
The Hidden Role of Non-Canonical Amyloid β Isoforms in Alzheimer’s Disease


Abstract
:1. Introduction
2. The Role of Glia in Neuroinflammation
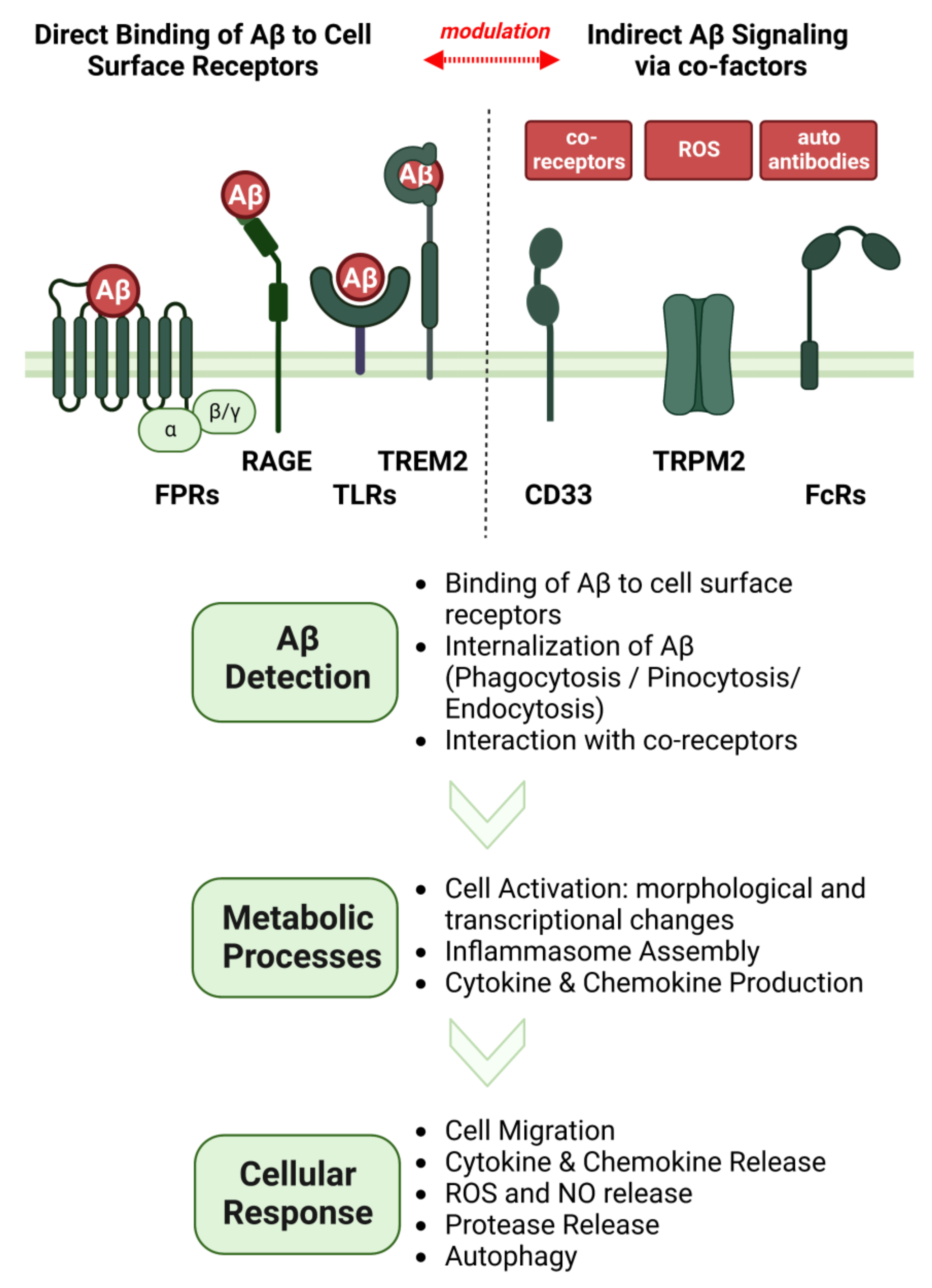
3. Processes That Lead to the Generation of Non-Canonical Aβ Variants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aβ Fragments | Modification | Source | References |
|---|---|---|---|
| C-ABRIDGED | |||
| 1–13 to 1–20 | - | CSF | [74,75] |
| 1–15 to 1–20 glyco-Y10 | glyco-Y10 | CSF | [76] |
| 1–16 to 1–17 | - | Brain, CSF | [74,75,77] |
| 1–20 | - | Brain | [70] |
| 1–28 | - | CSF | [74] |
| 1–30 | - | CSF | [74,75] |
| 1–31 | - | Brain | [70] |
| 1–33 to 1–34 | - | CSF | [74,75] |
| 1–37 to 1–40 | - | Brain, CSF | [74,75,78] |
| 1–37 ox-M35 to 1–40ox-M35 | ox-M35 | CSF | [74] |
| N-ABRIDGED | |||
| 2–42 to 11–42 | - | Brain | [78,79] |
| 2–40 | - | Brain | [79,80] |
| 3–40/42pyro-E3 | pyro-E3 | Brain | [78,80] |
| 4–42 ox-M35 to 5–42 ox-M35 | ox-M35 | Brain | [70] |
| 4–40 | - | Brain, CSF | [74,78] |
| 4–43 | - | Brain | [78] |
| 5–40 | - | Brain | [79,80] |
| 8–42 ox-M35 | ox-M35 | Brain | [70] |
| 9–40 | - | Brain | [78] |
| 11–42 | - | Brain | [80] |
| 11–42 ox-M35 | ox-M35 | Brain | [70] |
| 11–42 pyro-E11 | pyro-E11 | Brain | [78,79] |
| 11–42 pyro-E11, ox-M35 | pyro-E11, ox-M35 | Brain | [70] |
| 17–42 | - | Brain | [81] |
| C- & N-TRUNCATED | |||
| 2–14 | - | CSF | [75] |
| 2–16 | - | Brain | [77] |
| 3–15 to 3–17 | - | Brain | [74,77] |
| 3–15 to 4–15 glyco-Y10 | glyco-Y10 | CSF | [76] |
| 3–19 pyro-E3 to 3–20 pyro-E3 | pyro-E3 | Brain | [80] |
| 3–24 pyro-E3 | pyro-E3 | Brain | [80] |
| 4–16 to 5–16 | - | Brain | [77] |
| 4–17 to 5–17 glyco-Y10 | glyco-Y10 | CSF | [76] |
| 4–18 to 4–20 | - | Brain | [80] |
| 4–23 to 4 -25 | - | Brain | [80] |
| 4–34 | - | Brain | [80] |
| 4–37 | - | Brain | [80] |
| 4–37 ox-M35 to 4–40 ox-M35 | ox-M35 | Brain | [80] |
| 5–20 | - | Brain | [80] |
| 11–23 pyro-E11 to 11–25 pyro-E11 | pyro-E11 | Brain | [80] |
| 11–27 pyro-E11 | pyro-E11 | Brain | [80] |
| 11–30 | - | CSF | [74] |
| 11–34 | - | Brain | [70] |
| 25–35/40 race-D-S26 | race-D-S26 | Brain | [82] |
| CANONICAL FORMS | |||
| 1–38 to 1–40 | - | Brain, CSF | [74,78,79] |
| 1–42 | - | Brain, CSF | [74,78,79] |
| 1–43 | - | Brain | [83] |
| 1–40/42 ox-M35 | ox-M35 | Brain | [70] |
| 1–40/42race-D-S26 | race-D-S26 | Brain, CSF | [76,82] |
| 1–40/42 race-D-D7 | race-D-D7 | Brain | [84,85] |

3.1. N-Abridged Aβ Species
3.2. C-Terminal Variants
3.3. Post-Translational Aβ Modifications (PTMs)
3.4. Splice Variants of APP
3.5. APP Processing Is Different in Neurons, Astrocytes, Microglia, and Oligodendrocytes
4. Cellular Responses of Glial Cells towards Aβ
4.1. Contribution of Aβ Internalization and Degradation
4.2. Contribution of NLRP3 Signaling
4.3. Contribution of Oxidative Stress
4.4. Interaction of Aβ with Other Neuropathological Proteins
5. Genetic Variants Affecting Microglial Response to Aβ
6. Secondary Structure and Oligomerization Critically Affect Aβ Neurotoxicity
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akiyama, H.; Arai, T.; Kondo, H.; Tanno, E.; Haga, C.; Ikeda, K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis. Assoc. Disord. 2000, 14 (Suppl. 1), S47–S53. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Luber-Narod, J.; Styren, S.D.; Civin, W.H. Expression of immune system-associated antigens by cells of the human central nervous system: Relationship to the pathology of Alzheimer’s disease. Neurobiol. Aging 1988, 9, 339–349. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Occurrence of HLA-DR reactive microglia in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1988, 540, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, S.; McGeer, P.L.; Akiyama, H.; Zhu, S.; Selkoe, D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 1989, 24, 173–182. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Masters, C.L.; Multhaup, G.; Simms, G.; Pottgiesser, J.; Martins, R.N.; Beyreuther, K. Neuronal origin of a cerebral amyloid: Neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985, 4, 2757–2763. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D. If amyloid drives Alzheimer disease, why have anti-amyloid therapies not yet slowed cognitive decline? PLoS Biol. 2022, 20, e3001694. [Google Scholar] [CrossRef]
- Steiner, H.; Fukumori, A.; Tagami, S.; Okochi, M. Making the final cut: Pathogenic amyloid-beta peptide generation by gamma-secretase. Cell. Stress 2018, 2, 292–310. [Google Scholar] [CrossRef]
- Andreasson, U.; Portelius, E.; Andersson, M.E.; Blennow, K.; Zetterberg, H. Aspects of beta-amyloid as a biomarker for Alzheimer’s disease. Biomark. Med. 2007, 1, 59–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta. Pharm. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimers Res. 2014, 6, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doens, D.; Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflammation 2014, 11, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, L.; Vieten, S.; Brödel, S.; Endres, K.; Bufe, B. Emerging contributions of formyl peptide receptors to neurodegenerative diseases. Biol. Chem. 2022, 403, 27–41. [Google Scholar] [CrossRef]
- Gospodarska, E.; Kupniewska-Kozak, A.; Goch, G.; Dadlez, M. Binding studies of truncated variants of the Abeta peptide to the V-domain of the RAGE receptor reveal Abeta residues responsible for binding. Biochim. Biophys. Acta 2011, 1814, 592–609. [Google Scholar] [CrossRef]
- Wunderlich, P.; Glebov, K.; Kemmerling, N.; Tien, N.T.; Neumann, H.; Walter, J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma-secretase-dependent intramembranous cleavage. J. Biol. Chem. 2013, 288, 33027–33036. [Google Scholar] [CrossRef] [Green Version]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef]
- Okello, A.; Edison, P.; Archer, H.A.; Turkheimer, F.E.; Kennedy, J.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.; Rossor, M.; et al. Microglial activation and amyloid deposition in mild cognitive impairment: A PET study. Neurology 2009, 72, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Hamelin, L.; Lagarde, J.; Dorothee, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef]
- Femminella, G.D.; Dani, M.; Wood, M.; Fan, Z.; Calsolaro, V.; Atkinson, R.; Edginton, T.; Hinz, R.; Brooks, D.J.; Edison, P. Microglial activation in early Alzheimer trajectory is associated with higher gray matter volume. Neurology 2019, 92, e1331–e1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Tremblay, M.E.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The Role of Microglia in the Healthy Brain. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef] [Green Version]
- Brionne, T.C.; Tesseur, I.; Masliah, E.; Wyss-Coray, T. Loss of TGF-β1 Leads to Increased Neuronal Cell Death and Microgliosis in Mouse Brain. Neuron 2003, 40, 1133–1145. [Google Scholar] [CrossRef] [Green Version]
- Labandeira-Garcia, J.L.; Costa-Besada, M.A.; Labandeira, C.M.; Villar-Cheda, B.; Rodriguez-Perez, A.I. Insulin-Like Growth Factor-1 and Neuroinflammation. Front. Aging Neurosci. 2017, 9, 365. [Google Scholar] [CrossRef] [Green Version]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting Microglia Directly Monitor the Functional State of Synapses In Vivo and Determine the Fate of Ischemic Terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Harley, S.B.R.; Willis, E.F.; Shaikh, S.N.; Blackmore, D.G.; Sah, P.; Ruitenberg, M.J.; Bartlett, P.F.; Vukovic, J. Selective Ablation of BDNF from Microglia Reveals Novel Roles in Self-Renewal and Hippocampal Neurogenesis. J. Neurosci. 2021, 41, 4172–4186. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell. Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell. Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta. Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Heithoff, B.P.; George, K.K.; Phares, A.N.; Zuidhoek, I.A.; Munoz-Ballester, C.; Robel, S. Astrocytes are necessary for blood–brain barrier maintenance in the adult mouse brain. Glia 2021, 69, 436–472. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Jessen, N.A.; Munk, A.S.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends. Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, H.; Okamoto, T.; Hara, Y.; Komine, O.; Tamada, H.; Maeda, M.; Osako, F.; Kobayashi, M.; Nishiyama, A.; Kataoka, Y.; et al. Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. Embo. J. 2020, 39, e104464. [Google Scholar] [CrossRef]
- Gorina, R.; Font-Nieves, M.; Márquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011, 59, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Slowik, A.; Merres, J.; Elfgen, A.; Jansen, S.; Mohr, F.; Wruck, C.J.; Pufe, T.; Brandenburg, L.-O. Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE) - and amyloid beta 1-42-induced signal transduction in glial cells. Mol. Neurodegener. 2012, 7, 55. [Google Scholar] [CrossRef] [Green Version]
- Mitew, S.; Kirkcaldie, M.T.; Dickson, T.C.; Vickers, J.C. Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol. Aging 2013, 34, 2341–2351. [Google Scholar] [CrossRef]
- Yamamoto, N.; Ishikuro, R.; Tanida, M.; Suzuki, K.; Ikeda-Matsuo, Y.; Sobue, K. Insulin-signaling Pathway Regulates the Degradation of Amyloid β-protein via Astrocytes. Neuroscience 2018, 385, 227–236. [Google Scholar] [CrossRef]
- Sbai, O.; Ould-Yahoui, A.; Ferhat, L.; Gueye, Y.; Bernard, A.; Charrat, E.; Mehanna, A.; Risso, J.-J.; Chauvin, J.-P.; Fenouillet, E.; et al. Differential vesicular distribution and trafficking of MMP-2, MMP-9, and their inhibitors in astrocytes. Glia 2010, 58, 344–366. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Spampinato, S.F.; Merlo, S.; Fagone, E.; Fruciano, M.; Sano, Y.; Kanda, T.; Sortino, M.A. Reciprocal Interplay Between Astrocytes and CD4+ Cells Affects Blood-Brain Barrier and Neuronal Function in Response to β Amyloid. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, F.; Han, Z.; Yin, Z.; Ge, X.; Lei, P. Relationship Between Amyloid-β Deposition and Blood–Brain Barrier Dysfunction in Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, S.F.; Merlo, S.; Sano, Y.; Kanda, T.; Sortino, M.A. Astrocytes contribute to Aβ-induced blood–brain barrier damage through activation of endothelial MMP9. J. Neurochem. 2017, 142, 464–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Nave, K.-A.; Werner, H.B. Ensheathment and Myelination of Axons: Evolution of Glial Functions. Annu. Rev. Neurosci. 2021, 44, 197–219. [Google Scholar] [CrossRef]
- Church, J.S.; Kigerl, K.A.; Lerch, J.K.; Popovich, P.G.; McTigue, D.M. TLR4 Deficiency Impairs Oligodendrocyte Formation in the Injured Spinal Cord. J. Neurosci. 2016, 36, 6352–6364. [Google Scholar] [CrossRef] [Green Version]
- Grasselli, C.; Ferrari, D.; Zalfa, C.; Soncini, M.; Mazzoccoli, G.; Facchini, F.A.; Marongiu, L.; Granucci, F.; Copetti, M.; Vescovi, A.L.; et al. Toll-like receptor 4 modulation influences human neural stem cell proliferation and differentiation. Cell. Death Dis.. 2018, 9, 280. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Goswami, R.; Dawson, S.; Dawson, G. Expression of the receptor for advanced glycation end products in oligodendrocytes in response to oxidative stress. J. Neurosci. Res. 2008, 86, 2414–2422. [Google Scholar] [CrossRef] [Green Version]
- Cain, A.; Taga, M.; McCabe, C.; Green, G.; Hekselman, I.; White, C.C.; Lee, D.I.; Gaur, P.; Rozenblatt-Rosen, O.; Zhang, F.; et al. Multi-cellular communities are perturbed in the aging human brain and Alzheimer’s disease. bioRxiv 2022. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Leng, K.; Li, E.; Eser, R.; Piergies, A.; Sit, R.; Tan, M.; Neff, N.; Li, S.H.; Rodriguez, R.D.; Suemoto, C.K.; et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.-F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800–25809. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Shen, K.; Lee, S.H.; Shen, Y.A.; Wang, Y.; Otero-García, M.; Kotova, N.; Vito, S.T.; Laufer, B.I.; Newton, D.F.; et al. Disease-associated oligodendrocyte responses across neurodegenerative diseases. Cell. Rep. 2022, 40, 111189. [Google Scholar] [CrossRef]
- Caso, F.; Agosta, F.; Mattavelli, D.; Migliaccio, R.; Canu, E.; Magnani, G.; Marcone, A.; Copetti, M.; Falautano, M.; Comi, G.; et al. White Matter Degeneration in Atypical Alzheimer Disease. Radiology 2015, 277, 162–172. [Google Scholar] [CrossRef]
- Lee, S.; Viqar, F.; Zimmerman, M.E.; Narkhede, A.; Tosto, G.; Benzinger, T.L.; Marcus, D.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; et al. White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network. Ann. Neurol. 2016, 79, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Bartzokis, G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging 2011, 32, 1341–1371. [Google Scholar] [CrossRef] [Green Version]
- Depp, C.; Sun, T.; Sasmita, A.O.; Spieth, L.; Berghoff, S.A.; Steixner-Kumar, A.A.; Subramanian, S.; Möbius, W.; Göbbels, S.; Saher, G.; et al. Ageing-associated myelin dysfunction drives amyloid deposition in mouse models of Alzheimer’s disease. bioRxiv. 2021. [Google Scholar] [CrossRef]
