
Role of Mitochondrial Cytochrome P450 2E1 in Healthy and Diseased Liver


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Features of CYP2E1
2.1. Biotransformation of Endogenous and Exogenous Molecules
2.2. Regulation of CYP2E1 Expression and Activity
2.3. Role of CYP2E1 in Liver Diseases
3. mtCYP2E1 in Healthy Liver
3.1. Discovery and General Features of mtCYP2E1
3.2. Physiological Role of mtCYP2E1
3.3. Role of mtCYP2E1 in Xenobiotic Biotransformation and ROS Generation
3.4. Mechanisms of CYP2E1 Targeting to Mitochondria and Its Physiological Regulation
4. mtCYP2E1 in Liver Diseases
4.1. Ethanol Toxicity and AALD
4.2. Liver Injury Induced by Drugs and Other Xenobiotics
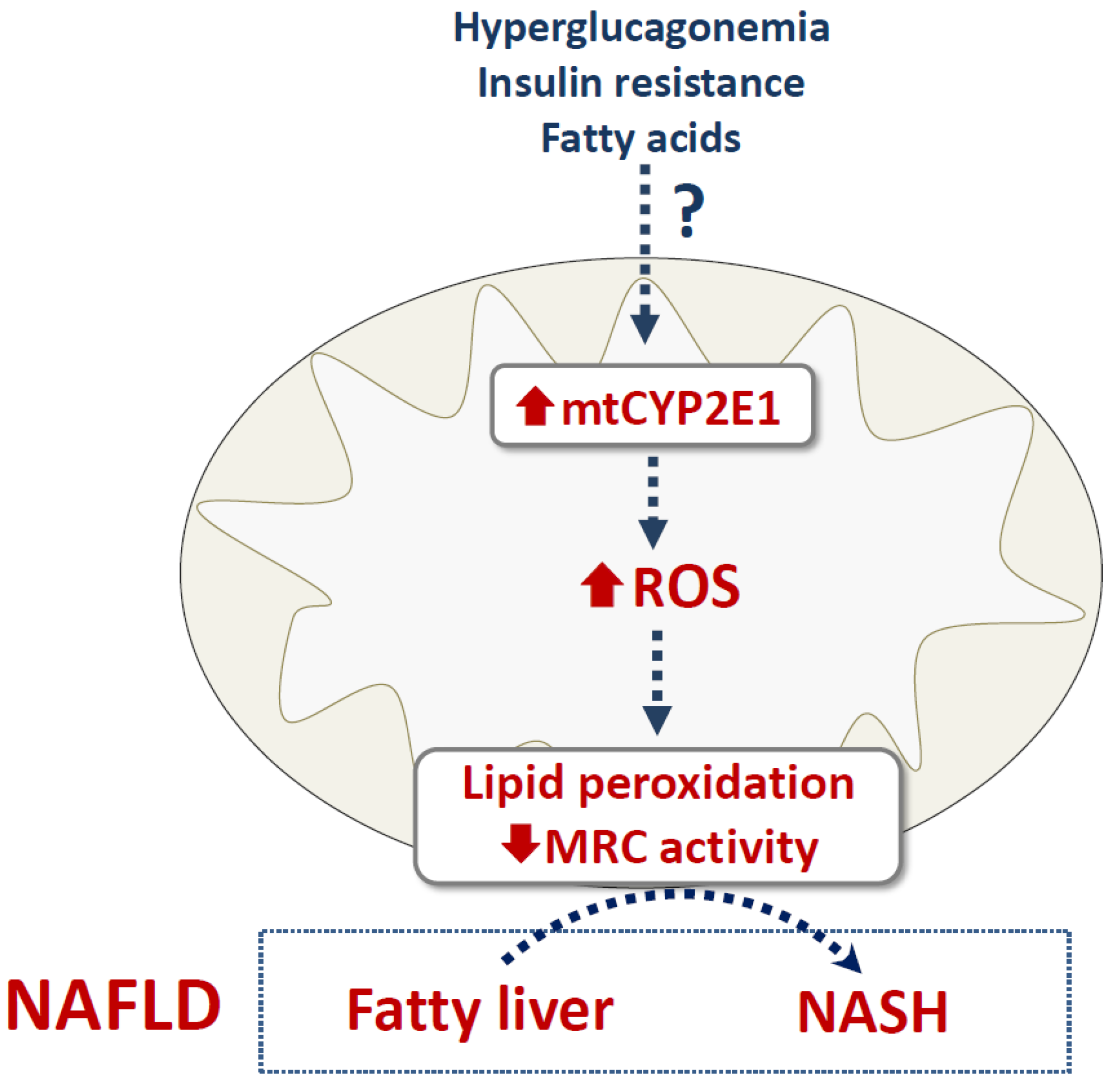
4.3. NAFLD
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guengerich, F.P. Cytochrome P450 2E1 and Its Roles in Disease. Chem. Biol. Interact. 2020, 322, 109056. [Google Scholar] [CrossRef] [PubMed]
- Harjumäki, R.; Pridgeon, C.S.; Ingelman-Sundberg, M. CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload. Int. J. Mol. Sci. 2021, 22, 8221. [Google Scholar] [CrossRef]
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.A.; Fromenty, B. Increased Expression of Cytochrome P450 2E1 in Nonalcoholic Fatty Liver Disease: Mechanisms and Pathophysiological Role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Robin, M.-A.; Anandatheerthavarada, H.K.; Biswas, G.; Sepuri, N.B.V.; Gordon, D.M.; Pain, D.; Avadhani, N.G. Bimodal Targeting of Microsomal CYP2E1 to Mitochondria through Activation of an N-Terminal Chimeric Signal by CAMP-Mediated Phosphorylation. J. Biol. Chem. 2002, 277, 40583–40593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knockaert, L.; Fromenty, B.; Robin, M.-A. Mechanisms of Mitochondrial Targeting of Cytochrome P450 2E1: Physiopathological Role in Liver Injury and Obesity: Mitochondrial CYP2E1. FEBS J. 2011, 278, 4252–4260. [Google Scholar] [CrossRef] [PubMed]
- Koop, D.R. Oxidative and Reductive Metabolism by Cytochrome P450 2E1. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1992, 6, 724–730. [Google Scholar] [CrossRef]
- Gonzalez, F.J. The 2006 Bernard B. Brodie Award Lecture. Cyp2e1. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 1–8. [Google Scholar] [CrossRef]
- Lu, Y.; Cederbaum, A.I. Cytochrome P450s and Alcoholic Liver Disease. Curr. Pharm. Des. 2018, 24, 1502–1517. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, S.; Wang, J.; Renukuntla, J.; Sirimulla, S.; Chen, J. A Comprehensive Review of Cytochrome P450 2E1 for Xenobiotic Metabolism. Drug Metab. Rev. 2019, 51, 178–195. [Google Scholar] [CrossRef]
