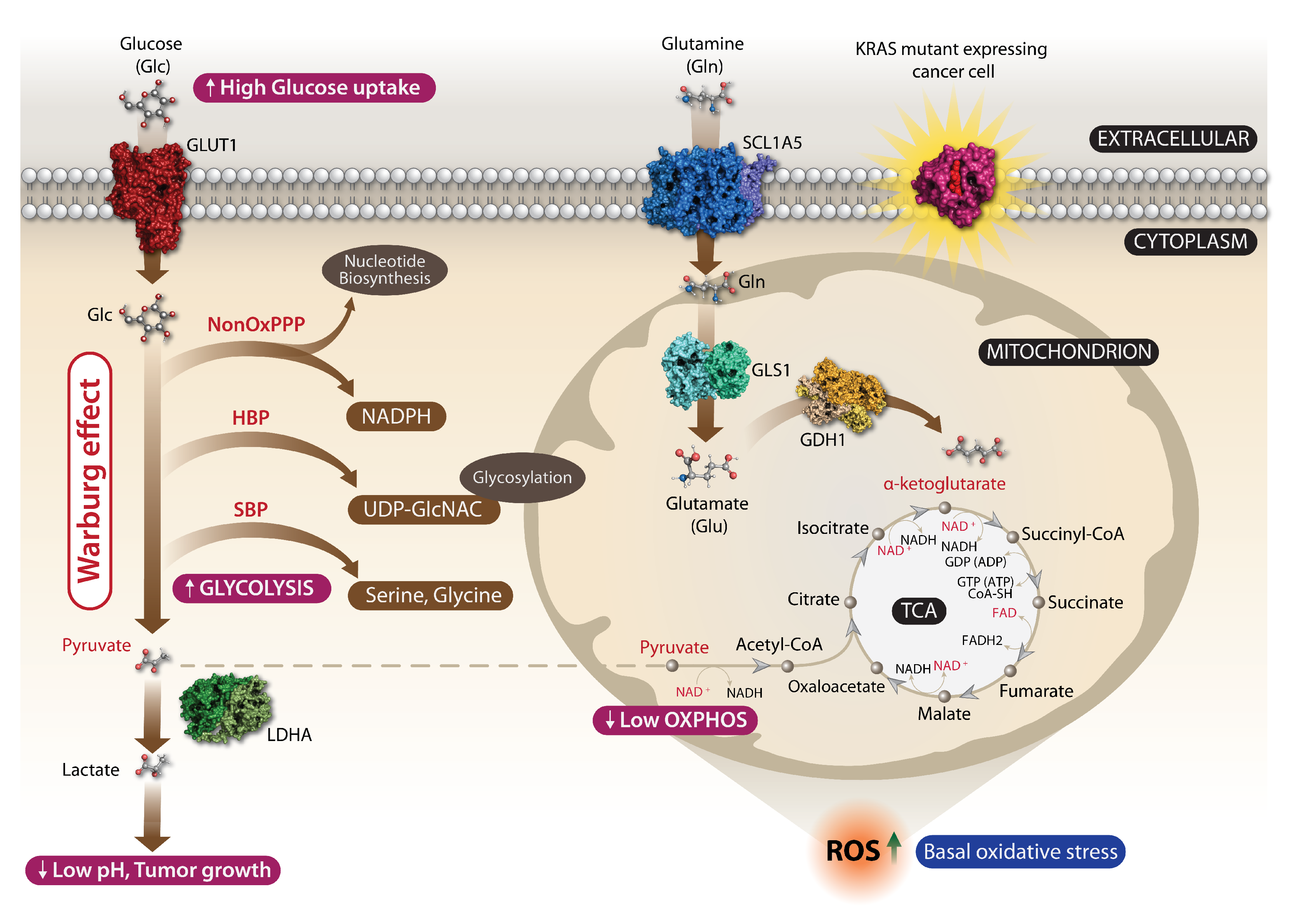
3.1. KRAS Cancer Mutations and Metabolic Reprogramming
3.2. A Basal Oxidative Stress Is a Liability of Malignant KRAS-Mutant Cancers
4. The Vitamin C Cancer Treatment Is Back: How a Famous Antioxidant Turns to a “Trojan Horse” Oxidant in KRAS-Mutant Cancer Cells
4.1. Vitamin C (Ascorbic Acid) in Cancer Treatment
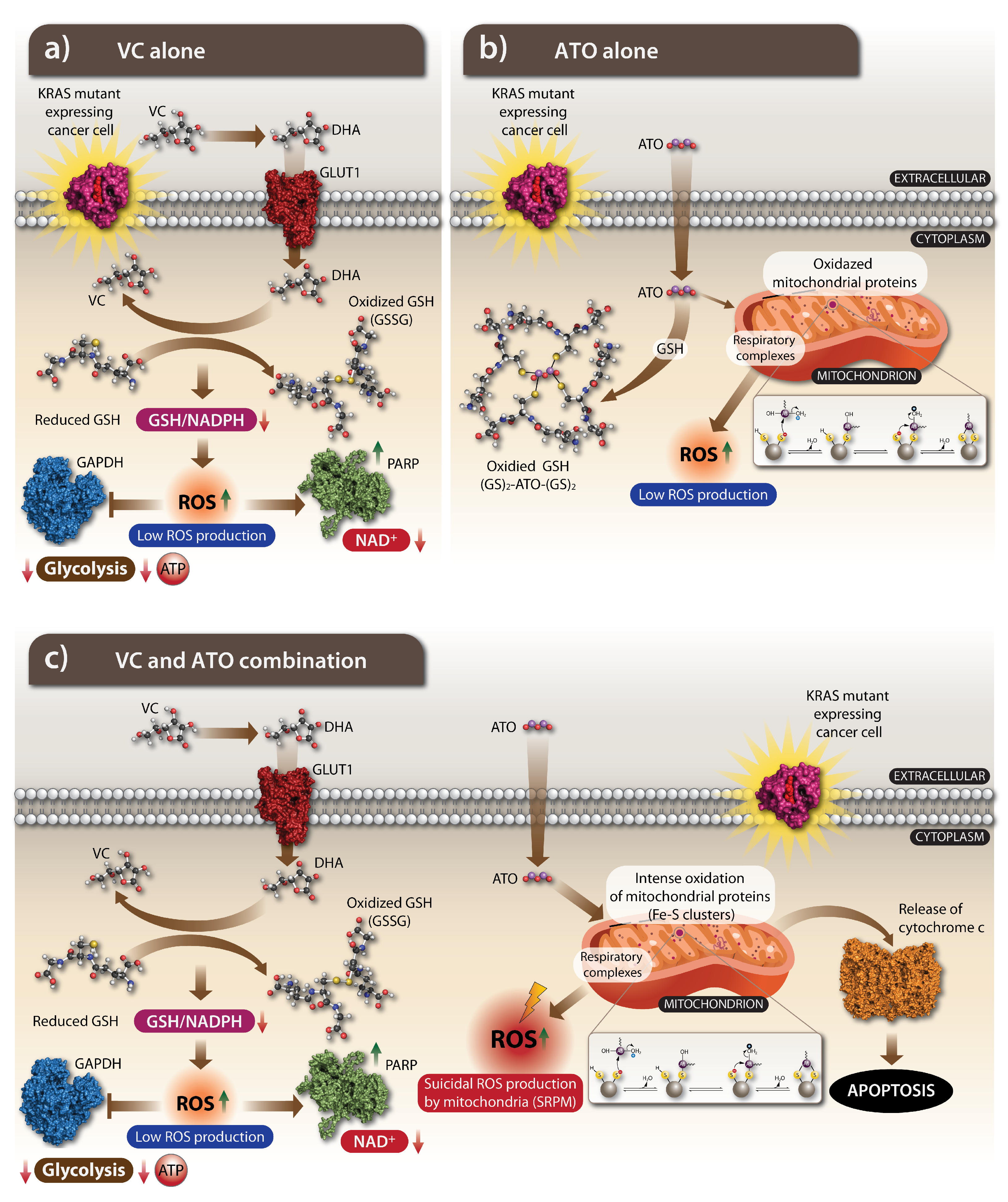
4.2. The Anti-Cancer Action of VC: How Does It Work
5. A Mild Oxidant ATO Is the Cancer Drug: The Mechanism of Action
5.1. The History of ATO: Harnessing the Poison for Cancer Treatment
5.2. The Oxidizing Impact of ATO
5.3. The Action of ATO on a Cellular Metabolism and Its Nuclear Effects
6. ATO Potentiates an Oxidative Effect of VC and the Combination of Both Drugs Is Effectively Killing KRAS-Mutant Cancer Cells
7. A Remarkable Turn: The VC’s Enantiomer D-VC Works Better in the Tumor Xenograft Animal Model
8. Conclusions
Funding
Conflicts of Interest
References
- Tsuchida, N.; Ryder, T.; Ohtsubo, E. Nucleotide sequence of the oncogene encoding the p21 transforming protein of Kirsten murine sarcoma virus. Science 1982, 217, 937–939. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, N.; Uesugi, S. Structure and functions of the Kirsten murine sarcoma virus genome: Molecular cloning of biologically active Kirsten murine sarcoma virus DNA. J. Virol. 1981, 38, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Hancock, J.; Cadwallader, K.; Paterson, H.; Marshall, C. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. Embo J. 1991, 10, 4033–4039. [Google Scholar] [CrossRef] [PubMed]
- Colicelli, J. Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, 2004, RE13. [Google Scholar] [CrossRef] [Green Version]
- Zenonos, K.; Kyprianou, K. RAS signaling pathways, mutations and their role in colorectal cancer. World J. Gastrointest. Oncol. 2013, 5, 97. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS proteins and their regulators in human disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Smit, V.T.; Boot, A.J.; Smits, A.M.; Fleuren, G.J.; Cornelisse, C.J.; Bos, J.L. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988, 16, 7773–7782. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Aviel-Ronen, S.; Blackhall, F.H.; Shepherd, F.A.; Tsao, M.-S. K-ras mutations in non-small-cell lung carcinoma: A review. Clin. Lung Cancer 2006, 8, 30–38. [Google Scholar] [CrossRef]
- Russo, M.; Di Nicolantonio, F.; Bardelli, A. Climbing RAS, the everest of oncogenes. Cancer Discov. 2014, 4, 19–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, B. RAS signaling and anti-RAS therapy: Lessons learned from genetically engineered mouse models, human cancer cells, and patient-related studies. Acta Biochim. Biophys. Sin. 2016, 48, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cekani, E.; Epistolio, S.; Dazio, G.; Cefalì, M.; Wannesson, L.; Frattini, M.; Froesch, P. Molecular Biology and Therapeutic Perspectives for K-Ras Mutant Non-Small Cell Lung Cancers. Cancers 2022, 14, 4103. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, Y.; Qian, L.; Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 2021, 14, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Vasan, N.; Boyer, J.L.; Herbst, R.S. A RAS renaissance: Emerging targeted therapies for KRAS-mutated non–small cell lung cancer. Clin. Cancer Res. 2014, 20, 3921–3930. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Mo, S.P.; Coulson, J.M.; Prior, I.A. RAS variant signalling. Biochem. Soc. Trans. 2018, 46, 1325. [Google Scholar] [CrossRef] [Green Version]
- Pantsar, T.; Rissanen, S.; Dauch, D.; Laitinen, T.; Vattulainen, I.; Poso, A. Assessment of mutation probabilities of KRAS G12 missense mutants and their long-timescale dynamics by atomistic molecular simulations and Markov state modeling. PLoS Comput. Biol. 2018, 14, e1006458. [Google Scholar] [CrossRef]
- Huynh, M.V.; Hobbs, G.A.; Schaefer, A.; Pierobon, M.; Carey, L.M.; Diehl, J.N.; DeLiberty, J.M.; Thurman, R.D.; Cooke, A.R.; Goodwin, C.M. Functional and biological heterogeneity of KRASQ61 mutations. Sci. Signal. 2022, 15, eabn2694. [Google Scholar] [CrossRef]
- Blons, H.; Emile, J.-F.; Le Malicot, K.; Julié, C.; Zaanan, A.; Tabernero, J.; Mini, E.; Folprecht, G.; Van Laethem, J.-L.; Thaler, J. Prognostic value of KRAS mutations in stage III colon cancer: Post hoc analysis of the PETACC8 phase III trial dataset. Ann. Oncol. 2014, 25, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-W.; Kim, K.J.; Han, S.-W.; Lee, H.J.; Rhee, Y.Y.; Bae, J.M.; Cho, N.-Y.; Lee, K.-H.; Kim, T.-Y.; Oh, D.-Y. KRAS mutation is associated with worse prognosis in stage III or high-risk stage II colon cancer patients treated with adjuvant FOLFOX. Ann. Surg. Oncol. 2015, 22, 187–194. [Google Scholar] [CrossRef]