- Wildburger, N.C.; Esparza, T.J.; Leduc, R.D.; Fellers, R.T.; Thomas, P.M.; Cairns, N.J.; Kelleher, N.L.; Bateman, R.J.; Brody, D.L. Diversity of Amyloid-beta Proteoforms in the Alzheimer’s Disease Brain. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Abraham, J.D.; Prome, S.; Salvetat, N.; Rubrecht, L.; Cobo, S.; du Paty, E.; Galea, P.; Mathieu-Dupas, E.; Ranaldi, S.; Caillava, C.; et al. Cerebrospinal Abeta11-x and 17-x levels as indicators of mild cognitive impairment and patients’ stratification in Alzheimer’s disease. Transl. Psychiatry 2013, 3, e281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, M.L.; Phillips, A.S.; Gaimster, K.; Paul, C.; Mudher, A.; Nicoll, J.A.R.; Boche, D. Pyroglutamate and Isoaspartate modified Amyloid-Beta in ageing and Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weggen, S.; Beher, D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimers Res. 2012, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmalm, G.; Portelius, E.; Öhrfelt, A.; Mattsson, N.; Persson, R.; Gustavsson, M.K.; Vite, C.H.; Gobom, J.; Månsson, J.E.; Nilsson, J.; et al. An online nano-LC-ESI-FTICR-MS method for comprehensive characterization of endogenous fragments from amyloid β and amyloid precursor protein in human and cat cerebrospinal fluid. J. Mass. Spectrom. JMS 2012, 47, 591–603. [Google Scholar] [CrossRef]
- Maddalena, A.S.; Papassotiropoulos, A.; Gonzalez-Agosti, C.; Signorell, A.; Hegi, T.; Pasch, T.; Nitsch, R.M.; Hock, C. Cerebrospinal fluid profile of amyloid β peptides in patients with Alzheimer’s disease determined by protein biochip technology. Neurodegener. Dis. 2004, 1, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Halim, A.; Brinkmalm, G.; Rüetschi, U.; Westman-Brinkmalm, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Larson, G.; Nilsson, J. Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid β-peptides in human cerebrospinal fluid. Proc. Natl. Acad. Sci. USA 2011, 108, 11848–11853. [Google Scholar] [CrossRef] [Green Version]
- Güntert, A.; Döbeli, H.; Bohrmann, B. High sensitivity analysis of amyloid-beta peptide composition in amyloid deposits from human and PS2APP mouse brain. Neuroscience 2006, 143, 461–475. [Google Scholar] [CrossRef]
- Portelius, E.; Bogdanovic, N.; Gustavsson, M.K.; Volkmann, I.; Brinkmalm, G.; Zetterberg, H.; Winblad, B.; Blennow, K. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010, 120, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Näslund, J.; Schierhorn, A.; Hellman, U.; Lannfelt, L.; Roses, A.D.; Tjernberg, L.O.; Silberring, J.; Gandy, S.E.; Winblad, B.; Greengard, P.; et al. Relative abundance of Alzheimer Aβ amyloid peptide variants in Alzheimer disease and normal aging. Proc. Natl. Acad. Sci. USA 1994, 91, 8378–8382. [Google Scholar] [CrossRef] [Green Version]
- Gkanatsiou, E.; Portelius, E.; Toomey, C.E.; Blennow, K.; Zetterberg, H.; Lashley, T.; Brinkmalm, G. A distinct brain beta amyloid signature in cerebral amyloid angiopathy compared to Alzheimer’s disease. Neurosci. Lett. 2019, 701, 125–131. [Google Scholar] [CrossRef]
- Gowing, E.; Roher, A.E.; Woods, A.S.; Cotter, R.J.; Chaney, M.; Little, S.P.; Ball, M.J. Chemical characterization of Aβ 17-42 peptide, a component of diffuse amyloid deposits of Alzheimer disease. J. Biol. Chem. 1994, 269, 10987–10990. [Google Scholar] [CrossRef]
- Kubo, T.; Kumagae, Y.; Miller, C.A.; Kaneko, I. β-amyloid racemized at the Ser26 residue in the brains of patients with Alzheimer disease: Implications in the pathogenesis of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2003, 62, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portelius, E.; Lashley, T.; Westerlund, A.; Persson, R.; Fox, N.C.; Blennow, K.; Revesz, T.; Zetterberg, H. Brain amyloid-beta fragment signatures in pathological ageing and alzheimer’s disease by hybrid immunoprecipitation mass spectrometry. Neurodegener. Dis. 2015, 15, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Hosaka, D.; Mochizuki, N.; Akatsu, H.; Tsutsumiuchi, K.; Hashizume, Y.; Matsukawa, N.; Yamamoto, T.; Toyo’Oka, T. Simultaneous determination of post-translational racemization and isomerization of N -terminal amyloid-β in alzheimer’s brain tissues by covalent chiral derivatized ultraperformance liquid chromatography tandem mass spectrometry. Anal. Chem. 2014, 86, 797–804. [Google Scholar] [CrossRef]
- Pekov, S.I.; Ivanov, D.G.; Bugrova, A.E.; Indeykina, M.I.; Zakharova, V.N.; Popov, I.A.; Kononikhin, A.S.; Kozin, S.A.; Makarov, A.A.; Nikolaev, E.N. Evaluation of MALDI-TOF/TOF Mass Spectrometry Approach for Quantitative Determination of Aspartate Residue Isomerization in the Amyloid-β Peptide. J. Am. Soc. Mass. Spectrom. 2019, 30, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Alzforum. Mutations Database. Available online: https://www.alzforum.org/mutations/search (accessed on 27 October 2022).
- Maler, J.M.; Spitzer, P.; Klafki, H.W.; Esselmann, H.; Lewczuk, P.; Kornhuber, J.; Herrmann, M.; Wiltfang, J. Distinct fractional Abeta release patterns in human mononuclear phagocytes. J. Neuroimmunol. 2009, 206, 1–4. [Google Scholar] [CrossRef]
- Giau, V.V.; Bagyinszky, E.; Yang, Y.S.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 2019, 9, 8368. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [Green Version]
- Armbrust, F.; Bickenbach, K.; Marengo, L.; Pietrzik, C.; Becker-Pauly, C. The Swedish dilemma—The almost exclusive use of APPswe-based mouse models impedes adequate evaluation of alternative β-secretases. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119164. [Google Scholar] [CrossRef] [PubMed]
- Kakuda, N.; Funamoto, S.; Yagishita, S.; Takami, M.; Osawa, S.; Dohmae, N.; Ihara, Y. Equimolar production of amyloid β-protein and amyloid precursor protein intracellular domain from β-carboxyl-terminal fragment by γ-secretase. J. Biol. Chem. 2006, 281, 14776–14786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, A.J.; Raskatov, J. Is the p3 Peptide (Abeta17-40, Abeta17-42) Relevant to the Pathology of Alzheimer’s Disease? J. Alzheimers. Dis. 2020, 74, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Bouter, Y.; Dietrich, K.; Wittnam, J.L.; Rezaei-Ghaleh, N.; Pillot, T.; Papot-Couturier, S.; Lefebvre, T.; Sprenger, F.; Wirths, O.; Zweckstetter, M.; et al. N-truncated amyloid beta (Abeta) 4-42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 2013, 126, 189–205. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, A.C.; Xue, R.; Gambetti, P. Amyloid precursor protein metabolism in primary cell cultures of neurons, astrocytes, and microglia. J. Neurochem. 1996, 66, 2300–2310. [Google Scholar] [CrossRef]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef]
- Oberstein, T.J.; Spitzer, P.; Klafki, H.W.; Linning, P.; Neff, F.; Knolker, H.J.; Lewczuk, P.; Wiltfang, J.; Kornhuber, J.; Maler, J.M. Astrocytes and microglia but not neurons preferentially generate N-terminally truncated Abeta peptides. Neurobiol. Dis. 2015, 73, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Bibl, M.; Gallus, M.; Welge, V.; Lehmann, S.; Sparbier, K.; Esselmann, H.; Wiltfang, J. Characterization of cerebrospinal fluid aminoterminally truncated and oxidized amyloid-β peptides. Proteom. Clin. Appl. 2012, 6, 163–169. [Google Scholar] [CrossRef]
- Portelius, E.; Price, E.; Brinkmalm, G.; Stiteler, M.; Olsson, M.; Persson, R.; Westman-Brinkmalm, A.; Zetterberg, H.; Simon, A.J.; Blennow, K. A novel pathway for amyloid precursor protein processing. Neurobiol. Aging 2011, 32, 1090–1098. [Google Scholar] [CrossRef]
- Marengo, L.; Armbrust, F.; Schoenherr, C.; Storck, S.E.; Schmitt, U.; Zampar, S.; Wirths, O.; Altmeppen, H.; Glatzel, M.; Kaether, C.; et al. Meprin beta knockout reduces brain Abeta levels and rescues learning and memory impairments in the APP/lon mouse model for Alzheimer’s disease. Cell. Mol. Life. Sci. 2022, 79, 168. [Google Scholar] [CrossRef]
- Schönherr, C.; Bien, J.; Isbert, S.; Wichert, R.; Prox, J.; Altmeppen, H.; Kumar, S.; Walter, J.; Lichtenthaler, S.F.; Weggen, S.; et al. Generation of aggregation prone N-terminally truncated amyloid β peptides by meprin β depends on the sequence specificity at the cleavage site. Mol. Neurodegener. 2016, 11, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savastano, A.; Klafki, H.; Haußmann, U.; Oberstein, T.J.; Muller, P.; Wirths, O.; Wiltfang, J.; Bayer, T.A. N-truncated Aβ2-X starting with position two in sporadic Alzheimer’s disease cases and two Alzheimer mouse models. J. Alzheimers Dis. 2016, 49, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Walter, S.; Kraus, I.; Klafki, H.W.; Stazi, M.; Oberstein, T.J.; Ghiso, J.; Wiltfang, J.; Bayer, T.A.; Weggen, S. N-truncated Aβ4–x peptides in sporadic Alzheimer’s disease cases and transgenic Alzheimer mouse models. Alzheimer’s Res. Ther. 2017, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Walter, S.; Jumpertz, T.; Hüttenrauch, M.; Ogorek, I.; Gerber, H.; Storck, S.E.; Zampar, S.; Dimitrov, M.; Lehmann, S.; Lepka, K.; et al. The metalloprotease ADAMTS4 generates N-truncated Aβ4-x species and marks oligodendrocytes as a source of amyloidogenic peptides in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Marr, R.A.; Guan, H.; Rockenstein, E.; Kindy, M.; Gage, F.H.; Verma, I.; Masliah, E.; Hersh, L.B. Neprilysin regulates amyloid Beta peptide levels. J. Mol. Neurosci. 2004, 22, 5–11. [Google Scholar] [CrossRef]
- Pike, C.J.; Overman, M.J.; Cotman, C.W. Amino-terminal deletions enhance aggregation of beta-amyloid peptides in vitro. J. Biol. Chem. 1995, 270, 23895–23898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostagno, A.; Cabrera, E.; Lashley, T.; Ghiso, J. N-terminally truncated Aβ4-x proteoforms and their relevance for Alzheimer’s pathophysiology. Transl. Neurodegener. 2022, 11, 30. [Google Scholar] [CrossRef]
- Huttenrauch, M.; Brauss, A.; Kurdakova, A.; Borgers, H.; Klinker, F.; Liebetanz, D.; Salinas-Riester, G.; Wiltfang, J.; Klafki, H.W.; Wirths, O. Physical activity delays hippocampal neurodegeneration and rescues memory deficits in an Alzheimer disease mouse model. Transl. Psychiatry 2016, 6, e800. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, C.P.; Berg, E.A.; Elliott-Bryant, R.; Fishman, J.B.; McKee, A.C.; Morin, P.J.; Shia, M.A.; Fine, R.E. Pyroglutamate-Aβ 3 and 11 colocalize in amyloid plaques in Alzheimer’s disease cerebral cortex with pyroglutamate-Aβ 11 forming the central core. Neurosci. Lett. 2011, 505, 109–112. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.; Zheng, J.; Sun, Y.; Dong, X. Seeding and Cross-Seeding Aggregations of Aβ40 and Its N-Terminal-Truncated Peptide Aβ11–40. Langmuir 2019, 35, 2821–2831. [Google Scholar] [CrossRef]
- Barritt, J.D.; Viles, J.H. Truncated Amyloid-beta(11-40/42) from Alzheimer Disease Binds Cu2+ with a Femtomolar Affinity and Influences Fiber Assembly. J. Biol. Chem. 2015, 290, 27791–27802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, L.; al Taleb, Z.; Tsai, Y.L.; Nguyen, V.T.; Lu, Q.; Synatschke, C.V.; Endres, K.; Bufe, B. Amyloid beta and its naturally occurring N-terminal variants are potent activators of human and mouse formyl peptide receptor. J. Biol. Chem. 2022, 102642. [Google Scholar] [CrossRef] [PubMed]
- Lalowski, M.; Golabek, A.; Lemere, C.A.; Selkoe, D.J.; Wisniewski, H.M.; Beavis, R.C.; Frangione, B.; Wisniewski, T. The “nonamyloidogenic” p3 fragment (amyloid beta17-42) is a major constituent of Down’s syndrome cerebellar preamyloid. J. Biol. Chem. 1996, 271, 33623–33631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, H.; Mori, H.; Saido, T.; Kondo, H.; Ikeda, K.; McGeer, P.L. Occurrence of the diffuse amyloid beta-protein (Abeta) deposits with numerous Abeta-containing glial cells in the cerebral cortex of patients with Alzheimer’s disease. Glia 1999, 25, 324–331. [Google Scholar] [CrossRef]
- Moghekar, A.; Rao, S.; Li, M.; Ruben, D.; Mammen, A.; Tang, X.; O’Brien, R.J. Large quantities of Abeta peptide are constitutively released during amyloid precursor protein metabolism in vivo and in vitro. J. Biol. Chem. 2011, 286, 15989–15997. [Google Scholar] [CrossRef] [Green Version]
- Hunter, S.; Brayne, C. Do anti-amyloid beta protein antibody cross reactivities confound Alzheimer disease research? J. Negat. Results Biomed. 2017, 16, 1. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, A.J.; Raskatov, J.A. A robust preparation method for the amyloidogenic and intrinsically disordered amyloid-alpha peptide. J. Pept. Sci. 2022, e3414. [Google Scholar] [CrossRef]
- Dulin, F.; Leveille, F.; Ortega, J.B.; Mornon, J.P.; Buisson, A.; Callebaut, I.; Colloc’h, N. P3 peptide, a truncated form of A beta devoid of synaptotoxic effect, does not assemble into soluble oligomers. FEBS. Lett. 2008, 582, 1865–1870. [Google Scholar] [CrossRef]
- Kuhn, A.J.; Abrams, B.S.; Knowlton, S.; Raskatov, J.A. Alzheimer’s Disease “Non-amyloidogenic” p3 Peptide Revisited: A Case for Amyloid-alpha. ACS. Chem. Neurosci. 2020, 11, 1539–1544. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Polymorphism of Alzheimer’s Abeta17-42 (p3) oligomers: The importance of the turn location and its conformation. Biophys. J. 2009, 97, 1168–1177. [Google Scholar] [CrossRef]
- Streltsov, V.A.; Varghese, J.N.; Masters, C.L.; Nuttall, S.D. Crystal structure of the amyloid-β p3 fragment provides a model for oligomer formation in Alzheimer’s disease. J. Neurosci. 2011, 31, 1419–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Norton, D.D.; Wang, X.; Kusiak, J.W. Abeta 17-42 in Alzheimer’s disease activates JNK and caspase-8 leading to neuronal apoptosis. Brain 2002, 125, 2036–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanik, A.M.; Rampe, D.; Ringheim, G.E. Amyloid-beta peptide fragments p3 and p4 induce pro-inflammatory cytokine and chemokine production in vitro and in vivo. J. Neurochem. 2001, 77, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Shoji, M.; Harigaya, Y.; Kawarabayashi, T.; Watanabe, M.; Kanai, M.; Hirai, S. Amyloid β-protein ending at Thr43 is a minor component of some diffuse plaques in the Alzheimer’s disease brain, but is not found in cerebrovascular amyloid. Brain. Res. 1995, 702, 275–278. [Google Scholar] [CrossRef]
- Jäkel, L.; Boche, D.; Nicoll, J.A.R.; Verbeek, M.M. Aβ43 in human Alzheimer’s disease: Effects of active Aβ42 immunization. Acta Neuropathol. Commun. 2019, 7, 141. [Google Scholar] [CrossRef] [Green Version]