- Porubsky, P.R.; Battaile, K.P.; Scott, E.E. Human Cytochrome P450 2E1 Structures with Fatty Acid Analogs Reveal a Previously Unobserved Binding Mode. J. Biol. Chem. 2010, 285, 22282–22290. [Google Scholar] [CrossRef] [Green Version]
- Dang, T.T.H.; Yun, J.W. Cytochrome P450 2E1 (CYP2E1) Positively Regulates Lipid Catabolism and Induces Browning in 3T3-L1 White Adipocytes. Life Sci. 2021, 278, 119648. [Google Scholar] [CrossRef]
- Bayoumy, A.B.; Mulder, C.J.J.; Mol, J.J.; Tushuizen, M.E. Gut Fermentation Syndrome: A Systematic Review of Case Reports. United Eur. Gastroenterol. J. 2021, 9, 332–342. [Google Scholar] [CrossRef]
- Bojić, M.; Sedgeman, C.A.; Nagy, L.D.; Guengerich, F.P. Aromatic Hydroxylation of Salicylic Acid and Aspirin by Human Cytochromes P450. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2015, 73, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Bello, M.; Mendieta-Wejebe, J.E.; Correa-Basurto, J. Structural and Energetic Analysis to Provide Insight Residues of CYP2C9, 2C11 and 2E1 Involved in Valproic Acid Dehydrogenation Selectivity. Biochem. Pharmacol. 2014, 90, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, J.; Kotnik, P.; Trontelj, J.; Knez, Ž.; Mašič, L.P. Bioactivation of Bisphenol A and Its Analogs (BPF, BPAF, BPZ and DMBPA) in Human Liver Microsomes. Toxicol. Vitro Int. J. Publ. Assoc. BIBRA 2013, 27, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Oh, W.-Y.; Yi, S.H.; Ku, B.; Lee, M.-Y.; Cho, Y.H.; Yang, M. Estimation of Bisphenol A-Human Toxicity by 3D Cell Culture Arrays, High Throughput Alternatives to Animal Tests. Toxicol. Lett. 2016, 259, 87–94. [Google Scholar] [CrossRef]
- Leung, T.-M.; Nieto, N. CYP2E1 and Oxidant Stress in Alcoholic and Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial Adaptations and Dysfunctions in Nonalcoholic Fatty Liver Disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Cheung, C.; Akiyama, T.E.; Kudo, G.; Gonzalez, F.J. Hepatic Expression of Cytochrome P450s in Hepatocyte Nuclear Factor 1-Alpha (HNF1alpha)-Deficient Mice. Biochem. Pharmacol. 2003, 66, 2011–2020. [Google Scholar] [CrossRef]
- Ge, W.; Wang, T.; Zhao, Y.; Yang, Y.; Sun, Q.; Yang, X.; Gao, Y.; Xu, X.; Zhang, J. Period1 Mediates Rhythmic Metabolism of Toxins by Interacting with CYP2E1. Cell Death Dis. 2021, 12, 76. [Google Scholar] [CrossRef]
- Sekine, S.; Lan, B.Y.-A.; Bedolli, M.; Feng, S.; Hebrok, M. Liver-Specific Loss of β-Catenin Blocks Glutamine Synthesis Pathway Activity and Cytochrome P450 Expression in Mice. Hepatology 2006, 43, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Ray, J.W.; Pathak, P.; Sweet, D.R.; Zhang, R.; Gao, H.; Jain, N.; Koritzinsky, E.H.; Matoba, K.; Xu, W.; et al. KLF15 Regulates Endobiotic and Xenobiotic Metabolism. Nat. Metab. 2019, 1, 422–430. [Google Scholar] [CrossRef]