- Yoon, H.H.; Tougeron, D.; Shi, Q.; Alberts, S.R.; Mahoney, M.R.; Nelson, G.D.; Nair, S.G.; Thibodeau, S.N.; Goldberg, R.M.; Sargent, D.J. KRAS Codon 12 and 13 Mutations in Relation to Disease-Free Survival in BRAF–Wild-Type Stage III Colon Cancers from an Adjuvant Chemotherapy Trial (N0147 Alliance) Prognostic Impact of Specific KRAS Mutations in Colon Cancer. Clin. Cancer Res. 2014, 20, 3033–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.E.; Yoo, S.-Y.; Cho, N.-Y.; Bae, J.M.; Han, S.-W.; Lee, H.S.; Park, K.J.; Kim, T.-Y.; Kang, G.H. Tumor microenvironment-adjusted prognostic implications of the KRAS mutation subtype in patients with stage III colorectal cancer treated with adjuvant FOLFOX. Sci. Rep. 2021, 11, 14609. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Addiction to oncogenes–the Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.; Wilson, M.B.; Crawford, Y.G.; Reynolds, P.A.; Sigaroudinia, M.; Tlsty, T.D. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 14867–14872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Nissley, D.V.; McCormick, F.; Stephens, R.M. ssGSEA score-based Ras dependency indexes derived from gene expression data reveal potential Ras addiction mechanisms with possible clinical implications. Sci. Rep. 2020, 10, 10258. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Sakai, D.; Taniguchi, H.; Sugimoto, N.; Tamura, T.; Nishina, T.; Hara, H.; Esaki, T.; Denda, T.; Sakamoto, T.; Okuda, H. Randomised phase II study of panitumumab plus irinotecan versus cetuximab plus irinotecan in patients with KRAS wild-type metastatic colorectal cancer refractory to fluoropyrimidine, irinotecan and oxaliplatin (WJOG 6510G). Eur. J. Cancer 2020, 135, 11–21. [Google Scholar] [CrossRef]
- Yokota, T. Are KRAS/BRAF mutations potent prognostic and/or predictive biomarkers in colorectal cancers? Anti-Cancer Agents Med. Chem. (Formerly Curr. Med.-Chem.-Anti-Cancer Agents) 2012, 12, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kaiser, C.E.; Frett, B.; Li, H.-Y. Targeting mutant KRAS for anticancer therapeutics: A review of novel small molecule modulators. J. Med. Chem. 2013, 56, 5219–5230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, T.; Garrenton, L.S.; Oh, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef] [Green Version]
- Mottini, C.; Tomihara, H.; Carrella, D.; Lamolinara, A.; Iezzi, M.; Huang, J.K.; Amoreo, C.A.; Buglioni, S.; Manni, I.; Robinson, F.S. Predictive Signatures Inform the Effective Repurposing of Decitabine to Treat KRAS–Dependent Pancreatic Ductal AdenocarcinomaDecitabine Inhibits KRAS–Dependent Growth of Selected PDAC. Cancer Res. 2019, 79, 5612–5625. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N. The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and PatientsTherapeutic Insight from the KRASG12C Inhibitor MRTX849. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Weng, J.; Fan, X.; Wang, E.; Zhu, Q.; Tao, L.; Han, Z.; Wang, Z.; Niu, H.; Jiang, Y. Discovery of D-1553, a novel and selective KRas-G12C inhibitor with potent anti-tumor activity in a broad spectrum of tumor cell lines and xenograft models. Cancer Res. 2021, 81, 932. [Google Scholar] [CrossRef]
- Shi, Z.; Weng, J.; Fan, X.; Zhu, Q.; Robb, E.; Moriarty, A.; Wick, M.; Jiang, Y.; Zhang, L.; Dai, X. Potent in vivo anti-tumor activity of D-1553 as a single agent and in combination with targeted therapeutics in a broad spectrum of patient-derived xenograft tumor models with KRas G12C mutation. Cancer Res. 2021, 81, 1056. [Google Scholar] [CrossRef]
- Weiss, A.; Lorthiois, E.; Barys, L.; Beyer, K.S.; Bomio-Confaglia, C.; Burks, H.; Chen, X.; Cui, X.; de Kanter, R.; Dharmarajan, L. Discovery, Preclinical Characterization, and Early Clinical Activity of JDQ443, a Structurally Novel, Potent, and Selective Covalent Oral Inhibitor of KRASG12C. Cancer Discov. 2022, 12, 1500–1517. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Voshol, H.; Porta, D.G.; Fedele, C.; Sterker, D.; De Kanter, R.; Stringer, R.; Widmer, T.; Loo, A.; Guthy, D.A. JDQ443, a covalent inhibitor of KRASG12C with a novel binding mode, shows broad antitumor activity in KRASG12C preclinical models as a single agent and in combination with inhibitors of SHP2, MEK or CDK4/6. Cancer Res. 2022, 82, 4026. [Google Scholar] [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J. Med. Chem. 2021, 65, 3123–3133. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Martin-Romano, P.; Cassier, P.; Johnson, M.; Haura, E.; Lenox, L.; Guo, Y.; Bandyopadhyay, N.; Russell, M.; Shearin, E. Phase I Study of JNJ-74699157 in Patients with Advanced Solid Tumors Harboring the KRAS G12C Mutation. Oncologist 2022, 27, e536–e553. [Google Scholar] [CrossRef]