- Welander, H.; Frånberg, J.; Graff, C.; Sundström, E.; Winblad, B.; Tjernberg, L.O. Aβ43 is more frequent than Aβ40 in amyloid plaque cores from Alzheimer disease brains. J. Neurochem. 2009, 110, 697–706. [Google Scholar] [CrossRef]
- Sandebring, A.; Welander, H.; Winblad, B.; Graff, C.; Tjernberg, L.O. The Pathogenic Aβ43 Is Enriched in Familial and Sporadic Alzheimer Disease. PLoS ONE 2013, 8, e55847. [Google Scholar] [CrossRef]
- Kakuda, N.; Takami, M.; Okochi, M.; Kasuga, K.; Ihara, Y.; Ikeuchi, T. Switched Aβ43 generation in familial Alzheimer’s disease with presenilin 1 mutation. Transl. Psychiatry 2021, 11, 558. [Google Scholar] [CrossRef]
- Trambauer, J.; Rodríguez Sarmiento, R.M.; Fukumori, A.; Feederle, R.; Baumann, K.; Steiner, H. Aβ43-producing PS1 FAD mutants cause altered substrate interactions and respond to γ-secretase modulation. EMBO Rep. 2020, 21, e47996. [Google Scholar] [CrossRef]
- Lauridsen, C.; Sando, S.B.; Møller, I.; Berge, G.; Pomary, P.K.; Grøntvedt, G.R.; Salvesen, Ø.; Bråthen, G.; White, L.R. Cerebrospinal Fluid Aβ43 Is Reduced in Early-Onset Compared to Late-Onset Alzheimer’s Disease, But Has Similar Diagnostic Accuracy to Aβ42. Front. Aging Neurosci. 2017, 9, 210. [Google Scholar] [CrossRef]
- Fu, L.; Sun, Y.; Guo, Y.; Chen, Y.; Yu, B.; Zhang, H.; Wu, J.; Yu, X.; Kong, W.; Wu, H. Comparison of neurotoxicity of different aggregated forms of Aβ40, Aβ42 and Aβ43 in cell cultures. J. Pept. Sci. 2017, 23, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Suemoto, T.; Brouwers, N.; Sleegers, K.; Funamoto, S.; Mihira, N.; Matsuba, Y.; Yamada, K.; Nilsson, P.; Takano, J.; et al. Potent amyloidogenicity and pathogenicity of Aβ43. Nat. Neurosci. 2011, 14, 1023–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Riquelme, A.; Mao, A.; Barghash, M.M.; Lau, H.H.C.; Stuart, E.; Kovacs, G.G.; Nilsson, K.P.R.; Fraser, P.E.; Schmitt-Ulms, G.; Watts, J.C. Aβ43 aggregates exhibit enhanced prion-like seeding activity in mice. Acta Neuropathol. Commun. 2021, 9, 83. [Google Scholar] [CrossRef]
- Moore, B.D.; Martin, J.; de Mena, L.; Sanchez, J.; Cruz, P.E.; Ceballos-Diaz, C.; Ladd, T.B.; Ran, Y.; Levites, Y.; Kukar, T.L.; et al. Short Aβ peptides attenuate Aβ42 toxicity in vivo. J. Exp. Med. 2018, 215, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Quartey, M.O.; Nyarko, J.N.K.; Maley, J.M.; Barnes, J.R.; Bolanos, M.A.C.; Heistad, R.M.; Knudsen, K.J.; Pennington, P.R.; Buttigieg, J.; De Carvalho, C.E.; et al. The Aβ(1-38) peptide is a negative regulator of the Aβ(1-42) peptide implicated in Alzheimer disease progression. Sci. Rep. 2021, 11, 431. [Google Scholar] [CrossRef]
- Wiltfang, J.; Esselmann, H.; Bibl, M.; Smirnov, A.; Otto, M.; Paul, S.; Schmidt, B.; Klafki, H.W.; Maler, M.; Dyrks, T.; et al. Highly conserved and disease-specific patterns of carboxyterminally truncated Abeta peptides 1-37/38/39 in addition to 1-40/42 in Alzheimer’s disease and in patients with chronic neuroinflammation. J. Neurochem. 2002, 81, 481–496. [Google Scholar] [CrossRef]
- Cabrera, E.; Mathews, P.; Mezhericher, E.; Beach, T.G.; Deng, J.; Neubert, T.A.; Rostagno, A.; Ghiso, J. Abeta truncated species: Implications for brain clearance mechanisms and amyloid plaque deposition. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 208–225. [Google Scholar] [CrossRef]
- Liebsch, F.; Kulic, L.; Teunissen, C.; Shobo, A.; Ulku, I.; Engelschalt, V.; Hancock, M.A.; van der Flier, W.M.; Kunach, P.; Rosa-Neto, P.; et al. Aβ34 is a BACE1-derived degradation intermediate associated with amyloid clearance and Alzheimer’s disease progression. Nat. Commun. 2019, 10, 2240. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Guillamon, M.; Mawhirt, S.; Blais, S.; Montaner, J.; Neubert, T.A.; Rostagno, A.; Ghiso, J. Sequential Amyloid-beta Degradation by the Matrix Metalloproteases MMP-2 and MMP-9. J. Biol. Chem. 2015, 290, 15078–15091. [Google Scholar] [CrossRef] [Green Version]
- Caillava, C.; Ranaldi, S.; Lauritzen, I.; Bauer, C.; Fareh, J.; Abraham, J.D.; Checler, F. Study on Aβ34 biology and detection in transgenic mice brains. Neurobiol. Aging 2014, 35, 1570–1581. [Google Scholar] [CrossRef]
- Giulian, D.; Haverkamp, L.J.; Yu, J.H.; Karshin, W.; Tom, D.; Li, J.; Kirkpatrick, J.; Kuo, L.M.; Roher, A.E. Specific domains of beta-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J. Neurosci. 1996, 16, 6021–6037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonyan, A.; Schlenzig, D.; Schilling, S.; Naumann, M.; Sharoyan, S.; Mardanyan, S.; Demuth, H.U. Concerted action of dipeptidyl peptidase IV and glutaminyl cyclase results in formation of pyroglutamate-modified amyloid peptides in vitro. Neurochem. Int. 2018, 113, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Cynis, H.; Schilling, S.; Bodnar, M.; Hoffmann, T.; Heiser, U.; Saido, T.C.; Demuth, H.U. Inhibition of glutaminyl cyclase alters pyroglutamate formation in mammalian cells. Biochim. Biophys. Acta 2006, 1764, 1618–1625. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, S.; Wirths, O.; Schilling, S.; Graubner, S.; Demuth, H.U.; Bayer, T.A. Overexpression of glutaminyl cyclase, the enzyme responsible for pyroglutamate A(1) formation, induces behavioral deficits, and glutaminyl cyclase knock-out rescues the behavioral phenotype in 5XFAD mice. J. Biol. Chem. 2011, 286, 4454–4460. [Google Scholar] [CrossRef] [Green Version]
- Schilling, S.; Appl, T.; Hoffmann, T.; Cynis, H.; Schulz, K.; Jagla, W.; Friedrich, D.; Wermann, M.; Buchholz, M.; Heiser, U.; et al. Inhibition of glutaminyl cyclase prevents pGlu-Abeta formation after intracortical/hippocampal microinjection in vivo/in situ. J. Neurochem. 2008, 106, 1225–1236. [Google Scholar] [CrossRef]
- Schlenzig, D.; Manhart, S.; Cinar, Y.; Kleinschmidt, M.; Hause, G.; Willbold, D.; Funke, S.A.; Schilling, S.; Demuth, H.U. Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry 2009, 48, 7072–7078. [Google Scholar] [CrossRef]
- Saido, T.C.; Iwatsubo, T.; Mann, D.M.; Shimada, H.; Ihara, Y.; Kawashima, S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 1995, 14, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Rijal Upadhaya, A.; Kosterin, I.; Kumar, S.; von Arnim, C.A.; Yamaguchi, H.; Fändrich, M.; Walter, J.; Thal, D.R. Biochemical stages of amyloid-β peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain 2014, 137, 887–903. [Google Scholar] [CrossRef]
- Wang, P.N.; Lin, K.J.; Liu, H.C.; Andreasson, U.; Blennow, K.; Zetterberg, H.; Yang, S.Y. Plasma pyroglutamate-modified amyloid beta differentiates amyloid pathology. Alzheimers Dement. 2020, 12, e12029. [Google Scholar] [CrossRef]
- Dammers, C.; Schwarten, M.; Buell, A.K.; Willbold, D. Pyroglutamate-modified Abeta(3-42) affects aggregation kinetics of Abeta(1-42) by accelerating primary and secondary pathways. Chem. Sci. 2017, 8, 4996–5004. [Google Scholar] [CrossRef]
- Hu, Z.W.; Au, D.F.; Cruceta, L.; Vugmeyster, L.; Qiang, W. N-Terminal Modified Abeta Variants Enable Modulations to the Structures and Cytotoxicity Levels of Wild-Type Abeta Fibrils through Cross-Seeding. ACS Chem. Neurosci. 2020, 11, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Neddens, J.; Daurer, M.; Flunkert, S.; Beutl, K.; Loeffler, T.; Walker, L.; Attems, J.; Hutter-Paier, B. Correlation of pyroglutamate amyloid beta and ptau Ser202/Thr205 levels in Alzheimer’s disease and related murine models. PLoS ONE 2020, 15, e0235543. [Google Scholar] [CrossRef] [PubMed]
- De Kimpe, L.; van Haastert, E.S.; Kaminari, A.; Zwart, R.; Rutjes, H.; Hoozemans, J.J.; Scheper, W. Intracellular accumulation of aggregated pyroglutamate amyloid beta: Convergence of aging and Abeta pathology at the lysosome. Age 2013, 35, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Baik, S.H.; Kang, S.; Son, S.M.; Mook-Jung, I. Microglia contributes to plaque growth by cell death due to uptake of amyloid beta in the brain of Alzheimer’s disease mouse model. Glia 2016, 64, 2274–2290. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-beta into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef] [Green Version]
- Grochowska, K.M.; Yuanxiang, P.; Bar, J.; Raman, R.; Brugal, G.; Sahu, G.; Schweizer, M.; Bikbaev, A.; Schilling, S.; Demuth, H.U.; et al. Posttranslational modification impact on the mechanism by which amyloid-beta induces synaptic dysfunction. EMBO Rep. 2017, 18, 962–981. [Google Scholar] [CrossRef]
- Balakrishnan, K.; Rijal Upadhaya, A.; Steinmetz, J.; Reichwald, J.; Abramowski, D.; Fandrich, M.; Kumar, S.; Yamaguchi, H.; Walter, J.; Staufenbiel, M.; et al. Impact of amyloid beta aggregate maturation on antibody treatment in APP23 mice. Acta Neuropathol. Commun. 2015, 3, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, J.L.; Le, K.X.; Cynis, H.; Ekpo, E.; Kleinschmidt, M.; Palmour, R.M.; Ervin, F.R.; Snigdha, S.; Cotman, C.W.; Saido, T.C.; et al. Pyroglutamate-3 amyloid-beta deposition in the brains of humans, non-human primates, canines, and Alzheimer disease-like transgenic mouse models. Am. J. Pathol. 2013, 183, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Bethge, T.; Marcello, A.; Harmeier, A.; Jawhar, S.; Lucassen, P.J.; Multhaup, G.; Brody, D.L.; Esparza, T.; Ingelsson, M.; et al. Pyroglutamate Abeta pathology in APP/PS1KI mice, sporadic and familial Alzheimer’s disease cases. J. Neural. Transm. 2010, 117, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crehan, H.; Liu, B.; Kleinschmidt, M.; Rahfeld, J.U.; Le, K.X.; Caldarone, B.J.; Frost, J.L.; Hettmann, T.; Hutter-Paier, B.; O’Nuallain, B.; et al. Effector function of anti-pyroglutamate-3 Aβ antibodies affects cognitive benefit, glial activation and amyloid clearance in Alzheimer’s-like mice. Alzheimers Res. 2020, 12, 12. [Google Scholar] [CrossRef]
- Mukherjee, S.; Perez, K.A.; Lago, L.C.; Klatt, S.; McLean, C.A.; Birchall, I.E.; Barnham, K.J.; Masters, C.L.; Roberts, B.R. Quantification of N-terminal amyloid-beta isoforms reveals isomers are the most abundant form of the amyloid-beta peptide in sporadic Alzheimer’s disease. Brain Commun. 2021, 3, fcab028. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, T.R.; Riggs, D.L.; Talbert, L.E.; Tang, J.; Coburn, E.; Kang, A.S.; Noll, J.; Augello, C.; Ford, B.D.; Julian, R.R. Spontaneous Isomerization of Long-Lived Proteins Provides a Molecular Mechanism for the Lysosomal Failure Observed in Alzheimer’s Disease. ACS Cent. Sci. 2019, 5, 1387–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, M.I.; Head, E.; Velazquez, P.; Cotman, C.W.; Tenner, A.J. The presence of isoaspartic acid in beta-amyloid plaques indicates plaque age. Exp. Neurol. 1999, 157, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zürcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [CrossRef]
- Fabian, H.; Szendrei, G.I.; Mantsch, H.H.; Greenberg, B.D.; ÖTvÖS Jr, L. Synthetic post-translationally modified human Aβ peptide exhibits a markedly increased tendency to form β-pleated sheets in vitro. Eur. J. Biochem. 1994, 221, 959–964. [Google Scholar] [CrossRef]
- Szendrei, G.I.; Fabian, H.; Mantsch, H.H.; Lovas, S.; Nyéki, O.; Schön, I.; Otvos, L., Jr. Aspartate-bond isomerization affects the major conformations of synthetic peptides. Eur. J. Biochem. 1994, 226, 917–924. [Google Scholar] [CrossRef] [Green Version]
- Barykin, E.P.; Garifulina, A.I.; Kruykova, E.V.; Spirova, E.N.; Anashkina, A.A.; Adzhubei, A.A.; Shelukhina, I.V.; Kasheverov, I.E.; Mitkevich, V.A.; Kozin, S.A.; et al. Isomerization of Asp7 in Beta-Amyloid Enhances Inhibition of the α7 Nicotinic Receptor and Promotes Neurotoxicity. Cells 2019, 8, 771. [Google Scholar] [CrossRef] [Green Version]
- Barykin, E.P.; Petrushanko, I.Y.; Burnysheva, K.M.; Makarov, A.A.; Mitkevich, V.A. [Isomerization of Asp7 increases the toxic effects of amyloid beta and its phosphorylated form in SH-SY5Y neuroblastoma cells]. Mol. Biol. 2016, 50, 863–869. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Webster, S.; Emmerling, M.R.; De Lima, N.; Roher, A.E. Irreversible dimerization/tetramerization and post-translational modifications inhibit proteolytic degradation of A beta peptides of Alzheimer’s disease. Biochim. Biophys. Acta. 1998, 1406, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Kapadia, A.; Theil, S.; Joshi, P.; Riffel, F.; Heneka, M.T.; Walter, J. Novel Phosphorylation-State Specific Antibodies Reveal Differential Deposition of Ser26 Phosphorylated Abeta Species in a Mouse Model of Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 619639. [Google Scholar] [CrossRef]
- Kumar, S.; Wirths, O.; Stüber, K.; Wunderlich, P.; Koch, P.; Theil, S.; Rezaei-Ghaleh, N.; Zweckstetter, M.; Bayer, T.A.; Brüstle, O.; et al. Phosphorylation of the amyloid β-peptide at Ser26 stabilizes oligomeric assembly and increases neurotoxicity. Acta Neuropathol. 2016, 131, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashby, E.L.; Miners, J.S.; Kumar, S.; Walter, J.; Love, S.; Kehoe, P.G. Investigation of Abeta phosphorylated at serine 8 (pAbeta) in Alzheimer’s disease, dementia with Lewy bodies and vascular dementia. Neuropathol. Appl. Neurobiol. 2015, 41, 428–444. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, S.; Hinze, D.; Josten, M.; Sahl, H.G.; Siepmann, M.; Walter, J. Phosphorylation of amyloid-β peptide at serine 8 attenuates its clearance via insulin-degrading and angiotensin-converting enzymes. J. Biol. Chem. 2012, 287, 8641–8651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaei-Ghaleh, N.; Amininasab, M.; Kumar, S.; Walter, J.; Zweckstetter, M. Phosphorylation modifies the molecular stability of beta-amyloid deposits. Nat. Commun. 2016, 7, 11359. [Google Scholar] [CrossRef] [Green Version]