- Lin, Y.; Ding, D.; Huang, Q.; Liu, Q.; Lu, H.; Lu, Y.; Chi, Y.; Sun, X.; Ye, G.; Zhu, H.; et al. Downregulation of MiR-192 Causes Hepatic Steatosis and Lipid Accumulation by Inducing SREBF1: Novel Mechanism for Bisphenol A-Triggered Non-Alcoholic Fatty Liver Disease. Biochim. Biophys. Acta BBA - Mol. Cell Biol. Lipids 2017, 1862, 869–882. [Google Scholar] [CrossRef]
- Ito, T.; Asakura, K.; Tougou, K.; Fukuda, T.; Kubota, R.; Nonen, S.; Fujio, Y.; Azuma, J. Regulation of Cytochrome P450 2E1 under Hypertonic Environment through TonEBP in Human Hepatocytes. Mol. Pharmacol. 2007, 72, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Moncion, A.; Truong, N.T.; Garrone, A.; Beaune, P.; Barouki, R.; de Waziers, I. Identification of a 16-Nucleotide Sequence That Mediates Post-Transcriptional Regulation of Rat CYP2E1 by Insulin. J. Biol. Chem. 2002, 277, 45904–45910. [Google Scholar] [CrossRef] [Green Version]
- Koop, D.R.; Tierney, D.J. Multiple Mechanisms in the Regulation of Ethanol-Inducible Cytochrome P450IIE1. BioEssays 1990, 12, 429–435. [Google Scholar] [CrossRef]
- Novak, R.F.; Woodcroft, K.J. The Alcohol-Inducible Form of Cytochrome P450 (CYP2E1): Role in Toxicology and Regulation of Expression. Arch. Pharm. Res. 2000, 23, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Oesch-Bartlomowicz, B.; Padma, P.R.; Becker, R.; Richter, B.; Hengstler, J.G.; Freeman, J.E.; Wolf, C.R.; Oesch, F. Differential Modulation of CYP2E1 Activity by CAMP-Dependent Protein Kinase upon Ser129Replacement. Exp. Cell Res. 1998, 242, 294–302. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, S.; Acharya, P.; Koop, D.R.; Liu, Y.; Liao, M.; Burlingame, A.L.; Correia, M.A. Ubiquitin-Dependent Proteasomal Degradation of Human Liver Cytochrome P450 2E1. J. Biol. Chem. 2011, 286, 9443–9456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasi, M.L.; Ramani, K.; Ryoo, M.; Cossu, C.; Floris, A.; Murray, B.J.; Iglesias-Ara, A.; Spissu, Y.; Mavila, N. SUMOylation Regulates Cytochrome P450 2E1 Expression and Activity in Alcoholic Liver Disease. FASEB J. 2018, 32, 3278–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osna, N.A.; Donohue, T.-M. Implication of Altered Proteasome Function in Alcoholic Liver Injury. World J. Gastroenterol. 2007, 13, 4931–4937. [Google Scholar] [CrossRef] [Green Version]
- Massart, J.; Begriche, K.; Fromenty, B. Cytochrome P450 2E1 Should Not Be Neglected for Acetaminophen-Induced Liver Injury in Metabolic Diseases with Altered Insulin Levels or Glucose Homeostasis. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101470. [Google Scholar] [CrossRef]