- Peng, S.-B.; Si, C.; Zhang, Y.; Van Horn, R.D.; Lin, X.; Gong, X.; Huber, L.; Donoho, G.; Curtis, C.; Strelow, J.M.; et al. Abstract 1259: Preclinical characterization of LY3537982, a novel, highly selective and potent KRAS-G12C inhibitor. Cancer Res. 2021, 81, 1259. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.J.; Yu, Z.; Griffith, K.; Hanada, K.-i.; Restifo, N.P.; Yang, J.C. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol. Res. 2016, 4, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Edkins, S.; O’Meara, S.; Parker, A.; Stevens, C.; Reis, M.; Jones, S.; Greenman, C.; Davies, H.; Dalgliesh, G.; Forbes, S. Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol. Ther. 2006, 5, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L. Targeting KRAS-mutant cancers with a covalent G12C-specific inhibitor. Cell 2018, 172, 578–589.e517. [Google Scholar] [CrossRef] [Green Version]
- Herdeis, L.; Gerlach, D.; McConnell, D.B.; Kessler, D. Stopping the beating heart of cancer: KRAS reviewed. Curr. Opin. Struct. Biol. 2021, 71, 136–147. [Google Scholar] [CrossRef]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Ballard, J.; Blake, J.F.; Bouhana, K.; Brandhuber, B.J.; Briere, D.M.; Burgess, L.E.; Burkard, M.R. Discovery of tetrahydropyridopyrimidines as irreversible covalent inhibitors of KRAS-G12C with in vivo activity. Acs Med. Chem. Lett. 2018, 9, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.; Rybkin, I.I.; Spira, A.; Riely, G.; Papadopoulos, K.; Sabari, J.; Johnson, M.; Heist, R.; Bazhenova, L.; Barve, M. KRYSTAL-1: Activity and safety of adagrasib (MRTX849) in advanced/metastatic non–small-cell lung cancer (NSCLC) harboring KRAS G12C mutation. Eur. J. Cancer 2020, 138, S1–S2. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F. Sotorasib for lung cancers with KRAS p. G12C mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Lu, J.; Li, L.; Feru, F.; Quan, C.; Gero, T.W.; Ficarro, S.B.; Xiong, Y.; Ambrogio, C.; Paranal, R.M. Potent and selective covalent quinazoline inhibitors of KRAS G12C. Cell Chem. Biol. 2017, 24, 1005–1016.e1003. [Google Scholar] [CrossRef] [Green Version]
- Qi, S.-M.; Dong, J.; Xu, Z.-Y.; Cheng, X.-D.; Zhang, W.-D.; Qin, J.-J. PROTAC: An effective targeted protein degradation strategy for cancer therapy. Front. Pharmacol. 2021, 12, 692574. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gerlach, D.; Misale, S.; Petronczki, M.; Kraut, N. Expanding the Reach of Precision Oncology by Drugging All KRAS-mutantsDrugging All KRAS-mutants. Cancer Discov. 2022, 12, 924–937. [Google Scholar] [CrossRef]
- Nagasaka, M.; Li, Y.; Sukari, A.; Ou, S.-H.I.; Al-Hallak, M.N.; Azmi, A.S. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat. Rev. 2020, 84, 101974. [Google Scholar] [CrossRef]
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.J.; Peng, D.; Ran, H.; Mita, P.; Geer, M.J.; Hattori, T.; Koide, A. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J. Exp. Med. 2021, 218, e20201414. [Google Scholar] [CrossRef]
- Koga, T.; Suda, K.; Fujino, T.; Ohara, S.; Hamada, A.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Arita, T. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, sotorasib and adagrasib, and overcoming strategies: Insights from in vitro experiments. J. Thorac. Oncol. 2021, 16, 1321–1332. [Google Scholar] [CrossRef]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRASG12C InhibitionCombined SHP2 and KRASG12C Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y. EGFR Blockade Reverts Resistance to KRASG12C Inhibition in Colorectal CancerOvercoming KRASG12C Inhibitor Resistance in Colorectal Cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, L.; Lang, J.; Cheng, K.; Wang, Y.; Li, X.; Shi, J.; Wang, Y.; Nie, G. A CRISPR-Cas13a system for efficient and specific therapeutic targeting of mutant KRAS for pancreatic cancer treatment. Cancer Lett. 2018, 431, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Pecot, C.V.; Wu, S.Y.; Bellister, S.; Filant, J.; Rupaimoole, R.; Hisamatsu, T.; Bhattacharya, R.; Maharaj, A.; Azam, S.; Rodriguez-Aguayo, C. Therapeutic Silencing of KRAS Using Systemically Delivered siRNAs in vivo KRAS Silencing with siRNA. Mol. Cancer Ther. 2014, 13, 2876–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, T.L.; Fellmann, C.; Lee, C.-S.; Ritchie, C.D.; Thapar, V.; Lee, L.C.; Hsu, D.J.; Grace, D.; Carver, J.O.; Zuber, J. Development of siRNA Payloads to Target KRAS-Mutant CancerRNAi Therapy for KRAS-Mutant Cancer. Cancer Discov. 2014, 4, 1182–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.-C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3, e99263. [Google Scholar] [CrossRef]