- Friedemann, M.; Helk, E.; Tiiman, A.; Zovo, K.; Palumaa, P.; Tougu, V. Effect of methionine-35 oxidation on the aggregation of amyloid-beta peptide. Biochem. Biophys. Rep. 2015, 3, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Razzokov, J.; Yusupov, M.; Bogaerts, A. Oxidation destabilizes toxic amyloid beta peptide aggregation. Sci. Rep. 2019, 9, 5476. [Google Scholar] [CrossRef] [Green Version]
- Head, E.; Garzon-Rodriguez, W.; Johnson, J.K.; Lott, I.T.; Cotman, C.W.; Glabe, C. Oxidation of Aβ and Plaque Biogenesis in Alzheimer’s Disease and Down Syndrome. Neurobiol. Dis. 2001, 8, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Tomiyama, T.; Asano, S.; Furiya, Y.; Shirasawa, T.; Endo, N.; Mori, H. Racemization of Asp23 residue affects the aggregation properties of Alzheimer amyloid beta protein analogues. J. Biol. Chem. 1994, 269, 10205–10208. [Google Scholar] [CrossRef]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. The amyloid precursor protein: A biochemical enigma in brain development, function and disease. FEBS Lett. 2013, 587, 2046–2054. [Google Scholar] [CrossRef]
- Panegyres, P.K.; Zafiris-Toufexis, K.; Kakulas, B.A. Amyloid precursor protein gene isoforms in Alzheimer’s disease and other neurodegenerative disorders. J. Neurol. Sci. 2000, 173, 81–92. [Google Scholar] [CrossRef]
- Beckmann, A.M.; Glebov, K.; Walter, J.; Merkel, O.; Mangold, M.; Schmidt, F.; Becker-Pauly, C.; Gütschow, M.; Stirnberg, M. The intact Kunitz domain protects the amyloid precursor protein from being processed by matriptase-2. Biol. Chem. 2016, 397, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Dawkins, E.; Small, D.H. Insights into the physiological function of the β-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Paganini, L.; Mucke, L.; Gordon, M.; Refolo, L.; Carman, M.; Sinha, S.; Oltersdorf, T.; Lieberburg, I.; McConlogue, L. Beta-secretase processing of the beta-amyloid precursor protein in transgenic mice is efficient in neurons but inefficient in astrocytes. J. Biol. Chem. 1996, 271, 31407–31411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Wang, Y.; McCarthy, D.; Wen, H.; Borchelt, D.R.; Price, D.L.; Wong, P.C. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 2001, 4, 233–234. [Google Scholar] [CrossRef]
- Koo, E.H.; Sisodia, S.S.; Archer, D.R.; Martin, L.J.; Weidemann, A.; Beyreuther, K.; Fischer, P.; Masters, C.L.; Price, D.L. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc. Natl. Acad. Sci. USA 1990, 87, 1561–1565. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Muller-Hill, B. Differential splicing of Alzheimer’s disease amyloid A4 precursor RNA in rat tissues: PreA4(695) mRNA is predominantly produced in rat and human brain. Biochem. Biophys. Res. Commun. 1990, 166, 1192–1200. [Google Scholar] [CrossRef]
- LeBlanc, A.C.; Chen, H.Y.; Autilio-Gambetti, L.; Gambetti, P. Differential APP gene expression in rat cerebral cortex, meninges, and primary astroglial, microglial and neuronal cultures. FEBS Lett. 1991, 292, 171–178. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, A.C.; Papadopoulos, M.; Belair, C.; Chu, W.; Crosato, M.; Powell, J.; Goodyer, C.G. Processing of amyloid precursor protein in human primary neuron and astrocyte cultures. J. Neurochem. 1997, 68, 1183–1190. [Google Scholar] [CrossRef]
- Laird, F.M.; Cai, H.; Savonenko, A.V.; Farah, M.H.; He, K.; Melnikova, T.; Wen, H.; Chiang, H.C.; Xu, G.; Koliatsos, V.E.; et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 2005, 25, 11693–11709. [Google Scholar] [CrossRef]
- Haass, C.; Hung, A.Y.; Selkoe, D.J. Processing of beta-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J. Neurosci. 1991, 11, 3783–3793. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Sergeant, N.; Bombois, S.; Ghestem, A.; Drobecq, H.; Kostanjevecki, V.; Missiaen, C.; Wattez, A.; David, J.P.; Vanmechelen, E.; Sergheraert, C.; et al. Truncated beta-amyloid peptide species in pre-clinical Alzheimer’s disease as new targets for the vaccination approach. J. Neurochem. 2003, 85, 1581–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberstein, T.J.; Utz, J.; Spitzer, P.; Klafki, H.W.; Wiltfang, J.; Lewczuk, P.; Kornhuber, J.; Maler, J.M. The Role of Cathepsin B in the Degradation of Abeta and in the Production of Abeta Peptides Starting With Ala2 in Cultured Astrocytes. Front. Mol. Neurosci. 2020, 13, 615740. [Google Scholar] [CrossRef] [PubMed]
- Bien, J.; Jefferson, T.; Causevic, M.; Jumpertz, T.; Munter, L.; Multhaup, G.; Weggen, S.; Becker-Pauly, C.; Pietrzik, C.U. The metalloprotease meprin beta generates amino terminal-truncated amyloid beta peptide species. J. Biol. Chem. 2012, 287, 33304–33313. [Google Scholar] [CrossRef] [Green Version]
- Howell, S.; Nalbantoglu, J.; Crine, P. Neutral endopeptidase can hydrolyze beta-amyloid(1-40) but shows no effect on beta-amyloid precursor protein metabolism. Peptides 1995, 16, 647–652. [Google Scholar] [CrossRef]
- Liao, M.C.; Ahmed, M.; Smith, S.O.; Van Nostrand, W.E. Degradation of amyloid beta protein by purified myelin basic protein. J. Biol. Chem. 2009, 284, 28917–28925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevalle, J.; Amoyel, A.; Robert, P.; Fournie-Zaluski, M.C.; Roques, B.; Checler, F. Aminopeptidase A contributes to the N-terminal truncation of amyloid beta-peptide. J. Neurochem. 2009, 109, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.A.; Wirths, O. Focusing the amyloid cascade hypothesis on N-truncated Abeta peptides as drug targets against Alzheimer’s disease. Acta Neuropathol. 2014, 127, 787–801. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, S.K.; Shnitka, T.K.; Elbrink, J. Reactive astrocytes--a review. Cytobios. 1990, 61, 133–160. [Google Scholar]
- Siman, R.; Card, J.P.; Nelson, R.B.; Davis, L.G. Expression of beta-amyloid precursor protein in reactive astrocytes following neuronal damage. Neuron 1989, 3, 275–285. [Google Scholar] [CrossRef]
- Mita, S.; Schon, E.A.; Herbert, J. Widespread expression of amyloid beta-protein precursor gene in rat brain. Am. J. Pathol. 1989, 134, 1253–1261. [Google Scholar] [PubMed]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Abeta production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflammation. 2011, 8, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banati, R.B.; Gehrmann, J.; Czech, C.; Monning, U.; Jones, L.L.; Konig, G.; Beyreuther, K.; Kreutzberg, G.W. Early and rapid de novo synthesis of Alzheimer beta A4-amyloid precursor protein (APP) in activated microglia. Glia 1993, 9, 199–210. [Google Scholar] [CrossRef]
- Bauer, J.; Konig, G.; Strauss, S.; Jonas, U.; Ganter, U.; Weidemann, A.; Monning, U.; Masters, C.L.; Volk, B.; Berger, M.; et al. In-vitro matured human macrophages express Alzheimer’s beta A4-amyloid precursor protein indicating synthesis in microglial cells. FEBS Lett. 1991, 282, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Monning, U.; Sandbrink, R.; Weidemann, A.; Banati, R.B.; Masters, C.L.; Beyreuther, K. Extracellular matrix influences the biogenesis of amyloid precursor protein in microglial cells. J. Biol. Chem. 1995, 270, 7104–7110. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Das, B.; Zhou, J.; Hu, X.; Yan, R. Targeted BACE-1 inhibition in microglia enhances amyloid clearance and improved cognitive performance. Sci. Adv. 2022, 8, eabo3610. [Google Scholar] [CrossRef]
- Gunner, G.; Cheadle, L.; Johnson, K.M.; Ayata, P.; Badimon, A.; Mondo, E.; Nagy, M.A.; Liu, L.; Bemiller, S.M.; Kim, K.W.; et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 2019, 22, 1075–1088. [Google Scholar] [CrossRef]
- Nadler, Y.; Alexandrovich, A.; Grigoriadis, N.; Hartmann, T.; Rao, K.S.; Shohami, E.; Stein, R. Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 2008, 56, 552–567. [Google Scholar] [CrossRef]
- Bitting, L.; Naidu, A.; Cordell, B.; Murphy, G.M., Jr. Beta-amyloid peptide secretion by a microglial cell line is induced by beta-amyloid-(25-35) and lipopolysaccharide. J. Biol. Chem. 1996, 271, 16084–16089. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.A.; Sens, M.A.; Combs, C.K. Amyloid Precursor Protein Mediates a Tyrosine Kinase-Dependent Activation Response in Endothelial Cells. J. Neurosci. 2009, 29, 14451–14462. [Google Scholar] [CrossRef] [PubMed]
- Sondag, C.M.; Combs, C.K. Amyloid precursor protein mediates proinflammatory activation of monocytic lineage cells. J. Biol. Chem. 2004, 279, 14456–14463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manocha, G.D.; Floden, A.M.; Rausch, K.; Kulas, J.A.; McGregor, B.A.; Rojanathammanee, L.; Puig, K.R.; Puig, K.L.; Karki, S.; Nichols, M.R.; et al. APP Regulates Microglial Phenotype in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2016, 36, 8471–8486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Ladona, F.J.; Huss, Y.; Frey, P.; Ghandour, M.S. Oligodendrocytes express different isoforms of beta-amyloid precursor protein in chemically defined cell culture conditions: In Situ hybridization and immunocytochemical detection. J. Neurosci. Res. 1997, 50, 50–61. [Google Scholar] [CrossRef]
- Sandbrink, R.; Masters, C.L.; Beyreuther, K. APP gene family. Alternative splicing generates functionally related isoforms. Ann. N. Y. Acad. Sci. 1996, 777, 281–287. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Ikeda, K.; Kim, S.U. Differential distribution of cellular forms of beta-amyloid precursor protein in murine glial cell cultures. Brain. Res. 1992, 584, 219–225. [Google Scholar] [CrossRef]
- Nihonmatsu-Kikuchi, N.; Yu, X.J.; Matsuda, Y.; Ozawa, N.; Ito, T.; Satou, K.; Kaname, T.; Iwasaki, Y.; Akagi, A.; Yoshida, M.; et al. Essential roles of plexin-B3(+) oligodendrocyte precursor cells in the pathogenesis of Alzheimer’s disease. Commun. Biol. 2021, 4, 870. [Google Scholar] [CrossRef]
- Card, J.P.; Meade, R.P.; Davis, L.G. Immunocytochemical localization of the precursor protein for beta-amyloid in the rat central nervous system. Neuron 1988, 1, 835–846. [Google Scholar] [CrossRef]
- Kawarabayashi, T.; Shoji, M.; Harigaya, Y.; Yamaguchi, H.; Hirai, S. Amyloid beta/A4 protein precursor is widely distributed in both the central and peripheral nervous systems of the mouse. Brain Res. 1991, 552, 1–7. [Google Scholar] [CrossRef]
- Palacios, G.; Palacios, J.M.; Mengod, G.; Frey, P. Beta-amyloid precursor protein localization in the Golgi apparatus in neurons and oligodendrocytes. An immunocytochemical structural and ultrastructural study in normal and axotomized neurons. Brain. Res. Mol. Brain Res. 1992, 15, 195–206. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Ikeda, K.; Kim, S.U. beta-Amyloid precursor protein of Alzheimer’s disease in cultured bovine oligodendrocytes. J. Neurosci. Res. 1992, 32, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Luo, J.; Redies, C. Differential expression of five members of the ADAM family in the developing chicken brain. Neuroscience 2008, 157, 360–375. [Google Scholar] [CrossRef]
- Jung, M.; Krämer, E.; Grzenkowski, M.; Tang, K.; Blakemore, W.; Aguzzi, A.; Khazaie, K.; Chlichlia, K.; von Blankenfeld, G.; Kettenmann, H.; et al. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur. J. Neurosci. 1995, 7, 1245–1265. [Google Scholar] [CrossRef]
- Sakry, D.; Neitz, A.; Singh, J.; Frischknecht, R.; Marongiu, D.; Biname, F.; Perera, S.S.; Endres, K.; Lutz, B.; Radyushkin, K.; et al. Oligodendrocyte precursor cells modulate the neuronal network by activity-dependent ectodomain cleavage of glial NG2. PLoS Biol. 2014, 12, e1001993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, L.O.; Konrad, M.; Wruck, C.; Koch, T.; Pufe, T.; Lucius, R. Involvement of formyl-peptide-receptor-like-1 and phospholipase D in the internalization and signal transduction of amyloid beta 1-42 in glial cells. Neuroscience 2008, 156, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Ruan, L.; Qian, L.; Liu, X.; Le, Y. Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J. Neurosci. 2010, 30, 11848–11857. [Google Scholar] [CrossRef] [Green Version]
- Yazawa, H.; Yu, Z.X.; Takeda, L.Y.; Gong, W.; Ferrans, V.J.; Oppenheim, J.J.; Li, C.C.; Wang, J.M. Beta amyloid peptide (Abeta42) is internalized via the G-protein-coupled receptor FPRL1 and forms fibrillar aggregates in macrophages. FASEB J. 2001, 15, 2454–2462. [Google Scholar] [CrossRef] [PubMed]
- Heurtaux, T.; Michelucci, A.; Losciuto, S.; Gallotti, C.; Felten, P.; Dorban, G.; Grandbarbe, L.; Morga, E.; Heuschling, P. Microglial activation depends on beta-amyloid conformation: Role of the formylpeptide receptor 2. J. Neurochem. 2010, 114, 576–586. [Google Scholar] [CrossRef]
- Le, Y.; Gong, W.; Tiffany, H.L.; Tumanov, A.; Nedospasov, S.; Shen, W.; Dunlop, N.M.; Gao, J.L.; Murphy, P.M.; Oppenheim, J.J.; et al. Amyloid (beta)42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J. Neurosci. 2001, 21, Rc123. [Google Scholar] [CrossRef]
- Zhang, H.; Su, Y.J.; Zhou, W.W.; Wang, S.W.; Xu, P.X.; Yu, X.L.; Liu, R.T. Activated scavenger receptor A promotes glial internalization of aβ. PLoS ONE 2014, 9, e94197. [Google Scholar] [CrossRef] [PubMed]
- Husemann, J.; Loike, J.D.; Kodama, T.; Silverstein, S.C. Scavenger receptor class B type I (SR-BI) mediates adhesion of neonatal murine microglia to fibrillar beta-amyloid. J. Neuroimmunol. 2001, 114, 142–150. [Google Scholar] [CrossRef]
- Coraci, I.S.; Husemann, J.; Berman, J.W.; Hulette, C.; Dufour, J.H.; Campanella, G.K.; Luster, A.D.; Silverstein, S.C.; El-Khoury, J.B. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am. J. Pathol. 2002, 160, 101–112. [Google Scholar] [CrossRef]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rube, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–1107. [Google Scholar] [CrossRef] [Green Version]