- Wang, J.; Hu, Y.; Nekvindova, J.; Ingelman-Sundberg, M.; Neve, E.P.A. IL-4-Mediated Transcriptional Regulation of Human CYP2E1 by Two Independent Signaling Pathways. Biochem. Pharmacol. 2010, 80, 1592–1600. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, A.; Jaeschke, H. Acetaminophen Hepatotoxicity. Semin. Liver Dis. 2019, 39, 221–234. [Google Scholar] [CrossRef] [Green Version]
- Knockaert, L.; Berson, A.; Ribault, C.; Prost, P.-E.; Fautrel, A.; Pajaud, J.; Lepage, S.; Lucas-Clerc, C.; Bégué, J.-M.; Fromenty, B.; et al. Carbon Tetrachloride-Mediated Lipid Peroxidation Induces Early Mitochondrial Alterations in Mouse Liver. Lab. Investig. 2012, 92, 396–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, H.K. The Role of Cytochrome P4502E1 in the Pathogenesis of Alcoholic Liver Disease and Carcinogenesis. Chem. Biol. Interact. 2020, 316, 108918. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.K.; Das, S.; Pourhoseini, S.; Dattaroy, D.; Igwe, S.; Ray, J.B.; Fan, D.; Michelotti, G.A.; Diehl, A.M.; Chatterjee, S. M1 Polarization Bias and Subsequent Nonalcoholic Steatohepatitis Progression Is Attenuated by Nitric Oxide Donor DETA NONOate via Inhibition of CYP2E1-Induced Oxidative Stress in Obese Mice. J. Pharmacol. Exp. Ther. 2015, 352, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmegeed, M.A.; Choi, Y.; Godlewski, G.; Ha, S.-K.; Banerjee, A.; Jang, S.; Song, B.-J. Cytochrome P450-2E1 Promotes Fast Food-Mediated Hepatic Fibrosis. Sci. Rep. 2017, 7, 39764. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J. Role of Cytochromes P450 in Chemical Toxicity and Oxidative Stress: Studies with CYP2E1. Mutat. Res. Mol. Mech. Mutagen. 2005, 569, 101–110. [Google Scholar] [CrossRef]
- Adas, F.; Salaün, J.P.; Berthou, F.; Picart, D.; Simon, B.; Amet, Y. Requirement for Omega and (Omega;-1)-Hydroxylations of Fatty Acids by Human Cytochromes P450 2E1 and 4A11. J. Lipid Res. 1999, 40, 1990–1997. [Google Scholar] [CrossRef]
- Anandatheerthavarada, H.K.; Addya, S.; Dwivedi, R.S.; Biswas, G.; Mullick, J.; Avadhani, N.G. Localization of Multiple Forms of Inducible Cytochromes P450 in Rat Liver Mitochondria: Immunological Characteristics and Patterns of Xenobiotic Substrate Metabolism. Arch. Biochem. Biophys. 1997, 339, 136–150. [Google Scholar] [CrossRef]
- Robin, M.-A.; Anandatheerthavarada, H.K.; Fang, J.-K.; Cudic, M.; Otvos, L.; Avadhani, N.G. Mitochondrial Targeted Cytochrome P450 2E1 (P450 MT5) Contains an Intact N Terminus and Requires Mitochondrial Specific Electron Transfer Proteins for Activity. J. Biol. Chem. 2001, 276, 24680–24689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, J.H.; Miller, G.P.; Caro, A.A.; Byrum, S.D.; Orr, L.M.; Mackintosh, S.G.; Tackett, A.J.; MacMillan-Crow, L.A.; Hallberg, L.M.; Ameredes, B.T.; et al. 1,3-Butadiene-Induced Mitochondrial Dysfunction Is Correlated with Mitochondrial CYP2E1 Activity in Collaborative Cross Mice. Toxicology 2017, 378, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Cederbaum, A.I. Overexpression of CYP2E1 in Mitochondria Sensitizes HepG2 Cells to the Toxicity Caused by Depletion of Glutathione. J. Biol. Chem. 2006, 281, 5128–5136. [Google Scholar] [CrossRef] [Green Version]