- Brea, E.J.; Oh, C.Y.; Manchado, E.; Budhu, S.; Gejman, R.S.; Mo, G.; Mondello, P.; Han, J.E.; Jarvis, C.A.; Ulmert, D. Kinase Regulation of Human MHC Class I Molecule Expression on Cancer CellsKinase Regulation of MHC-I in Tumors. Cancer Immunol. Res. 2016, 4, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 1–48. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J. Acquired resistance to KRASG12C inhibition in cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M. Clinical Acquired Resistance to KRASG12C Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS–MAPK ReactivationClinical Acquired Resistance to KRASG12C Inhibition. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L. Rapid non-uniform adaptation to conformation-specific KRAS (G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef]
- Dunnett-Kane, V.; Nicola, P.; Blackhall, F.; Lindsay, C. Mechanisms of Resistance to KRASG12C Inhibitors. Cancers 2021, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Yonesaka, K.; Teramura, T.; Takehara, T.; Kato, R.; Sakai, H.; Haratani, K.; Tanizaki, J.; Kawakami, H.; Hayashi, H. KRAS Inhibitor Resistance in MET-Amplified KRAS G12C Non–Small Cell Lung Cancer Induced By RAS-and Non–RAS-Mediated Cell Signaling Mechanisms. Clin. Cancer Res. 2021, 27, 5697–5707. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Y.; Zhang, J.; Liu, P.; Jiao, B.; Wang, Z.; Ren, R. Focal Adhesion Kinase (FAK) Inhibition Synergizes with KRAS G12C Inhibitors in Treating Cancer through the Regulation of the FAK–YAP Signaling. Adv. Sci. 2021, 8, 2100250. [Google Scholar] [CrossRef] [PubMed]
- Akhave, N.S.; Biter, A.B.; Hong, D.S. Mechanisms of Resistance to KRASG12C-Targeted TherapyResistance to KRASG12C Inhibitors. Cancer Discov. 2021, 11, 1345–1352. [Google Scholar] [CrossRef]
- Chaft, J.E.; Litvak, A.; Arcila, M.E.; Patel, P.; D’Angelo, S.P.; Krug, L.M.; Rusch, V.; Mattson, A.; Coeshott, C.; Park, B. Phase II study of the GI-4000 KRAS vaccine after curative therapy in patients with stage I-III lung adenocarcinoma harboring a KRAS G12C, G12D, or G12V mutation. Clin. Lung Cancer 2014, 15, 405–410. [Google Scholar] [CrossRef]
- Liu, Y.; Xiang, F.; Huang, Y.; Shi, L.; Hu, C.; Yang, Y.; Wang, D.; He, N.; Tao, K.; Wu, K. Interleukin-22 promotes aerobic glycolysis associated with tumor progression via targeting hexokinase-2 in human colon cancer cells. Oncotarget 2017, 8, 25372. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Fang, X. Advances in glucose metabolism research in colorectal cancer. Biomed. Rep. 2016, 5, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Gillies, R.J.; Gatenby, R.A. Adaptive landscapes and emergent phenotypes: Why do cancers have high glycolysis? J. Bioenerg. Biomembr. 2007, 39, 251–257. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, S.Y.; Johnson, C.; Washburn, J.G.; Cruz-Correa, M.R.; Dang, D.T.; Dang, L.H. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1α and HIF-2α target genes. Mol. Cancer 2010, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Oncogene-directed alterations in cancer cell metabolism. Trends Cancer 2016, 2, 365–377. [Google Scholar] [CrossRef] [Green Version]
- Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 1955, 122, 501–504. [Google Scholar] [CrossRef]
- Kovačević, Z.; Morris, H. The role of glutamine in the oxidative metabolism of malignant cells. Cancer Res. 1972, 32, 326–333. [Google Scholar]
- Hutton, J.E.; Wang, X.; Zimmerman, L.J.; Slebos, R.J.; Trenary, I.A.; Young, J.D.; Li, M.; Liebler, D.C. Oncogenic KRAS and BRAF drive metabolic reprogramming in colorectal cancer. Mol. Cell. Proteom. 2016, 15, 2924–2938. [Google Scholar] [CrossRef] [Green Version]
- Varshavi, D.; Varshavi, D.; McCarthy, N.; Veselkov, K.; Keun, H.C.; Everett, J.R. Metabolic characterization of colorectal cancer cells harbouring different KRAS mutations in codon 12, 13, 61 and 146 using human SW48 isogenic cell lines. Metabolomics 2020, 16, 51. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsushita, J.; Lambeth, J.D.; Kamata, T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004, 64, 3580–3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, E.; McCoy III, J.W.; Macina, R.A.; Liu, W.; Cheng, G.; Robine, S.; Papkoff, J.; Lambeth, J.D. Nox1 is over-expressed in human colon cancers and correlates with activating mutations in K-Ras. Int. J. Cancer 2008, 123, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H. K-rasG12V transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Meng, N.; Glorieux, C.; Zhang, Y.; Liang, L.; Zeng, P.; Lu, W.; Huang, P. Oncogenic K-ras induces mitochondrial OPA3 expression to promote energy metabolism in pancreatic cancer cells. Cancers 2019, 12, 65. [Google Scholar] [CrossRef] [Green Version]