- Richard, K.L.; Filali, M.; Prefontaine, P.; Rivest, S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J. Neurosci. 2008, 28, 5784–5793. [Google Scholar] [CrossRef] [Green Version]
- Tahara, K.; Kim, H.D.; Jin, J.J.; Maxwell, J.A.; Li, L.; Fukuchi, K.I. Role of toll-like receptor signalling in A uptake and clearance. Brain 2006, 129, 3006–3019. [Google Scholar] [CrossRef] [Green Version]
- Li, H.Q.; Chen, C.; Dou, Y.; Wu, H.J.; Liu, Y.J.; Lou, H.F.; Zhang, J.M.; Li, X.M.; Wang, H.; Duan, S. P2Y4 receptor-mediated pinocytosis contributes to amyloid beta-induced self-uptake by microglia. Mol. Cell. Biol. 2013, 33, 4282–4293. [Google Scholar] [CrossRef] [Green Version]
- Nazere, K.; Takahashi, T.; Hara, N.; Muguruma, K.; Nakamori, M.; Yamazaki, Y.; Morino, H.; Maruyama, H. Amyloid Beta Is Internalized via Macropinocytosis, an HSPG- and Lipid Raft-Dependent and Rac1-Mediated Process. Front. Mol. Neurosci. 2022, 15. [Google Scholar] [CrossRef]
- Cho, M.H.; Cho, K.; Kang, H.J.; Jeon, E.Y.; Kim, H.S.; Kwon, H.J.; Kim, H.M.; Kim, D.H.; Yoon, S.Y. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef] [Green Version]
- Prakash, P.; Jethava, K.P.; Korte, N.; Izquierdo, P.; Favuzzi, E.; Rose, I.V.L.; Guttenplan, K.A.; Manchanda, P.; Dutta, S.; Rochet, J.C.; et al. Monitoring phagocytic uptake of amyloid beta into glial cell lysosomes in real time. Chem. Sci. 2021, 12, 10901–10918. [Google Scholar] [CrossRef] [PubMed]
- Yim, W.W.-Y.; Mizushima, N. Lysosome biology in autophagy. Cell. Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell. Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Sosna, J.; Philipp, S.; Albay, R., 3rd; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef] [Green Version]
- Pomilio, C.; Gorojod, R.M.; Riudavets, M.; Vinuesa, A.; Presa, J.; Gregosa, A.; Bentivegna, M.; Alaimo, A.; Alcon, S.P.; Sevlever, G.; et al. Microglial autophagy is impaired by prolonged exposure to beta-amyloid peptides: Evidence from experimental models and Alzheimer’s disease patients. Geroscience 2020, 42, 613–632. [Google Scholar] [CrossRef]
- Liang, T.; Zhang, Y.; Wu, S.; Chen, Q.; Wang, L. The Role of NLRP3 Inflammasome in Alzheimer’s Disease and Potential Therapeutic Targets. Front. Pharm. 2022, 13, 845185. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [Green Version]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, R.; Fusco, R.; Cordaro, M.; Siracusa, R.; Peritore, A.F.; Gugliandolo, E.; Crupi, R.; Scuto, M.; Cuzzocrea, S.; Di Paola, R.; et al. Modulation of NLRP3 Inflammasome through Formyl Peptide Receptor 1 (Fpr-1) Pathway as a New Therapeutic Target in Bronchiolitis Obliterans Syndrome. Int. J. Mol. Sci. 2020, 2144. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Kaneko, N.; Takeda, H.; Sawasaki, T.; Morikawa, S.; Zhou, W.; Kurata, M.; Yamamoto, T.; Akbar, S.M.F.; Zako, T.; et al. Amyloid beta directly interacts with NLRP3 to initiate inflammasome activation: Identification of an intrinsic NLRP3 ligand in a cell-free system. Inflamm. Regen. 2018, 38, 27. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.; Sheng, J.G.; Roberts, G.W.; Mrak, R.E. Interleukin-1 expression in different plaque types in Alzheimer’s disease: Significance in plaque evolution. J. Neuropathol. Exp. Neurol. 1995, 54, 276–281. [Google Scholar] [CrossRef]
- Ciallella, J.R.; Ikonomovic, M.D.; Paljug, W.R.; Wilbur, Y.I.; Dixon, C.E.; Kochanek, P.M.; Marion, D.W.; DeKosky, S.T. Changes in expression of amyloid precursor protein and interleukin-1beta after experimental traumatic brain injury in rats. J. Neurotrauma. 2002, 19, 1555–1567. [Google Scholar] [CrossRef]
- Donnelly, R.J.; Friedhoff, A.J.; Beer, B.; Blume, A.J.; Vitek, M.P. Interleukin-1 stimulates the beta-amyloid precursor protein promoter. Cell. Mol. Neurobiol. 1990, 10, 485–495. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Oishi, M.; Chen, H.I.; Pinkas-Kramarski, R.; Jaffe, E.A.; Gandy, S.E.; Greengard, P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer beta/A4 amyloid protein precursor. Proc. Natl. Acad. Sci. USA 1992, 89, 10075–10078. [Google Scholar] [CrossRef] [Green Version]
- Kong, Q.; Peterson, T.S.; Baker, O.; Stanley, E.; Camden, J.; Seye, C.I.; Erb, L.; Simonyi, A.; Wood, W.G.; Sun, G.Y.; et al. Interleukin-1beta enhances nucleotide-induced and alpha-secretase-dependent amyloid precursor protein processing in rat primary cortical neurons via up-regulation of the P2Y(2) receptor. J. Neurochem. 2009, 109, 1300–1310. [Google Scholar] [CrossRef] [Green Version]
- Tachida, Y.; Nakagawa, K.; Saito, T.; Saido, T.C.; Honda, T.; Saito, Y.; Murayama, S.; Endo, T.; Sakaguchi, G.; Kato, A.; et al. Interleukin-1β up-regulates TACE to enhance α-cleavage of APP in neurons: Resulting decrease in Aβ production. J. Neurochem. 2008, 104, 1387–1393. [Google Scholar] [CrossRef]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; Miller, J.N.; Johnson, R.E.; O’Banion, M.K. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Invest. 2007, 117, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Zhu, M.; Li, D.; Wu, Y.; Huang, X.; Wu, M. TREM-2 promotes macrophage-mediated eradication of Pseudomonas aeruginosa via a PI3K/Akt pathway. Scand. J. Immunol. 2014, 79, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Tiffany, H.L.; Lavigne, M.C.; Cui, Y.H.; Wang, J.M.; Leto, T.L.; Gao, J.L.; Murphy, P.M. Amyloid-beta induces chemotaxis and oxidant stress by acting at formylpeptide receptor 2, a G protein-coupled receptor expressed in phagocytes and brain. J. Biol. Chem. 2001, 276, 23645–23652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 5010. [Google Scholar] [CrossRef] [PubMed]
- Skoumalová, A.; Hort, J. Blood markers of oxidative stress in Alzheimer’s disease. J. Cell. Mol. Med. 2012, 16, 2291–2300. [Google Scholar] [CrossRef]
- Galasko, D.; Montine, T.J. Biomarkers of oxidative damage and inflammation in Alzheimer’s disease. Biomark. Med. 2010, 4, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, H.; Hanyu, H.; Hirose, D.; Fukusawa, R.; Namioka, N.; Iwamoto, T. Peripheral Oxidative Stress Markers in Individuals with Alzheimer’s Disease with or without Cerebrovascular Disease. J. Am. Geriatr. Soc. 2015, 63, 1472–1474. [Google Scholar] [CrossRef]
- Sakakibara, R.; Kawai, T. Cerebrospinal fluid oxidative stress markers in Alzheimer’s disease. Neurol. Clin. Neurosci. 2020, 8, 232–240. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Gupta, S.; Parrino, T.E.; Knight, A.G.; Ebenezer, P.J.; Weidner, A.M.; LeVine, H., 3rd; Keller, J.N.; Markesbery, W.R. NOX activity is increased in mild cognitive impairment. Antioxid. Redox. Signal. 2010, 12, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Fan, L.M.; Liu, F.; Smith, C.; Li, J.M. Nox2 dependent redox-regulation of microglial response to amyloid-β stimulation and microgliosis in aging. Sci. Rep. 2020, 10, 1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourdel-Marchasson, I.; Delmas-Beauvieux, M.C.; Peuchant, E.; Richard-Harston, S.; Decamps, A.; Reignier, B.; Emeriau, J.P.; Rainfray, M. Antioxidant defences and oxidative stress markers in erythrocytes and plasma from normally nourished elderly Alzheimer patients. Age. Ageing 2001, 30, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, S.; Kawakami, S.; Hara, Y.; Wakamori, M.; Itoh, E.; Minami, T.; Takada, Y.; Kume, T.; Katsuki, H.; Mori, Y.; et al. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J. Pharm. Sci. 2006, 101, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.M.; Cho, H.J.; Jung, E.S.; Shim, M.Y.; Mook-Jung, I. DNA damage-inducing agents elicit γ-secretase activation mediated by oxidative stress. Cell. Death Differ. 2008, 15, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Muche, A.; Arendt, T.; Schliebs, R. Oxidative stress affects processing of amyloid precursor protein in vascular endothelial cells. PLoS ONE 2017, 12, e0178127. [Google Scholar] [CrossRef] [Green Version]
- Tamagno, E.; Bardini, P.; Obbili, A.; Vitali, A.; Borghi, R.; Zaccheo, D.; Pronzato, M.A.; Danni, O.; Smith, M.A.; Perry, G.; et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol. Dis. 2002, 10, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Tamagno, E.; Guglielmotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A.; et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008, 104, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Jo, D.G.; Arumugam, T.V.; Woo, H.N.; Park, J.S.; Tang, S.C.; Mughal, M.; Hyun, D.H.; Park, J.H.; Choi, Y.H.; Gwon, A.R.; et al. Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Hay, D.L.; Chen, S.; Lutz, T.A.; Parkes, D.G.; Roth, J.D. Amylin: Pharmacology, Physiology, and Clinical Potential. Pharmacol. Rev. 2015, 67, 564. [Google Scholar] [CrossRef] [Green Version]
- Ly, H.; Verma, N.; Sharma, S.; Kotiya, D.; Despa, S.; Abner, E.L.; Nelson, P.T.; Jicha, G.A.; Wilcock, D.M.; Goldstein, L.B.; et al. The association of circulating amylin with β-amyloid in familial Alzheimer’s disease. Alzheimers Dement. 2021, 7, e12130. [Google Scholar] [CrossRef] [PubMed]
- Ly, H.; Verma, N.; Kotiya, D.; Goldstein, L.B.; Jicha, G.A.; Despa, F. Amyloid-β removal from the brain is blocked by circulating amyloid-forming amylin secreted by the pancreas. Alzheimer’s Dement. 2021, 17, e053782. [Google Scholar] [CrossRef]
- Jhamandas, J.H.; Mactavish, D. β-Amyloid protein (Aβ) and human amylin regulation of apoptotic genes occurs through the amylin receptor. Apoptosis 2012, 17, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Solomon, T.; Sahoo, B.R.; Ignasiak, K.; Gaskin, S.; Rowles, J.; Verdile, G.; Howard, M.J.; Bond, C.S.; Ramamoorthy, A.; et al. Amylin and beta amyloid proteins interact to form amorphous heterocomplexes with enhanced toxicity in neuronal cells. Sci. Rep. 2020, 10, 10356. [Google Scholar] [CrossRef]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrPC): Its physiological function and role in disease. Biochim. Biophys. Acta. (BBA.) Mol. Basis. Dis. 2007, 1772, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.; Colello, R.J.; Pott, U.; Oesch, B. Developmental expression of the prion protein gene in glial cells. Neuron 1995, 14, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Zanusso, G.; Liu, D.; Ferrari, S.; Hegyi, I.; Yin, X.; Aguzzi, A.; Hornemann, S.; Liemann, S.; Glockshuber, R.; Manson, J.C.; et al. Prion protein expression in different species: Analysis with a panel of new mAbs. Proc. Natl. Acad. Sci. USA 1998, 95, 8812–8816. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Gatti, J.-L.; Métayer, S.; Moudjou, M.; Andréoletti, O.; Lantier, F.d.r.; Dacheux, J.-L.; Sarradin, P. Prion Protein Is Secreted in Soluble Forms in the Epididymal Fluid and Proteolytically Processed and Transported in Seminal Plasma1. Biol. Reprod. 2002, 67, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Tagliavini, F.; Prelli, F.; Porro, M.; Salmona, M.; Bugiani, O.; Frangione, B. A soluble form of prion protein in human cerebrospinal fluid: Implications for prion-related encephalopathies. Biochem. Biophys. Res. Commun. 1992, 184, 1398–1404. [Google Scholar] [CrossRef]
- Robinson, S.W.; Nugent, M.L.; Dinsdale, D.; Steinert, J.R. Prion protein facilitates synaptic vesicle release by enhancing release probability. Hum. Mol. Genet. 2014, 23, 4581–4596. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Whittington, M.A.; Sidle, K.C.; Smith, C.J.; Palmer, M.S.; Clarke, A.R.; Jefferys, J.G. Prion protein is necessary for normal synaptic function. Nature 1994, 370, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Kanaani, J.; Prusiner, S.B.; Diacovo, J.; Baekkeskov, S.; Legname, G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J. Neurochem. 2005, 95, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Calella, A.M. Prions: Protein Aggregation and Infectious Diseases. Physiol. Rev. 2009, 89, 1105–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Lindquist, S. Conversion of PrP to a Self-Perpetuating PrPSc-like Conformation in the Cytosol. Science 2002, 298, 1785–1788. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Blanco, R.; Carmona, M.; Puig, B.; Ribera, R.; Rey, M.J.; Ribalta, T. Prion protein expression in senile plaques in Alzheimer’s disease. Acta Neuropathol. 2001, 101, 49–56. [Google Scholar] [CrossRef]
- Schwarze-Eicker, K.; Keyvani, K.; Görtz, N.; Westaway, D.; Sachser, N.; Paulus, W. Prion protein (PrPc) promotes β-amyloid plaque formation. Neurobiol. Aging 2005, 26, 1177–1182. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Yokotsuka, M.; Tobiume, M.; Sato, Y.; Hasegawa, H.; Nagao, T.; Gouras, G.K. Accumulation of cellular prion protein within β-amyloid oligomer plaques in aged human brains. Brain. Pathol. 2021, 31, e12941. [Google Scholar] [CrossRef]
- Dohler, F.; Sepulveda-Falla, D.; Krasemann, S.; Altmeppen, H.; Schlüter, H.; Hildebrand, D.; Zerr, I.; Matschke, J.; Glatzel, M. High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer’s disease. Brain 2014, 137, 873–886. [Google Scholar] [CrossRef]