- Neve, E.P.; Ingelman-Sundberg, M. A Soluble NH(2)-Terminally Truncated Catalytically Active Form of Rat Cytochrome P450 2E1 Targeted to Liver Mitochondria(1). FEBS Lett. 1999, 460, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I. Methodology to Assay CYP2E1 Mixed Function Oxidase Catalytic Activity and Its Induction. Redox Biol. 2014, 2, 1048–1054. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Dong, H.; Balaz, M.; Slyper, M.; Drokhlyansky, E.; Colleluori, G.; Giordano, A.; Kovanicova, Z.; Stefanicka, P.; Balazova, L.; et al. SnRNA-Seq Reveals a Subpopulation of Adipocytes That Regulates Thermogenesis. Nature 2020, 587, 98–102. [Google Scholar] [CrossRef]
- Hartman, J.H.; Martin, H.C.; Caro, A.A.; Pearce, A.R.; Miller, G.P. Subcellular Localization of Rat CYP2E1 Impacts Metabolic Efficiency toward Common Substrates. Toxicology 2015, 338, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Quesnot, N.; Bucher, S.; Gade, C.; Vlach, M.; Vene, E.; Valença, S.; Gicquel, T.; Holst, H.; Robin, M.-A.; Loyer, P. Production of Chlorzoxazone Glucuronides via Cytochrome P4502E1 Dependent and Independent Pathways in Human Hepatocytes. Arch. Toxicol. 2018, 92, 3077–3091. [Google Scholar] [CrossRef]
- Sangar, M.C.; Bansal, S.; Avadhani, N.G. Bimodal Targeting of Microsomal Cytochrome P450s to Mitochondria: Implications in Drug Metabolism and Toxicity. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1231–1251. [Google Scholar] [CrossRef] [Green Version]
- Hartman, J.H.; Miller, G.P.; Meyer, J.N. Toxicological Implications of Mitochondrial Localization of CYP2E1. Toxicol. Res. 2017, 6, 273–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, H.; Prabu, S.K.; Robin, M.-A.; Avadhani, N.G. Elevated Mitochondrial Cytochrome P450 2E1 and Glutathione S-Transferase A4-4 in Streptozotocin-Induced Diabetic Rats: Tissue-Specific Variations and Roles in Oxidative Stress. Diabetes 2004, 53, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-C.; Rupnik, M.S.; Karimian, N.; Herrera, P.L.; Gilon, P.; Feng, Z.-P.; Gaisano, H.Y. In Situ Electrophysiological Examination of Pancreatic Cells in the Streptozotocin-Induced Diabetes Model, Revealing the Cellular Basis of Glucagon Hypersecretion. Diabetes 2013, 62, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Wewer Albrechtsen, N.J.; Kuhre, R.E.; Pedersen, J.; Knop, F.K.; Holst, J.J. The Biology of Glucagon and the Consequences of Hyperglucagonemia. Biomark. Med. 2016, 10, 1141–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robin, M.