- Graham, N.A.; Tahmasian, M.; Kohli, B.; Komisopoulou, E.; Zhu, M.; Vivanco, I.; Teitell, M.A.; Wu, H.; Ribas, A.; Lo, R.S. Glucose deprivation activates a metabolic and signaling amplification loop leading to cell death. Mol. Syst. Biol. 2012, 8, 589. [Google Scholar] [CrossRef]
- Ullah, M.; H Bhat, S.; Hussain, E.; Abu-Duhier, F.; Ahmad, A.; M Hadi, S. Ascorbic acid in cancer chemoprevention: Translational perspectives and efficacy. Curr. Drug Targets 2012, 13, 1757–1771. [Google Scholar] [CrossRef]
- Lee, K.W.; Lee, H.J.; Surh, Y.-J.; Lee, C.Y. Vitamin C and cancer chemoprevention: Reappraisal. Am. J. Clin. Nutr. 2003, 78, 1074–1078. [Google Scholar] [CrossRef] [Green Version]
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 3685–3689. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, C.; Hutton, B.; Ng, T.; Shorr, R.; Clemons, M. Is there a role for oral or intravenous ascorbate (vitamin C) in treating patients with cancer? A systematic review. Oncologist 2015, 20, 210–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, N.; Creagan, E.; Witzig, T.; Levine, M. Ascorbic acid in cancer treatment: Let the phoenix fly. Cancer Cell 2018, 34, 700–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef]
- Hoffer, L.; Levine, M.; Assouline, S.; Melnychuk, D.; Padayatty, S.; Rosadiuk, K.; Rousseau, C.; Robitaille, L.; Miller Jr, W. Phase I clinical trial of iv ascorbic acid in advanced malignancy. Ann. Oncol. 2008, 19, 1969–1974. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Padayatty, S.J.; Espey, M.G. Vitamin C: A concentration-function approach yields pharmacology and therapeutic discoveries. Adv. Nutr. 2011, 2, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Hickey, S.; Roberts, H.J.; Miller, N.J. Pharmacokinetics of oral vitamin C. J. Nutr. Environ. Med. 2008, 17, 169–177. [Google Scholar] [CrossRef]
- Davis, J.L.; Paris, H.L.; Beals, J.W.; Binns, S.E.; Giordano, G.R.; Scalzo, R.L.; Schweder, M.M.; Blair, E.; Bell, C. Liposomal-encapsulated ascorbic acid: Influence on vitamin C bioavailability and capacity to protect against ischemia—Reperfusion injury. Nutr. Metab. Insights 2016, 9, NMI-S39764. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.S.; Wilkes, J.G.; Schroeder, S.R.; Buettner, G.R.; Wagner, B.A.; Du, J.; Gibson-Corley, K.; O’Leary, B.R.; Spitz, D.R.; Buatti, J.M. Pharmacologic ascorbate reduces radiation-induced normal tissue toxicity and enhances tumor radiosensitization in pancreatic cancer. Cancer Res. 2018, 78, 6838–6851. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Jeong, J.-H.; Lee, I.H.; Lee, J.; Jung, J.H.; Park, H.Y.; Lee, D.H.; Chae, Y.S. Effect of high-dose vitamin C combined with anti-cancer treatment on breast cancer cells. Anticancer. Res. 2019, 39, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Magrì, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chilà, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707. [Google Scholar] [CrossRef] [PubMed]
- Gerecke, C.; Schumacher, F.; Edlich, A.; Wetzel, A.; Yealland, G.; Neubert, L.K.; Scholtka, B.; Homann, T.; Kleuser, B. Vitamin C promotes decitabine or azacytidine induced DNA hydroxymethylation and subsequent reactivation of the epigenetically silenced tumour suppressor CDKN1A in colon cancer cells. Oncotarget 2018, 9, 32822. [Google Scholar] [CrossRef] [Green Version]
- Hamaguchi, R.; Narui, R.; Morikawa, H.; Wada, H. Improved Chemotherapy Outcomes of Patients With Small-cell Lung Cancer Treated With Combined Alkalization Therapy and Intravenous Vitamin C. Cancer Diagn. Progn. 2021, 1, 157. [Google Scholar] [CrossRef] [PubMed]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 583, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Raffaghello, L.; Brandhorst, S.; Safdie, F.M.; Bianchi, G.; Martin-Montalvo, A.; Pistoia, V.; Wei, M.; Hwang, S.; Merlino, A. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci. Transl. Med. 2012, 4, 124ra127. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, G.; Martella, R.; Ravera, S.; Marini, C.; Capitanio, S.; Orengo, A.; Emionite, L.; Lavarello, C.; Amaro, A.; Petretto, A. Fasting induces anti-Warburg effect that increases respiration but reduces ATP-synthesis to promote apoptosis in colon cancer models. Oncotarget 2015, 6, 11806. [Google Scholar] [CrossRef] [Green Version]