- Falker, C.; Hartmann, A.; Guett, I.; Dohler, F.; Altmeppen, H.; Betzel, C.; Schubert, R.; Thurm, D.; Wegwitz, F.; Joshi, P.; et al. Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity. J. Neurochem. 2016, 137, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Lauren, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory Impairment in Transgenic Alzheimer Mice Requires Cellular Prion Protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, S.V.; Gallardo, C.; Kaufman, A.C.; Herber, C.S.; Haas, L.T.; Robinson, S.; Manson, J.C.; Lee, M.K.; Strittmatter, S.M. Conditional Deletion of Prnp Rescues Behavioral and Synaptic Deficits after Disease Onset in Transgenic Alzheimer’s Disease. J. Neurosci. 2017, 37, 9207–9221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkin, E.T.; Watt, N.T.; Hussain, I.; Eckman, E.A.; Eckman, C.B.; Manson, J.C.; Baybutt, H.N.; Turner, A.J.; Hooper, N.M. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2007, 104, 11062–11067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, H.H.; Whitehouse, I.J.; Baybutt, H.; Brown, D.; Kellett, K.A.; Jackson, C.D.; Turner, A.J.; Piccardo, P.; Manson, J.C.; Hooper, N.M. Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J. Biol. Chem. 2011, 286, 33489–33500. [Google Scholar] [CrossRef] [Green Version]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [Green Version]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Vukojevic, V.; Patel, A.; Soudy, R.; MacTavish, D.; Westaway, D.; Kaur, K.; Goncharuk, V.; Jhamandas, J. Role of microglial amylin receptors in mediating beta amyloid (Abeta)-induced inflammation. J. Neuroinflammation. 2017, 14, 199. [Google Scholar] [CrossRef] [Green Version]
- Bailey, R.J.; Bradley, J.W.; Poyner, D.R.; Rathbone, D.L.; Hay, D.L. Functional characterization of two human receptor activity-modifying protein 3 variants. Peptides 2010, 31, 579–584. [Google Scholar] [CrossRef]
- Carrasquillo, M.M.; Belbin, O.; Hunter, T.A.; Ma, L.; Bisceglio, G.D.; Zou, F.; Crook, J.E.; Pankratz, V.S.; Sando, S.B.; Aasly, J.O.; et al. Replication of EPHA1 and CD33 associations with late-onset Alzheimer’s disease: A multi-centre case-control study. Mol. Neurodegener. 2011, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, L.R.; Pimassoni, L.H.S.; Sena, G.G.S.; Camporez, D.; Belcavello, L.; Trancozo, M.; Morelato, R.L.; Errera, F.I.V.; Bueno, M.R.P.; de Paula, F. Validating GWAS Variants from Microglial Genes Implicated in Alzheimer’s Disease. J. Mol. Neurosci. 2017, 62, 215–221. [Google Scholar] [CrossRef]
- Zhou, L.; Li, H.Y.; Wang, J.H.; Deng, Z.Z.; Shan, Y.L.; Tan, S.; Shi, Y.H.; Zhang, M.X.; Liu, S.X.; Zhang, B.J.; et al. Correlation of gene polymorphisms of CD36 and ApoE with susceptibility of Alzheimer disease: A case-control study. Medicine 2018, 97, e12470. [Google Scholar] [CrossRef]
- Sery, O.; Janoutova, J.; Ewerlingova, L.; Halova, A.; Lochman, J.; Janout, V.; Khan, N.A.; Balcar, V.J. CD36 gene polymorphism is associated with Alzheimer’s disease. Biochimie 2017, 135, 46–53. [Google Scholar] [CrossRef]
- Peng, L.; Yu, Y.; Liu, J.; Li, S.; He, H.; Cheng, N.; Ye, R.D. The chemerin receptor CMKLR1 is a functional receptor for amyloid-beta peptide. J. Alzheimers Dis. 2015, 43, 227–242. [Google Scholar] [CrossRef]
- Brandenburg, L.O.; Konrad, M.; Wruck, C.J.; Koch, T.; Lucius, R.; Pufe, T. Functional and physical interactions between formyl-peptide-receptors and scavenger receptor MARCO and their involvement in amyloid beta 1-42-induced signal transduction in glial cells. J. Neurochem. 2010, 113, 749–760. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, X.; Qian, L.; Chen, S.H.; Zhou, H.; Wilson, B.; Miller, D.S.; Hong, J.S. Microglial MAC1 receptor and PI3K are essential in mediating beta-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J. Neuroinflammation 2011, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Roos, D.; Law, S.K. Hematologically important mutations: Leukocyte adhesion deficiency. Blood Cells Mol. Dis. 2001, 27, 1000–1004. [Google Scholar] [CrossRef]
- Alarcon, R.; Fuenzalida, C.; Santibanez, M.; von Bernhardi, R. Expression of scavenger receptors in glial cells. Comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound beta-amyloid. J. Biol. Chem. 2005, 280, 30406–30415. [Google Scholar] [CrossRef] [PubMed]
- Morkuniene, R.; Cizas, P.; Jankeviciute, S.; Petrolis, R.; Arandarcikaite, O.; Krisciukaitis, A.; Borutaite, V. Small Abeta1-42 oligomer-induced membrane depolarization of neuronal and microglial cells: Role of N-methyl-D-aspartate receptors. J. Neurosci. Res. 2015, 93, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Jia, J. Association between NR2B subunit gene (GRIN2B) promoter polymorphisms and sporadic Alzheimer’s disease in the North Chinese population. Neurosci. Lett. 2009, 450, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.P.; Lin, W.Y.; Liu, S.H.; Wang, W.F.; Tsai, C.H.; Wu, B.T.; Wang, C.K.; Tsai, F.J. Genetic variation in N-methyl-D-aspartate receptor subunit NR3A but not NR3B influences susceptibility to Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2009, 28, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Andreoli, V.; De Marco, E.V.; Trecroci, F.; Cittadella, R.; Di Palma, G.; Gambardella, A. Potential involvement of GRIN2B encoding the NMDA receptor subunit NR2B in the spectrum of Alzheimer’s disease. J. Neural. Transm. 2014, 121, 533–542. [Google Scholar] [CrossRef]
- Stein, J.L.; Hua, X.; Morra, J.H.; Lee, S.; Hibar, D.P.; Ho, A.J.; Leow, A.D.; Toga, A.W.; Sul, J.H.; Kang, H.M.; et al. Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer’s disease. Neuroimage 2010, 51, 542–554. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Li, X.; Wang, T.; Wang, H.H.; Fu, Y.; Zhang, L.; Xiao, S.F. Association between NMDA receptor subunit 2b gene polymorphism and Alzheimer’s disease in Chinese Han population in Shanghai. Neurosci. Bull. 2010, 26, 395–400. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.J.; Liu, H.C.; Liu, T.Y.; Cheng, C.Y.; Hong, C.J. Association analysis for the genetic variants of the NMDA receptor subunit 2b and Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2002, 13, 91–94. [Google Scholar] [CrossRef]
- Ozawa, D.; Nakamura, T.; Koike, M.; Hirano, K.; Miki, Y.; Beppu, M. Shuttling Protein Nucleolin Is a Microglia Receptor for Amyloid Beta Peptide 1-42. Biol. Pharm. Bull. 2013, 36, 1587–1593. [Google Scholar] [CrossRef] [Green Version]
- Lue, L.F.; Walker, D.G.; Brachova, L.; Beach, T.G.; Rogers, J.; Schmidt, A.M.; Stern, D.M.; Yan, S.D. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: Identification of a cellular activation mechanism. Exp. Neurol. 2001, 171, 29–45. [Google Scholar] [CrossRef]
- Li, K.; Dai, D.; Zhao, B.; Yao, L.; Yao, S.; Wang, B.; Yang, Z. Association between the RAGE G82S polymorphism and Alzheimer’s disease. J. Neural. Transm. 2010, 117, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Daborg, J.; von Otter, M.; Sjolander, A.; Nilsson, S.; Minthon, L.; Gustafson, D.R.; Skoog, I.; Blennow, K.; Zetterberg, H. Association of the RAGE G82S polymorphism with Alzheimer’s disease. J. Neural. Transm. 2010, 117, 861–867. [Google Scholar] [CrossRef] [Green Version]
- Atac, Z.S.; Alaylioglu, M.; Dursun, E.; Gezen-Ak, D.; Yilmazer, S.; Gurvit, H. G82S polymorphism of receptor for advanced glycation end products gene and serum soluble RAGE levels in mild cognitive impairment and dementia of Alzheimer’s type patients in Turkish population. J. Clin. Neurosci. 2019, 59, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Pericak-Vance, M.A.; Bass, M.P.; Yamaoka, L.H.; Gaskell, P.C.; Scott, W.K.; Terwedow, H.A.; Menold, M.M.; Conneally, P.M.; Small, G.W.; Vance, J.M.; et al. Complete genomic screen in late-onset familial Alzheimer disease. Evidence for a new locus on chromosome 12. JAMA 1997, 278, 1237–1241. [Google Scholar] [CrossRef] [PubMed]
- Luedecking-Zimmer, E.; DeKosky, S.T.; Nebes, R.; Kamboh, M.I. Association of the 3′ UTR transcription factor LBP-1c/CP2/LSF polymorphism with late-onset Alzheimer’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 117B, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Vergeer, M.; Korporaal, S.J.; Franssen, R.; Meurs, I.; Out, R.; Hovingh, G.K.; Hoekstra, M.; Sierts, J.A.; Dallinga-Thie, G.M.; Motazacker, M.M.; et al. Genetic variant of the scavenger receptor BI in humans. N. Engl. J. Med. 2011, 364, 136–145. [Google Scholar] [CrossRef]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Ohya, W.; Funakoshi, H.; Sakaguchi, G.; Kato, A.; Takeda, M.; Kudo, T.; Nakamura, T. Possible role of scavenger receptor SRCL in the clearance of amyloid-beta in Alzheimer’s disease. J. Neurosci. Res. 2006, 84, 874–890. [Google Scholar] [CrossRef]
- Fassbender, K.; Walter, S.; Kuhl, S.; Landmann, R.; Ishii, K.; Bertsch, T.; Stalder, A.K.; Muehlhauser, F.; Liu, Y.; Ulmer, A.J.; et al. The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J. 2004, 18, 203–205. [Google Scholar] [CrossRef]
- Hamann, L.; Bustami, J.; Iakoubov, L.; Szwed, M.; Mossakowska, M.; Schumann, R.R.; Puzianowska-Kuznicka, M. TLR-6 SNP P249S is associated with healthy aging in nonsmoking Eastern European Caucasians—A cohort study. Immun. Ageing 2016, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, X.; Deng, X.; Ma, Y.; Huang, S.; Wang, Y. Association of CD14-260 (-159) C/T and Alzheimer’s disease: Systematic review and trial sequential analyses. J. Neural. Transm. 2018, 125, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and Toll-Like Receptors 2 and 4 Are Required for Fibrillar A beta-Stimulated Microglial Activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Zhang, Y.D.; Gao, Q.; Zhou, J.S.; Zhu, X.C.; Lu, H.; Shi, J.Q.; Tan, L.; Chen, Q.; Yu, J.T. TREM1 facilitates microglial phagocytosis of amyloid beta. Acta Neuropathol. 2016, 132, 667–683. [Google Scholar] [CrossRef]
- Zhong, L.; Wang, Z.; Wang, D.; Wang, Z.; Martens, Y.A.; Wu, L.; Xu, Y.; Wang, K.; Li, J.; Huang, R.; et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 2018, 13, 15. [Google Scholar] [CrossRef]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccio, L.; Deming, Y.; Del-Aguila, J.L.; Ghezzi, L.; Holtzman, D.M.; Fagan, A.M.; Fenoglio, C.; Galimberti, D.; Borroni, B.; Cruchaga, C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016, 131, 925–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Lupton, M.K.; et al. TREM2 Variants in Alzheimer’s Disease. New Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Slattery, C.F.; Beck, J.A.; Harper, L.; Adamson, G.; Abdi, Z.; Uphill, J.; Campbell, T.; Druyeh, R.; Mahoney, C.J.; Rohrer, J.D.; et al. R47H TREM2 variant increases risk of typical early-onset Alzheimer’s disease but not of prion or frontotemporal dementia. Alzheimers Dement. 2014, 10, 602–608. [Google Scholar] [CrossRef] [Green Version]
- Kleinberger, G.; Yamanishi, Y.; Suarez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra286. [Google Scholar] [CrossRef]
- Benitez, B.A.; Cooper, B.; Pastor, P.; Jin, S.C.; Lorenzo, E.; Cervantes, S.; Cruchaga, C. TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol. Aging 2013, 34. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Hou, J.K.; Gao, Q.; Yu, J.T.; Zhou, J.S.; Zhao, H.D.; Zhang, Y.D. TREM2 p.H157Y Variant and the Risk of Alzheimer’s Disease: A Meta-Analysis Involving 14,510 Subjects. Curr. Neurovasc. Res. 2016, 13, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, X.; He, P.F. The most prevalent rare coding variants of TREM2 conferring risk of Alzheimer’s disease: A systematic review and meta-analysis. Exp. Med. 2021, 21. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht, J.; Landreth, G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J. Neurosci. 2004, 24, 9838–9846. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-Like Receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Kielian, T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J. Neurosci. Res. 2006, 83, 711–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Jin, J.; Lim, J.E.; Kou, J.; Pattanayak, A.; Rehman, J.A.; Kim, H.D.; Tahara, K.; Lalonde, R.; Fukuchi, K. TLR4 mutation reduces microglial activation, increases Abeta deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflammation 2011, 8, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schüffer, W.; et al. Role of the Toll-Like Receptor 4 in Neuroinflammation in Alzheimer’s Disease. Cell. Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef]
- Jin, J.J.; Kim, H.D.; Maxwell, J.A.; Li, L.; Fukuchi, K. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J. Neuroinflammation 2008, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.L.; Hennessy, E.; Rubio-Araiz, A.; Keogh, B.; McCormack, W.; McGuirk, P.; Reilly, M.; Lynch, M.A. Inhibiting TLR2 activation attenuates amyloid accumulation and glial activation in a mouse model of Alzheimer’s disease. Brain Behav. Immun. 2016, 58, 191–200. [Google Scholar] [CrossRef]
- Glatz, J.C.; Luiken, J.F.P. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid. Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Ioghen, O.; Chitoiu, L.; Gherghiceanu, M.; Ceafalan, L.C.; Hinescu, M.E. CD36—A novel molecular target in the neurovascular unit. Eur. J. Neurosci. 2021, 53, 2500–2510. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A Cell Surface Receptor Complex for Fibrillar β-Amyloid Mediates Microglial Activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Ricciarelli, R.; D’Abramo, C.; Zingg, J.M.; Giliberto, L.; Markesbery, W.; Azzi, A.; Marinari, U.M.; Pronzato, M.A.; Tabaton, M. CD36 overexpression in human brain correlates with beta-amyloid deposition but not with Alzheimer’s disease. Free Radic. Biol. Med. 2004, 36, 1018–1024. [Google Scholar] [CrossRef]
- Egana-Gorrono, L.; Lopez-Diez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights From Human Subjects and Animal Models. Front. Cardiovasc. Med. 2020, 7, 37. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Vianna, M.; Gerlach, M.; Brett, J.; Ryan, J.; Kao, J.; Esposito, C.; Hegarty, H.; Hurley, W.; Clauss, M. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J. Biol. Chem. 1992, 267, 14987–14997. [Google Scholar] [CrossRef]