-A.; Sauvage, I.; Grandperret, T.; Descatoire, V.; Pessayre, D.; Fromenty, B. Ethanol Increases Mitochondrial Cytochrome P450 2E1 in Mouse Liver and Rat Hepatocytes. FEBS Lett. 2005, 579, 6895–6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, S.; Anandatheerthavarada, H.K.; Prabu, G.K.; Milne, G.L.; Martin, M.V.; Guengerich, F.P.; Avadhani, N.G. Human Cytochrome P450 2E1 Mutations That Alter Mitochondrial Targeting Efficiency and Susceptibility to Ethanol-Induced Toxicity in Cellular Models. J. Biol. Chem. 2013, 288, 12627–12644. [Google Scholar] [CrossRef] [Green Version]
- Fromenty, B.; Pessayre, D. Inhibition of Mitochondrial Beta-Oxidation as a Mechanism of Hepatotoxicity. Pharmacol. Ther. 1995, 67, 101–154. [Google Scholar] [CrossRef]
- Singal, A.K.; Mathurin, P. Diagnosis and Treatment of Alcohol-Associated Liver Disease: A Review. JAMA 2021, 326, 165. [Google Scholar] [CrossRef]
- Barrera, G.; Pizzimenti, S.; Daga, M.; Dianzani, C.; Arcaro, A.; Cetrangolo, G.P.; Giordano, G.; Cucci, M.A.; Graf, M.; Gentile, F. Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders. Antioxidants 2018, 7, 102. [Google Scholar] [CrossRef] [Green Version]
- Fromenty, B. Alteration of Mitochondrial DNA Homeostasis in Drug-Induced Liver Injury. Food Chem. Toxicol. 2020, 135, 110916. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Maeda, S. Molecular Mechanisms of Liver Injury and Hepatocarcinogenesis: Focusing on the Role of Stress-Activated MAPK. Pathol. Res. Int. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Lieber, C.S.; Cao, Q.; DeCarli, L.M.; Leo, M.A.; Mak, K.M.; Ponomarenko, A.; Ren, C.; Wang, X. Role of Medium-Chain Triglycerides in the Alcohol-Mediated Cytochrome P450 2E1 Induction of Mitochondria. Alcohol. Clin. Exp. Res. 2007, 31, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.; Liu, C.-P.; Sepuri, N.B.V.; Anandatheerthavarada, H.K.; Selvaraj, V.; Hoek, J.; Milne, G.L.; Guengerich, F.P.; Avadhani, N.G. Mitochondria-Targeted Cytochrome P450 2E1 Induces Oxidative Damage and Augments Alcohol-Mediated Oxidative Stress. J. Biol. Chem. 2010, 285, 24609–24619. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Qiang, X.; Zhang, M.; Ma, D.; Zhao, Z.; Zhou, C.; Liu, X.; Li, R.; Chen, H.; Zhang, Y. Demethyleneberberine, a Natural Mitochondria-Targeted Antioxidant, Inhibits Mitochondrial Dysfunction, Oxidative Stress, and Steatosis in Alcoholic Liver Disease Mouse Model. J. Pharmacol. Exp. Ther. 2015, 352, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Raza, H.; John, A. 4-Hydroxynonenal Induces Mitochondrial Oxidative Stress, Apoptosis and Expression of Glutathione S-Transferase A4-4 and Cytochrome P450 2E1 in PC12 Cells. Toxicol. Appl. Pharmacol. 2006, 216, 309–318. [Google Scholar] [CrossRef]