- Di Tano, M.; Raucci, F.; Vernieri, C.; Caffa, I.; Buono, R.; Fanti, M.; Brandhorst, S.; Curigliano, G.; Nencioni, A.; de Braud, F. Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Kaźmierczak-Barańska, J.; Boguszewska, K.; Adamus-Grabicka, A.; Karwowski, B.T. Two faces of vitamin C—Antioxidative and pro-oxidative agent. Nutrients 2020, 12, 1501. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.-H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M. Vitamin C uncouples the Warburg metabolic switch in KRAS-mutant colon cancer. Oncotarget 2016, 7, 47954. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.R.; O’Leary, B.R.; Du, J.; Sarsour, E.H.; Kalen, A.L.; Wagner, B.A.; Stolwijk, J.M.; Falls-Hubert, K.C.; Alexander, M.S.; Carroll, R.S. Dual Oxidase-Induced Sustained Generation of Hydrogen Peroxide Contributes to Pharmacologic Ascorbate-Induced CytotoxicityPharmacologic Ascorbate Induces Dual Oxidases. Cancer Res. 2020, 80, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Hejleh, T.A. O2.- and H2O2-mediated disruption of Fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell 2017, 31, 487–500.e8. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Cenigaonandia-Campillo, A.; Serna-Blasco, R.; Gómez-Ocabo, L.; Solanes-Casado, S.; Baños-Herraiz, N.; Del Puerto-Nevado, L.; Cañas, J.A.; Aceñero, M.J.; García-Foncillas, J.; Aguilera, Ó. Vitamin C activates pyruvate dehydrogenase (PDH) targeting the mitochondrial tricarboxylic acid (TCA) cycle in hypoxic KRAS-mutant colon cancer. Theranostics 2021, 11, 3595. [Google Scholar] [CrossRef]
- Wilkes, J.G.; O’Leary, B.R.; Du, J.; Klinger, A.R.; Sibenaller, Z.A.; Doskey, C.M.; Gibson-Corley, K.N.; Alexander, M.S.; Tsai, S.; Buettner, G.R. Pharmacologic ascorbate (P-AscH-) suppresses hypoxia-inducible Factor-1α (HIF-1α) in pancreatic adenocarcinoma. Clin. Exp. Metastasis 2018, 35, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhang, Y.; Yun, Z.; He, B.; Zhang, Q.; Hu, L.; Jiang, G. Speciation and bioaccessibility of arsenic in traditional Chinese medicines and assessment of its potential health risk. Sci. Total Environ. 2018, 619, 1088–1097. [Google Scholar] [CrossRef]
- Kwong, Y.; Todd, D. Delicious poison: Arsenic trioxide for the treatment of leukemia. Blood J. Am. Soc. Hematol. 1997, 89, 3487. [Google Scholar] [CrossRef]
- Aronson, S.M. Arsenic and old myths. RI Med. 1994, 77, 233–234. [Google Scholar]
- Forkner, C.E.; Scott, T.M. Arsenic as a therapeutic agent in chronic myelogenous leukemia: Preliminary report. J. Am. Med. Assoc. 1931, 97, 3–5. [Google Scholar] [CrossRef]
- Niu, C.; Yan, H.; Yu, T.; Sun, H.-P.; Liu, J.-X.; Li, X.-S.; Wu, W.; Zhang, F.-Q.; Chen, Y.; Zhou, L. Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: Remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood J. Am. Soc. Hematol. 1999, 94, 3315–3324. [Google Scholar] [CrossRef]
- Soignet, S.; Frankel, S.; Tallman, M.; Douer, D.; Kantarjian, H.; Stone, R.; Sievers, E.; Kalayeio, M.; Coutre, S.; Ellison, R. US multicenter trial of arsenic trioxide (AT) in acute promyelocytic leukemia (APL). Blood 1999, 94, 698a. [Google Scholar]
- Soignet, S.L.; Maslak, P.; Wang, Z.-G.; Jhanwar, S.; Calleja, E.; Dardashti, L.J.; Corso, D.; DeBlasio, A.; Gabrilove, J.; Scheinberg, D.A. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N. Engl. J. Med. 1998, 339, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Podolsky, L.; Oh, M.; Subbarayan, P.R.; Francheschi, D.; Livingstone, A.; Ardalan, B. 5-Fluorouracil/Leucovorin and arsenic trioxide for patients with refractory/relapsed colorectal carcinoma: A clinical experience. Acta Oncol. 2011, 50, 602–605. [Google Scholar] [CrossRef]
- Ardalan, B.; Subbarayan, P.R.; Ramos, Y.; Gonzalez, M.; Fernandez, A.; Mezentsev, D.; Reis, I.; Duncan, R.; Podolsky, L.; Lee, K. A Phase I Study of 5-Fluorouracil/Leucovorin and Arsenic Trioxide for Patients with Refractory/Relapsed Colorectal CarcinomaArsenic Trioxide and 5-FU/Leucovorin for CRC. Clin. Cancer Res. 2010, 16, 3019–3027. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free. Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Bak, D.W.; Pizzagalli, M.D.; Weerapana, E. Identifying functional cysteine residues in the mitochondria. Acs Chem. Biol. 2017, 12, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, T.; Kobayashi, Y.; Cui, X.; Hirano, S. A new metabolic pathway of arsenite: Arsenic–glutathione complexes are substrates for human arsenic methyltransferase Cyt19. Arch. Toxicol. 2005, 79, 183–191. [Google Scholar] [CrossRef]
- Scott, N.; Hatlelid, K.M.; MacKenzie, N.E.; Carter, D.E. Reactions of arsenic (III) and arsenic (V) species with glutathione. Chem. Res. Toxicol. 1993, 6, 102–106. [Google Scholar] [CrossRef]
- Kulshrestha, A.; Jarouliya, U.; Prasad, G.; Flora, S.; Bisen, P.S. Arsenic-induced abnormalities in glucose metabolism: Biochemical basis and potential therapeutic and nutritional interventions. World J. Transl. Med. 2014, 3, 96–111. [Google Scholar] [CrossRef]