- Schlueter, C.; Hauke, S.; Flohr, A.M.; Rogalla, P.; Bullerdiek, J. Tissue-specific expression patterns of the RAGE receptor and its soluble forms—A result of regulated alternative splicing? Biochim. Biophys. Acta 2003, 1630, 1–6. [Google Scholar] [CrossRef]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and their role in Cancer and Inflammation. J. Transl. Med. 2009, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Chellappa, R.C.; Palanisamy, R.; Swaminathan, K. RAGE Isoforms, its Ligands and their Role in Pathophysiology of Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 1262–1279. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, F.; Yu, Q.; Arancio, O.; Chen, D.; Gore, S.S.; Yan, S.S.; Yan, S.F. RAGE mediates Abeta accumulation in a mouse model of Alzheimer’s disease via modulation of beta- and gamma-secretase activity. Hum. Mol. Genet. 2018, 27, 1002–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedini, A.; Cao, P.; Plesner, A.; Zhang, J.; He, M.; Derk, J.; Patil, S.A.; Rosario, R.; Lonier, J.; Song, F.; et al. RAGE binds preamyloid IAPP intermediates and mediates pancreatic β cell proteotoxicity. J. Clin. Investig. 2018, 128, 682–698. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Lue, L.F.; Yan, S.; Xu, H.; Luddy, J.S.; Chen, D.; Walker, D.G.; Stern, D.M.; Yan, S.; Schmidt, A.M.; et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010, 24, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.L.; Chen, H.; Filie, J.D.; Kozak, C.A.; Murphy, P.M. Differential expansion of the N-formylpeptide receptor gene cluster in human and mouse. Genomics 1998, 51, 270–276. [Google Scholar] [CrossRef]
- Migeotte, I.; Communi, D.; Parmentier, M. Formyl peptide receptors: A promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006, 17, 501–519. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Ammendola, R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem. Res. 2010, 35, 2018–2026. [Google Scholar] [CrossRef]
- Bloes, D.A.; Kretschmer, D.; Peschel, A. Enemy attraction: Bacterial agonists for leukocyte chemotaxis receptors. Nat. Rev. Microbiol. 2015, 13, 95–104. [Google Scholar] [CrossRef]
- Dahlgren, C.; Gabl, M.; Holdfeldt, A.; Winther, M.; Forsman, H. Basic characteristics of the neutrophil receptors that recognize formylated peptides, a danger-associated molecular pattern generated by bacteria and mitochondria. Biochem. Pharmacol. 2016, 114, 22–39. [Google Scholar] [CrossRef]
- Brandenburg, L.-O.; Koch, T.; Sievers, J.; Lucius, R. Internalization of PrP106–126 by the formyl-peptide-receptor-like-1 in glial cells. J. Neurochem. 2007, 101, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gong, H.; Ge, Y.; Ye, R.D. Biased allosteric modulation of formyl peptide receptor 2 leads to distinct receptor conformational states for pro- and anti-inflammatory signaling. Pharm. Res. 2020, 161, 105117. [Google Scholar] [CrossRef] [PubMed]
- Schröder, N.; Schaffrath, A.; Welter, J.A.; Putzka, T.; Griep, A.; Ziegler, P.; Brandt, E.; Samer, S.; Heneka, M.T.; Kaddatz, H.; et al. Inhibition of formyl peptide receptors improves the outcome in a mouse model of Alzheimer disease. J. Neuroinflammation 2020, 17, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arredouani, M.S.; Palecanda, A.; Koziel, H.; Huang, Y.C.; Imrich, A.; Sulahian, T.H.; Ning, Y.Y.; Yang, Z.; Pikkarainen, T.; Sankala, M.; et al. MARCO is the major binding receptor for unopsonized particles and bacteria on human alveolar macrophages. J. Immunol. 2005, 175, 6058–6064. [Google Scholar] [CrossRef] [Green Version]
- Van der Laan, L.J.W.; Döpp, E.A.; Haworth, R.; Pikkarainen, T.; Kangas, M.; Elomaa, O.; Dijkstra, C.D.; Gordon, S.; Tryggvason, K.; Kraal, G. Regulation and Functional Involvement of Macrophage Scavenger Receptor MARCO in Clearance of Bacteria In Vivo. J. Immunol. 1999, 162, 939. [Google Scholar]
- Thelen, T.; Hao, Y.; Medeiros, A.I.; Curtis, J.L.; Serezani, C.H.; Kobzik, L.; Harris, L.H.; Aronoff, D.M. The class A scavenger receptor, macrophage receptor with collagenous structure, is the major phagocytic receptor for Clostridium sordellii expressed by human decidual macrophages. J. Immunol. 2010, 185, 4328–4335. [Google Scholar] [CrossRef] [Green Version]
- Sankala, M.; Brannstrom, A.; Schulthess, T.; Bergmann, U.; Morgunova, E.; Engel, J.; Tryggvason, K.; Pikkarainen, T. Characterization of recombinant soluble macrophage scavenger receptor MARCO. J. Biol. Chem. 2002, 277, 33378–33385. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Varin, A.; Chen, Y.; Liu, B.; Tryggvason, K.; Gordon, S. SR-A/MARCO-mediated ligand delivery enhances intracellular TLR and NLR function, but ligand scavenging from cell surface limits TLR4 response to pathogens. Blood 2011, 117, 1319–1328. [Google Scholar] [CrossRef]
- Yun, H.; Dumbell, R.; Hanna, K.; Bowen, J.; McLean, S.L.; Kantamneni, S.; Pors, K.; Wu, Q.F.; Helfer, G. The Chemerin-CMKLR1 Axis is Functionally important for Central Regulation of Energy Homeostasis. Front. Physiol. 2022, 13, 897105. [Google Scholar] [CrossRef]
- Arita, M.; Ohira, T.; Sun, Y.P.; Elangovan, S.; Chiang, N.; Serhan, C.N. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 2007, 178, 3912–3917. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lin, A.; Gong, P.; Chen, Y.; Ye, R.D.; Qian, F.; Zhang, Y.; Yu, Y. The Chemokine-like Receptor 1 Deficiency Improves Cognitive Deficits of AD Mice and Attenuates Tau Hyperphosphorylation via Regulating Tau Seeding. J. Neurosci. 2020, 40, 6991–7007. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.L.; Simonato, M.; Spolaore, B.; Franchin, C.; Arrigoni, G.; Marin, O.; Monturiol-Gross, L.; Fernandez, J.; Lomonte, B.; Tonello, F. Cell surface nucleolin interacts with and internalizes Bothrops asper Lys49 phospholipase A2 and mediates its toxic activity. Sci. Rep. 2018, 8, 10619. [Google Scholar] [CrossRef] [PubMed]
- Tajrishi, M.M.; Tuteja, R.; Tuteja, N. Nucleolin: The most abundant multifunctional phosphoprotein of nucleolus. Commun. Integr. Biol. 2011, 4, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, S.H.E.; Malter, J.S. Nucleolin and Heterogeneous Nuclear Ribonucleoprotein C Proteins Specifically Interact with the 3′-Untranslated Region of Amyloid Protein Precursor mRNA. J. Biol. Chem. 1995, 270, 17292–17298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westmark, C.J.; Malter, J.S. The regulation of AβPP expression by RNA-binding proteins. Ageing Res. Rev. 2012, 11, 450–459. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, L.E.; Westmark, C.J.; Jarzembowski, J.A.; Malter, J.S. hnRNP C increases amyloid precursor protein (APP) production by stabilizing APP mRNA. Nucleic Acids Res. 1998, 26, 3418–3423. [Google Scholar] [CrossRef] [Green Version]
- Belrose, J.C.; Jackson, M.F. TRPM2: A candidate therapeutic target for treating neurological diseases. Acta Pharm. Sin. 2018, 39, 722–732. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.-H.; McNaughton, P.A. The TRPM2 ion channel is required for sensitivity to warmth. Nature 2016, 536, 460–463. [Google Scholar] [CrossRef] [Green Version]
- Malko, P.; Syed Mortadza, S.A.; McWilliam, J.; Jiang, L.H. TRPM2 Channel in Microglia as a New Player in Neuroinflammation Associated With a Spectrum of Central Nervous System Pathologies. Front. Pharm. 2019, 10, 239. [Google Scholar] [CrossRef] [Green Version]
- Fonfria, E.; Marshall, I.C.; Benham, C.D.; Boyfield, I.; Brown, J.D.; Hill, K.; Hughes, J.P.; Skaper, S.D.; McNulty, S. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br. J. Pharm. 2004, 143, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Aminzadeh, M.; Roghani, M.; Sarfallah, A.; Riazi, G.H. TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer’s disease. Int. Immunopharmacol. 2018, 54, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Alawieyah Syed Mortadza, S.; Sim, J.A.; Neubrand, V.E.; Jiang, L.H. A critical role of TRPM2 channel in Abeta42 -induced microglial activation and generation of tumor necrosis factor-alpha. Glia 2018, 66, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Ostapchenko, V.G.; Chen, M.; Guzman, M.S.; Xie, Y.F.; Lavine, N.; Fan, J.; Beraldo, F.H.; Martyn, A.C.; Belrose, J.C.; Mori, Y.; et al. The Transient Receptor Potential Melastatin 2 (TRPM2) Channel Contributes to beta-Amyloid Oligomer-Related Neurotoxicity and Memory Impairment. J. Neurosci. 2015, 35, 15157–15169. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Yu, J.T.; Hu, N.; Tan, M.S.; Zhu, X.C.; Tan, L. CD33 in Alzheimer’s disease. Mol. Neurobiol. 2014, 49, 529–535. [Google Scholar] [CrossRef]
- Zhao, L. CD33 in Alzheimer’s Disease—Biology, Pathogenesis, and Therapeutics: A Mini-Review. Gerontology 2019, 65, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Wißfeld, J.; Nozaki, I.; Mathews, M.; Raschka, T.; Ebeling, C.; Hornung, V.; Brüstle, O.; Neumann, H. Deletion of Alzheimer’s disease-associated CD33 results in an inflammatory human microglia phenotype. Glia 2021, 69, 1393–1412. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Federico, A.N.; Natasan, J.; Forte, A.M.; McGinty, D.; Nguyen, H.; Volak, A.; LeRoy, S.; Gandhi, S.; Lerner, E.P.; et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet. 2020, 29, 2920–2935. [Google Scholar] [CrossRef]
- Peress, N.S.; Fleit, H.B.; Perillo, E.; Kuljis, R.; Pezzullo, C. Identification of Fc gamma RI, II and III on normal human brain ramified microglia and on microglia in senile plaques in Alzheimer’s disease. J. Neuroimmunol. 1993, 48, 71–79. [Google Scholar] [CrossRef]
- Janus, C.; Pearson, J.; McLaurin, J.; Mathews, P.M.; Jiang, Y.; Schmidt, S.D.; Chishti, M.A.; Horne, P.; Heslin, D.; French, J.; et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 2000, 408, 979–982. [Google Scholar] [CrossRef]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söllvander, S.; Ekholm-Pettersson, F.; Brundin, R.M.; Westman, G.; Kilander, L.; Paulie, S.; Lannfelt, L.; Sehlin, D. Increased Number of Plasma B Cells Producing Autoantibodies Against Aβ42 Protofibrils in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Bulati, M.; Buffa, S.; Martorana, A.; Gervasi, F.; Camarda, C.; Azzarello, D.M.; Monastero, R.; Caruso, C.; Colonna-Romano, G. Double negative (IgG+IgD-CD27-) B cells are increased in a cohort of moderate-severe Alzheimer’s disease patients and show a pro-inflammatory trafficking receptor phenotype. J. Alzheimers Dis. 2015, 44, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Bacskai, B.J.; Kajdasz, S.T.; McLellan, M.E.; Games, D.; Seubert, P.; Schenk, D.; Hyman, B.T. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J. Neurosci. 2002, 22, 7873–7878. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Escott-Price, V. Genes, pathways and risk prediction in Alzheimer’s disease. Hum. Mol. Genet. 2019, 28, R235–R240. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Geng, R.; Li, C.; Meng, F.; Zhao, H.; Liu, J.; Dai, J.; Wang, X. Dysregulated gene-associated biomarkers for Alzheimer’s disease and aging. Transl. Neurosci. 2021, 12, 83–95. [Google Scholar] [CrossRef]
- Feuerbach, D.; Schindler, P.; Barske, C.; Joller, S.; Beng-Louka, E.; Worringer, K.A.; Kommineni, S.; Kaykas, A.; Ho, D.J.; Ye, C.; et al. ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157. Neurosci. Lett. 2017, 660, 109–114. [Google Scholar] [CrossRef]
- Schlepckow, K.; Kleinberger, G.; Fukumori, A.; Feederle, R.; Lichtenthaler, S.F.; Steiner, H.; Haass, C. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO. Mol. Med. 2017, 9, 1356–1365. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F.; Tschirner, S.K.; Steiner, H. Secretases in Alzheimer’s disease: Novel insights into proteolysis of APP and TREM2. Curr. Opin. Neurobiol. 2022, 72, 101–110. [Google Scholar] [CrossRef]
- Zheng, H.; Jia, L.; Liu, C.C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.F.; Fryer, J.D.; Wang, X.; Zhang, Y.W.; et al. TREM2 Promotes Microglial Survival by Activating Wnt/beta-Catenin Pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Chen, X.F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzmeier, N.; Suarez-Calvet, M.; Frontzkowski, L.; Moore, A.; Hohman, T.J.; Morenas-Rodriguez, E.; Nuscher, B.; Shaw, L.; Trojanowski, J.Q.; Dichgans, M.; et al. Higher CSF sTREM2 attenuates ApoE4-related risk for cognitive decline and neurodegeneration. Mol. Neurodegener. 2020, 15, 57. [Google Scholar] [CrossRef] [PubMed]
- Thornton, P.; Sevalle, J.; Deery, M.J.; Fraser, G.; Zhou, Y.; Stahl, S.; Franssen, E.H.; Dodd, R.B.; Qamar, S.; Gomez Perez-Nievas, B.; et al. TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol. Med. 2017, 9, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Shen, C.; Liu, Y.H.; Li, J.; Zhu, J.; Wang, Y.R.; Yan, J.C.; Gao, C.Y.; Zhou, H.D.; Deng, J.; et al. Genetic Association Between APP, ADAM10 Gene Polymorphism, and Sporadic Alzheimer’s Disease in the Chinese Population. Neurotox. Res. 2015, 27, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.H.; Chen, W.; Jiang, L.Y.; Yang, Y.X.; Yao, L.F.; Li, K.S. Influence of ADAM10 Polymorphisms on Plasma Level of Soluble Receptor for Advanced Glycation End Products and The Association With Alzheimer’s Disease Risk. Front. Genet. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criscuolo, C.; Fontebasso, V.; Middei, S.; Stazi, M.; Ammassari-Teule, M.; Yan, S.S.; Origlia, N. Entorhinal Cortex dysfunction can be rescued by inhibition of microglial RAGE in an Alzheimer’s disease mouse model. Sci. Rep. 2017, 7, 42370. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Bukulin, M.; Kojro, E.; Roth, A.; Metz, V.V.; Fahrenholz, F.; Nawroth, P.P.; Bierhaus, A.; Postina, R. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J. Biol. Chem. 2008, 283, 35507–35516. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Kim, J.Y.; Kang, S.M.; Kim, J.S.; Chae, J.S.; Kim, O.Y.; Koh, S.J.; Lee, H.C.; Ahn, C.W.; Song, Y.D.; et al. Association of the Gly82Ser polymorphism in the receptor for advanced glycation end products (RAGE) gene with circulating levels of soluble RAGE and inflammatory markers in nondiabetic and nonobese Koreans. Metabolism 2007, 56, 199–205. [Google Scholar] [CrossRef]