- Diehl, A.M.; Bisgaard, H.C.; Kren, B.T.; Steer, C.J. Ethanol Interferes with Regeneration-Associated Changes in Biotransforming Enzymes: A Potential Mechanism Underlying Ethanol’s Carcinogenicity? Hepatol. Baltim. Md 1991, 13, 722–727. [Google Scholar] [CrossRef]
- Ronis, M.J.; Huang, J.; Crouch, J.; Mercado, C.; Irby, D.; Valentine, C.R.; Lumpkin, C.K.; Ingelman-Sundberg, M.; Badger, T.M. Cytochrome P450 CYP 2E1 Induction during Chronic Alcohol Exposure Occurs by a Two-Step Mechanism Associated with Blood Alcohol Concentrations in Rats. J. Pharmacol. Exp. Ther. 1993, 264, 944–950. [Google Scholar]
- Hugbart, C.; Verres, Y.; Le Daré, B.; Bucher, S.; Vène, E.; Bodin, A.; Lagente, V.; Fromenty, B.; Bouvet, R.; Morel, I.; et al. Non-Oxidative Ethanol Metabolism in Human Hepatic Cells in Vitro: Involvement of Uridine Diphospho-Glucuronosyltransferase 1A9 in Ethylglucuronide Production. Toxicol. In Vitro 2020, 66, 104842. [Google Scholar] [CrossRef]
- Badger, T.M.; Huang, J.; Ronis, M.; Lumpkin, C.K. Induction of Cytochrome P450 2E1 during Chronic Ethanol Exposure Occurs via Transcription of the Cyp 2E1 Gene When Blood Alcohol Concentrations Are High. Biochem. Biophys. Res. Commun. 1993, 190, 780–785. [Google Scholar] [CrossRef]
- Elnagdy, M.; Barve, S.; McClain, C.; Gobejishvili, L. CAMP Signaling in Pathobiology of Alcohol Associated Liver Disease. Biomolecules 2020, 10, 1433. [Google Scholar] [CrossRef] [PubMed]
- Knockaert, L.; Descatoire, V.; Vadrot, N.; Fromenty, B.; Robin, M.-A. Mitochondrial CYP2E1 Is Sufficient to Mediate Oxidative Stress and Cytotoxicity Induced by Ethanol and Acetaminophen. Toxicol. In Vitro 2011, 25, 475–484. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Williams, C.D.; Xie, Y.; Ramachandran, A.; Jaeschke, H. Acetaminophen-Induced Liver Injury in Rats and Mice: Comparison of Protein Adducts, Mitochondrial Dysfunction, and Oxidative Stress in the Mechanism of Toxicity. Toxicol. Appl. Pharmacol. 2012, 264, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.H.; Sung, S.H.; Lee, H.E.; Kang, H.T.; Lee, S.K.; Oh, S.Y.; Woo, H.A.; Kil, I.S.; Rhee, S.G. Peroxiredoxin III and Sulfiredoxin Together Protect Mice from Pyrazole-Induced Oxidative Liver Injury. Antioxid. Redox Signal. 2012, 17, 1351–1361. [Google Scholar] [CrossRef]
- Lake, B.G. Coumarin Metabolism, Toxicity and Carcinogenicity: Relevance for Human Risk Assessment. Food Chem. Toxicol. 1999, 37, 423–453. [Google Scholar] [CrossRef]