- You, B.R.; Park, W.H. Arsenic trioxide induces human pulmonary fibroblast cell death via increasing ROS levels and GSH depletion. Oncol. Rep. 2012, 28, 749–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Y.; Dai, J.; Chalmers-Redman, R.M.; Tatton, W.G.; Waxman, S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood J. Am. Soc. Hematol. 1999, 94, 2102–2111. [Google Scholar]
- Akao, Y.; Nakagawa, Y.; Akiyama, K. Arsenic trioxide induces apoptosis in neuroblastoma cell lines through the activation of caspase 3 in vitro. FEBS Lett. 1999, 455, 59–62. [Google Scholar] [CrossRef]
- Zhang, H.-N.; Yang, L.; Ling, J.-Y.; Czajkowsky, D.M.; Wang, J.-F.; Zhang, X.-W.; Zhou, Y.-M.; Ge, F.; Yang, M.-K.; Xiong, Q. Systematic identification of arsenic-binding proteins reveals that hexokinase-2 is inhibited by arsenic. Proc. Natl. Acad. Sci. USA 2015, 112, 15084–15089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, N.; De Melo, J.; Tang, D. PKM2, a central point of regulation in cancer metabolism. Int. J. Cell Biol. 2013, 2013, 242513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Lu, H.; Li, W.; Hu, R.; Chen, Z. Identification of arsenic direct-binding proteins in acute promyelocytic leukaemia cells. Int. J. Mol. Sci. 2015, 16, 26871–26879. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, F.; Shim, J.-Y.; Kirk, K.L.; Anderson, D.E.; Chen, X. Identification of arsenic-binding proteins in human breast cancer cells. Cancer Lett. 2007, 255, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ding, X.; Adrian, T.E. Arsenic trioxide causes redistribution of cell cycle, caspase activation, and GADD expression in human colonic, breast, and pancreatic cancer cells. Cancer Investig. 2004, 22, 389–400. [Google Scholar] [CrossRef]
- Liu, Q.; Hilsenbeck, S.; Gazitt, Y. Arsenic Trioxide—Induced apoptosis in myeloma cells: P53-dependent G1 or G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL: Presented in preliminary form at the 43rd annual meeting of the American Society of Hematology, Orlando, FL, December 8, 2001.42. Blood J. Am. Soc. Hematol. 2003, 101, 4078–4087. [Google Scholar]
- Huang, X.-J.; Wiernik, P.H.; Klein, R.S.; Gallagher, R.E. Arsenic trioxide induces apoptosis of myeloid leukemia cells by activation of caspases. Med. Oncol. 1999, 16, 58–64. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Heo, J.S.; Kim, P.; Lian, Z.; Lee, S.; Park, J.; Hong, E.; Pang, K.; Park, Y.; Ooshima, A. Tetraarsenic hexoxide enhances generation of mitochondrial ROS to promote pyroptosis by inducing the activation of caspase-3/GSDME in triple-negative breast cancer cells. Cell Death Dis. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Meakin, C.J.; Martin, E.M.; Fry, R.C. Epigenetic mechanisms underlying arsenic-induced toxicity. Curr. Opin. Toxicol. 2017, 6, 1–9. [Google Scholar] [CrossRef]
- Fu, H.-Y.; Shen, J.-Z.; Wu, Y.; Shen, S.-F.; Zhou, H.-R.; Fan, L.-P. Arsenic trioxide inhibits DNA methyltransferase and restores expression of methylation-silenced CDKN2B/CDKN2A genes in human hematologic malignant cells. Oncol. Rep. 2010, 24, 335–343. [Google Scholar]
- Eyvani, H.; Moghaddaskho, F.; Kabuli, M.; Zekri, A.; Momeny, M.; Tavakkoly-Bazzaz, J.; Alimoghaddam, K.; Ghavamzadeh, A.; Ghaffari, S.H. Arsenic trioxide induces cell cycle arrest and alters DNA methylation patterns of cell cycle regulatory genes in colorectal cancer cells. Life Sci. 2016, 167, 67–77. [Google Scholar] [CrossRef]
- Maimaitiyiming, Y.; Wang, Q.Q.; Hsu, C.-H.; Naranmandura, H. Arsenic induced epigenetic changes and relevance to treatment of acute promyelocytic leukemia and beyond. Toxicol. Appl. Pharmacol. 2020, 406, 115212. [Google Scholar] [CrossRef]
- Gu, J.; Zhu, X.; Li, Y.; Dong, D.; Yao, J.; Lin, C.; Huang, K.; Hu, H.; Fei, J. miRNA-21 regulates arsenic-induced anti-leukemia activity in myelogenous cell lines. Med. Oncol. 2011, 28, 211–218. [Google Scholar] [CrossRef]
- Liu, L.; Chen, R.; Huang, S.; Wu, Y.; Li, G.; Zhang, B.; Liu, Q.; Yin, D.; Liang, Y. miR-153 sensitized the K562 cells to As2O3-induced apoptosis. Med. Oncol. 2012, 29, 243–247. [Google Scholar] [CrossRef]
- Ghaffari, S.H.; Momeny, M.; Bashash, D.; Mirzaei, R.; Ghavamzadeh, A.; Alimoghaddam, K. Cytotoxic effect of arsenic trioxide on acute promyelocytic leukemia cells through suppression of NFkβ-dependent induction of hTERT due to down-regulation of Pin1 transcription. Hematology 2012, 17, 198–206. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, B.; Sun, Y.F.; Huang, X.W.; Cheng, J.W.; Huang, A.; Zeng, H.Y.; Qiu, S.J.; Cao, Y.; Fan, J. Arsenic trioxide induces differentiation of cancer stem cells in hepatocellular carcinoma through inhibition of LIF/JAK1/STAT3 and NF-kB signaling pathways synergistically. Clin. Transl. Med. 2021, 11, e335. [Google Scholar] [CrossRef]