- Cohn, W.; Melnik, M.; Huang, C.; Teter, B.; Chandra, S.; Zhu, C.; McIntire, L.B.; John, V.; Gylys, K.H.; Bilousova, T. Multi-Omics Analysis of Microglial Extracellular Vesicles From Human Alzheimer’s Disease Brain Tissue Reveals Disease-Associated Signatures. Front. Pharm. 2021, 12, 766082. [Google Scholar] [CrossRef]
- Yang, H.; Wang, Y.; Kar, S. Effects of cholesterol transport inhibitor U18666A on APP metabolism in rat primary astrocytes. Glia. 2017, 65, 1728–1743. [Google Scholar] [CrossRef]
- Stavropoulou, A.V.; Mavrofrydi, O.; Saftig, P.; Efthimiopoulos, S. Serum Starvation Induces BACE1 Processing and Secretion. Curr. Alzheimer Res. 2017, 14, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.A.; Lin, K.; van Lengerich, B.; Lianoglou, S.; Przybyla, L.; Davis, S.S.; Llapashtica, C.; Wang, J.; Kim, D.J.; Xia, D.; et al. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 2020, 105, 837–854 e839. [Google Scholar] [CrossRef] [PubMed]
- Erten-Lyons, D.; Woltjer, R.L.; Dodge, H.; Nixon, R.; Vorobik, R.; Calvert, J.F.; Leahy, M.; Montine, T.; Kaye, J. Factors associated with resistance to dementia despite high Alzheimer disease pathology. Neurology 2009, 72, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Sloane, J.A.; Pietropaolo, M.F.; Rosene, D.L.; Moss, M.B.; Peters, A.; Kemper, T.; Abraham, C.R. Lack of correlation between plaque burden and cognition in the aged monkey. Acta. Neuropathol. 1997, 94, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Aisen, P.; Boeve, B.F.; Geda, Y.E.; Ivnik, R.J.; Knopman, D.S.; Mielke, M.; Pankratz, V.S.; Roberts, R.; Rocca, W.A.; et al. Mild cognitive impairment due to Alzheimer disease in the community. Ann. Neurol. 2013, 74, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Klyubin, I.; Betts, V.; Welzel, A.T.; Blennow, K.; Zetterberg, H.; Wallin, A.; Lemere, C.A.; Cullen, W.K.; Peng, Y.; Wisniewski, T.; et al. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: Prevention by systemic passive immunization. J. Neurosci. 2008, 28, 4231–4237. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Tomiyama, T.; Shimada, H. APP Osaka Mutation in Familial Alzheimer’s Disease-Its Discovery, Phenotypes, and Mechanism of Recessive Inheritance. Int. J. Mol. Sci. 2020, 21, 1413. [Google Scholar] [CrossRef] [Green Version]
- Faucher, P.; Mons, N.; Micheau, J.; Louis, C.; Beracochea, D.J. Hippocampal Injections of Oligomeric Amyloid β-peptide (1–42) Induce Selective Working Memory Deficits and Long-lasting Alterations of ERK Signaling Pathway. Front. Aging Neurosci. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Piller, C. Blots on a field? Science 2022, 377, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Brinkmalm, G.; Hong, W.; Wang, Z.; Liu, W.; O’Malley, T.T.; Sun, X.; Frosch, M.P.; Selkoe, D.J.; Portelius, E.; Zetterberg, H.; et al. Identification of neurotoxic cross-linked amyloid-β dimers in the Alzheimer’s brain. Brain 2019, 142, 1441–1457. [Google Scholar] [CrossRef]
- Winkler, J.; Connor, D.J.; Frautschy, S.A.; Behl, C.; Waite, J.J.; Cole, G.M.; Thal, L.J. Lack of long-term effects after β-amyloid protein injections in rat brain. Neurobiol. Aging 1994, 15, 601–607. [Google Scholar] [CrossRef]
- Clemens, J.A.; Stephenson, D.T. Implants containing β-amyloid protein are not neurotoxic to young and old rat brain. Neurobiol. Aging 1992, 13, 581–586. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Dahlgren, K.N.; Manelli, A.M.; Stine, W.B., Jr.; Baker, L.K.; Krafft, G.A.; LaDu, M.J. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 2002, 277, 32046–32053. [Google Scholar] [CrossRef] [Green Version]
- Sgourakis, N.G.; Yan, Y.; McCallum, S.A.; Wang, C.; Garcia, A.E. The Alzheimer’s peptides Abeta40 and 42 adopt distinct conformations in water: A combined MD/NMR study. J. Mol. Biol. 2007, 368, 1448–1457. [Google Scholar] [CrossRef] [Green Version]
- Vivekanandan, S.; Brender, J.R.; Lee, S.Y.; Ramamoorthy, A. A partially folded structure of amyloid-beta(1-40) in an aqueous environment. Biochem. Biophys. Res. Commun. 2011, 411, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Balbach, J.J.; Ishii, Y.; Antzutkin, O.N.; Leapman, R.D.; Rizzo, N.W.; Dyda, F.; Reed, J.; Tycko, R. Amyloid fibril formation by A beta 16-22, a seven-residue fragment of the Alzheimer’s beta-amyloid peptide, and structural characterization by solid state NMR. Biochemistry 2000, 39, 13748–13759. [Google Scholar] [CrossRef] [PubMed]
- Jahn, T.R.; Makin, O.S.; Morris, K.L.; Marshall, K.E.; Tian, P.; Sikorski, P.; Serpell, L.C. The common architecture of cross-beta amyloid. J. Mol. Biol. 2010, 395, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ø.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Faller, P.; Hureau, C. Reproducibility Problems of Amyloid-β Self-Assembly and How to Deal With Them. Front. Chem. 2021, 8, 611227. [Google Scholar] [CrossRef]
- Kheterpal, I.; Lashuel, H.A.; Hartley, D.M.; Walz, T.; Lansbury, P.T., Jr.; Wetzel, R. Abeta protofibrils possess a stable core structure resistant to hydrogen exchange. Biochemistry 2003, 42, 14092–14098. [Google Scholar] [CrossRef] [Green Version]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell. Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Armen, R.S.; DeMarco, M.L.; Alonso, D.O.; Daggett, V. Pauling and Corey’s alpha-pleated sheet structure may define the prefibrillar amyloidogenic intermediate in amyloid disease. Proc. Natl. Acad. Sci. USA 2004, 101, 11622–11627. [Google Scholar] [CrossRef] [Green Version]
- Shea, D.; Hsu, C.-C.; Bi, T.M.; Paranjapye, N.; Childers, M.C.; Cochran, J.; Tomberlin, C.P.; Wang, L.; Paris, D.; Zonderman, J.; et al. α-Sheet secondary structure in amyloid β-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 8895–8900. [Google Scholar] [CrossRef] [Green Version]
- Bi, T.M.; Daggett, V. The Role of α-sheet in Amyloid Oligomer Aggregation and Toxicity. Yale J. Biol. Med. 2018, 91, 247–255. [Google Scholar]
- Kasim, J.K.; Kavianinia, I.; Harris, P.W.R.; Brimble, M.A. Three Decades of Amyloid Beta Synthesis: Challenges and Advances. Front. Chem. 2019, 7, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrow, C.J.; Yasuda, A.; Kenny, P.T.; Zagorski, M.G. Solution conformations and aggregational properties of synthetic amyloid beta-peptides of Alzheimer’s disease. Analysis of circular dichroism spectra. J. Mol. Biol. 1992, 225, 1075–1093. [Google Scholar] [CrossRef]
- Jao, S.-C.; Ma, K.; Talafous, J.; Orlando, R.; Zagorski, M.G. Trifluoroacetic acid pretreatment reproducibly disaggregates the amyloid β-peptide. Amyloid 1997, 4, 240–252. [Google Scholar] [CrossRef]
- Foley, A.R.; Raskatov, J.A. Assessing Reproducibility in Amyloid β Research: Impact of Aβ Sources on Experimental Outcomes. ChemBioChem 2020, 21, 2425–2430. [Google Scholar] [CrossRef] [PubMed]
- Stine, W.B.; Jungbauer, L.; Yu, C.; LaDu, M.J. Preparing synthetic Aβ in different aggregation states. Methods Mol. Biol. 2011, 670, 13–32. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.M.; Caine, J.; Mertens, H.D.; Kirby, N.; Nigro, J.; Breheney, K.; Waddington, L.J.; Streltsov, V.A.; Curtain, C.; Masters, C.L.; et al. Ammonium hydroxide treatment of Aβ produces an aggregate free solution suitable for biophysical and cell culture characterization. PeerJ 2013, 1, e73. [Google Scholar] [CrossRef] [Green Version]
- Cruz, L.; Urbanc, B.; Borreguero, J.M.; Lazo, N.D.; Teplow, D.B.; Stanley, H.E. Solvent and mutation effects on the nucleation of amyloid β-protein folding. Proc. Natl. Acad. Sci. USA 2005, 102, 18258–18263. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Shea, J.-E. Effects of Solvent on the Structure of the Alzheimer Amyloid-β(25–35) Peptide. Biophys. J. 2006, 91, 1638–1647. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]




| Receptor | Alternative Receptor Names | Binding to Aβ Demonstrated in | Ref. | Function | Polymorphisms/ Mutations Associated with AD | Ref. |
|---|---|---|---|---|---|---|
| Amylin receptor | calcitonin receptor and receptor activity modifying protein 3 | human fetal microglial (HFM) cultures and BV-2, oligomeric soluble Aβ1-42 | [310] | Increase IL-6 secretion | No association with AD described Cys40Trp/Phe100Ser/Leu147Pro variant associated with reduced Amylin potency * | [311] |
| CD33 | SIGLEC3 | No direct binding | - | Blockade of TREM2 | rs3865444C | [312,313,314,315] |
| CD36 | Platelet glycoprotein 4 Fatty acid translocase (FAT) Glycoprotein IIIb (GPIIIB) PAS IV | Primary murine microglia, fibrillary Aβ 1-42 | [235] | Increase ROS production and Aβ phagocytosis | rs7755 rs3211956 rs3211892 | [316] [317] |
| CMKLR1 (Chemokine-like receptor 1) | Chemerin-like receptor 1 1 G-protein-coupled receptor ChemR23 G-protein-coupled receptor DEZ | stably transfected rat basophilic leukemia (RBL) cells, primary microglia, N9 cells, Aβ-1-42 | [318] | Activation of G proteins and β-arrestin pathways | No association with AD described | - |
| FPR1/2 (fMet-Leu-Phe receptors 1/2) | N-formyl peptide receptor (FPR) N-formylpeptide chemoattractant receptor | Rat primary astrocytes and microglia, | [319] | Intracellular calcium mobilization, cell migration and superoxide anion release | No association with AD described | - |
| human Aβ1–42 | [113] | |||||
| transfected HEK293 cells and glial U87 cells, Aβ42 and N-truncated forms | ||||||
| MAC1 (Macrophage antigen complex 1) | integrin CD11b/CD18 receptor CR3 | Primary microglia enriched culture, mice, Aβ-42 | [320] | Adhesive interactions mediation of the uptake of complement-coated particles and pathogens | No association with AD described | |
| [321] | ||||||
| Various polymorphisms/mutations in CD18 causing leukocyte adhesion deficiency, e.g., rs552407409 * | ||||||
| MARCO (Macrophage receptor with collagenous structure) | SCARA2 | Microglia from neonatal rats, fibrillary and non-fibrillary Aβ-42 | [319,322] | Inflammatory response | No association with AD described | - |
| NMDA-R (NMDA receptor) | glial cells in rat cerebellar granule cell cultures, small Aβ1-42 oligomers | [323] | Decrease in plasma membrane potential | -421C/A in sporadic AD, North Han Chinese populations | [324] [325] | |
| 3723 G/A (rs3739722), Taiwanese population | [326] | |||||
| rs1806201 T, Southern Italy population | [327] | |||||
| rs10845840, US populations | [328] | |||||
| C2664T, Chinese Han population | not found in [329] | |||||
| Nucleolin | Protein C23 | EOC2 cells, transfected HEK cells, monomeric and fibrillary Aβ1-42 | [330] | Chromatin decondensation, pre-rRNA transcription, and ribosome assembly | No association with AD described | - |
| RAGE (Receptor for advanced glycosylation end products) | - | Human primary microglia, soluble Aβ and plaques | [331] | instigates pro-inflammatory mediators | G82S | [332,333,334] |
| cAI (Scavenger receptor type AI) | Macrophage scavenger receptor types I and II Macrophage acetylated LDL receptor I and II CD204 | human fetal microglia, microglia from newborn mice, fibrillary aβ | [233] | Phagocytosis of soluble and fibrillar Aβ | No association with AD described | - |
| SRBI (Scavenger receptor class B member 1) | CD36 and LIMPII analogous 1 (CLA-1) CD36 antigen-like 1 Collagen type I receptor, thrombospondin receptor-like 1 | human fetal microglia, microglia from newborn mice, fibrillary aβ | [233] | Decreases amyloid fibrillar and plaque formation | Gene is included in a region on chromosome 12 with linkage to AD [335], polymorphisms were not found associated | [336] |
| rs387906791, rs74830677, impact on cholesterol metabolism * | [337] [338] | |||||
| SRCL (scavenger receptor with C-type lectin) | Collectin-12 Collectin placenta protein 1 (CL-P1; hCL-P1) Nurse cell scavenger receptor 2 Scavenger receptor class A member 4 | CHO-K1 cells, fibrillary A-β | [339] | Aβ and Gram-positive, Gram-negative bacteria and yeast phagocytosis | No association with AD described | - |
| TLR2/ TLR4/ CD14 (Toll-like receptor 2/4/6/ CD14) | CD282/CD284/ Monocyte differentiation antigen CD14 | CD14: soluble murine, fibrillary human Aβ-42 | [340] | Instigates inflammatory response and Aβ phagocytosis | P249S (TLR6) 260C/T (CD14) | [341] [342] |
| CD14/TLR2/4: BV2 microglia, fibrillary Aβ | [343] | |||||
| TREM1 (Triggering receptor expressed on myeloid cells 1) | CD354 | mouse primary microglia, monomeric Aβ1-42 | [65] | Amplifying inflammatory responses | rs6910730G | [344] |
| TREM2 (Triggering receptor expressed on myeloid cells 2) | immobilized TREM2-FC, oligomeric and monomeric Aβ1-42 | [307] [345] | Regulates microglial activity, chemotaxis, and process outgrowth | R47H R62H | [346,347,348,349] [350,351] [352] [353] | |
| H157Y D87N | ||||||
| α6β1 integrin | CD49 antigen-like family member F VLA-6 CD49f/ Fibronectin receptor subunit β Glycoprotein IIa (GPIIA) VLA-4 subunit β CD29 | BV-2 cells, fibrillary Aβ25-35 and Aβ1-42 | [354] | increase ROS production and Aβ phagocytosis | No association with AD described |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Busch, L.; Eggert, S.; Endres, K.; Bufe, B. The Hidden Role of Non-Canonical Amyloid β Isoforms in Alzheimer’s Disease. Cells 2022, 11, 3421. https://doi.org/10.3390/cells11213421
Busch L, Eggert S, Endres K, Bufe B. The Hidden Role of Non-Canonical Amyloid β Isoforms in Alzheimer’s Disease. Cells. 2022; 11(21):3421. https://doi.org/10.3390/cells11213421
Chicago/Turabian StyleBusch, Lukas, Simone Eggert, Kristina Endres, and Bernd Bufe. 2022. "The Hidden Role of Non-Canonical Amyloid β Isoforms in Alzheimer’s Disease" Cells 11, no. 21: 3421. https://doi.org/10.3390/cells11213421