- Wu, X.; Park, M.; Sarbassova, D.A.; Ying, H.; Lee, M.G.; Bhattacharya, R.; Ellis, L.; Peterson, C.B.; Hung, M.C.; Lin, H.K. A chirality-dependent action of vitamin C in suppressing Kirsten rat sarcoma mutant tumor growth by the oxidative combination: Rationale for cancer therapeutics. Int. J. Cancer 2020, 146, 2822–2828. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Wang, Z.; Tang, N.-n.; Li, J.-t.; Liu, Y.; Chu, W.-F.; Yang, B.-F. Ascorbic acid sensitizes colorectal carcinoma to the cytotoxicity of arsenic trioxide via promoting reactive oxygen species-dependent apoptosis and pyroptosis. Front. Pharmacol. 2020, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Ohnsorge, P. Influence of mitochondrion-toxic agents on the cardiovascular system. Regul. Toxicol. Pharmacol. 2013, 67, 434–445. [Google Scholar] [CrossRef]
- Pace, C.; Dagda, R.; Angermann, J. Antioxidants protect against arsenic induced mitochondrial cardio-toxicity. Toxics 2017, 5, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yih, L.-H.; Huang, H.; Jan, K.; Lee, T.-C. Sodium arsenite induces ATP depletion and mitochondrial damage in HeLa cells. Cell Biol. Int. Rep. 1991, 15, 253–264. [Google Scholar] [CrossRef]
- Read, A.D.; Bentley, R.E.; Archer, S.L.; Dunham-Snary, K.J. Mitochondrial iron–sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox Biol. 2021, 47, 102164. [Google Scholar] [CrossRef]
- Leung, P.Y.; Miyashita, K.; Young, M.; Tsao, C. Cytotoxic effect of ascorbate and its derivatives on cultured malignant and nonmalignant cell lines. Anticancer. Res. 1993, 13, 475–480. [Google Scholar] [PubMed]
- Sauberlich, H.E.; Tamura, T.; Craig, C.B.; Freeberg, L.E.; Liu, T. Effects of erythorbic acid on vitamin C metabolism in young women. Am. J. Clin. Nutr. 1996, 64, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Baker, R.A.; Bruemmer, J. Oxidation of ascorbic acid by enzyme preparations from orange. Fla. State Hortic. Soc. Proc. 1968, 269–275. [Google Scholar]
- Bahlis, N.J.; McCafferty-Grad, J.; Jordan-McMurry, I.; Neil, J.; Reis, I.; Kharfan-Dabaja, M.; Eckman, J.; Goodman, M.; Fernandez, H.F.; Boise, L.H. Feasibility and correlates of arsenic trioxide combined with ascorbic acid-mediated depletion of intracellular glutathione for the treatment of relapsed/refractory multiple myeloma. Clin. Cancer Res. 2002, 8, 3658–3668. [Google Scholar]
- Campbell, R.A.; Chen, H.; Zhu, D.; Santos, J.C.; Bonavida, B.; Pang, S.; Said, J.; Berenson, J.R. Ascorbic Acid Overcomes Drug Resistance in Myeloma and Significantly Increases the Anti-Myeloma Effects of both Arsenic Trioxide and Melphalan in vitro and in vivo. Blood 2004, 104, 2470. [Google Scholar] [CrossRef]
- Subbarayan, P.R.; Lima, M.; Ardalan, B. Arsenic trioxide/ascorbic acid therapy in patients with refractory metastatic colorectal carcinoma: A clinical experience. Acta Oncol. 2007, 46, 557–561. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Marker | Reference |
|---|---|---|
| AMG510/Sotorasib (Amgen) | KRAS G12C | [37] |
| MRTX849/Adagrasib (Mirati) | KRAS G12C | [38] |
| D-1553 (Iventis Bio) | KRAS G12C | [39,40] |
| JDQ443 (Novartis) | KRAS G12C | [41,42] |
| MRTX1133 (Mirati) | KRAS G12D | [43] |
| JNJ-74699157 (Johnson & Johnson) | KRAS G12C | [44] |
| LY3499446 (Eli Lilly & Company) | KRAS G12C | [45] NCT04165031 discontinued |
| iExosomes | KRAS G12D | [46] |
| Anti-KRAS G12D mTCR PBL | KRAS G12D | [47] |
| Anti-KRAS G12V mTCR PBL | KRAS G12V | [47] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burska, A.N.; Ilyassova, B.; Dildabek, A.; Khamijan, M.; Begimbetova, D.; Molnár, F.; Sarbassov, D.D. Enhancing an Oxidative “Trojan Horse” Action of Vitamin C with Arsenic Trioxide for Effective Suppression of KRAS-Mutant Cancers: A Promising Path at the Bedside. Cells 2022, 11, 3454. https://doi.org/10.3390/cells11213454
Burska AN, Ilyassova B, Dildabek A, Khamijan M, Begimbetova D, Molnár F, Sarbassov DD. Enhancing an Oxidative “Trojan Horse” Action of Vitamin C with Arsenic Trioxide for Effective Suppression of KRAS-Mutant Cancers: A Promising Path at the Bedside. Cells. 2022; 11(21):3454. https://doi.org/10.3390/cells11213454
Chicago/Turabian StyleBurska, Agata N., Bayansulu Ilyassova, Aruzhan Dildabek, Medina Khamijan, Dinara Begimbetova, Ferdinand Molnár, and Dos D. Sarbassov. 2022. "Enhancing an Oxidative “Trojan Horse” Action of Vitamin C with Arsenic Trioxide for Effective Suppression of KRAS-Mutant Cancers: A Promising Path at the Bedside" Cells 11, no. 21: 3454. https://doi.org/10.3390/cells11213454