- Tanaka, Y.; Fujii, W.; Hori, H.; Kitagawa, Y.; Ozaki, K. Relationship between Coumarin-Induced Hepatocellular Toxicity and Mitochondrial Function in Rats. Food Chem. Toxicol. 2016, 90, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Yoo, S.-H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.-J. PPARα Expression Protects Male Mice from High Fat–Induced Nonalcoholic Fatty Liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef]
- Junker, A.E.; Gluud, L.; Holst, J.J.; Knop, F.K.; Vilsbøll, T. Diabetic and Nondiabetic Patients with Nonalcoholic Fatty Liver Disease Have an Impaired Incretin Effect and Fasting Hyperglucagonaemia. J. Intern. Med. 2016, 279, 485–493. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Rygg, M.O.; Kristiansen, V.B.; Olsen, B.H.; Serizawa, R.R.; Holst, J.J.; Madsbad, S.; Gluud, L.L.; Bendtsen, F.; Wewer Albrechtsen, N.J. Nonalcoholic Fatty Liver Disease Impairs the Liver–Alpha Cell Axis Independent of Hepatic Inflammation and Fibrosis. Hepatol. Commun. 2020, 4, 1610–1623. [Google Scholar] [CrossRef]
- Jia, J.; Chen, S.; Pan, W.; Yu, S.; Zhao, X.; Hao, Y.; Shen, Y.; Cheng, Y.; Wei, C.; Tian, F.; et al. Mechanism of Subchronic Vinyl Chloride Exposure Combined with a High-fat Diet on Hepatic Steatosis. J. Appl. Toxicol. 2021, jat.4234. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Charkoftaki, G.; Orlicky, D.J.; Davidson, E.; Wan, F.; Ginsberg, G.; Thompson, D.C.; Vasiliou, V. Oxidative Stress and Genotoxicity in 1,4-Dioxane Liver Toxicity as Evidenced in a Mouse Model of Glutathione Deficiency. Sci. Total Environ. 2022, 806, 150703. [Google Scholar] [CrossRef] [PubMed]
- Oropeza-Hernández, L.F.; López-Romero, R.; Albores, A. Hepatic CYP1A, 2B, 2C, 2E and 3A Regulation by Methoxychlor in Male and Female Rats. Toxicol. Lett. 2003, 144, 93–103. [Google Scholar] [CrossRef]
- Zhang, J.; Song, W.; Sun, Y.; Shan, A. Effects of Phoxim-Induced Hepatotoxicity on SD Rats and the Protection of Vitamin E. Environ. Sci. Pollut. Res. 2017, 24, 24916–24927. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, A.D.; Breckenridge, C.B.; Yi, K.D.; Sawhney Coder, P.; Wanders, D.; Judd, R.L.; Foradori, C.D. Changes in Hepatic Phase I and Phase II Biotransformation Enzyme Expression and Glutathione Levels Following Atrazine Exposure in Female Rats. Xenobiotica 2018, 48, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Yadav, A.; Singh, C. Autonomous Regulation of Inducible Nitric Oxide Synthase and Cytochrome P450 2E1-Mediated Oxidative Stress in Maneb- and Paraquat-Treated Rat Polymorphs. Pestic. Biochem. Physiol. 2021, 178, 104944. [Google Scholar] [CrossRef]
- Sharma, R.; Upadhyay, G.; Siddiqi, N.; Sharma, B. Pesticides-Induced Biochemical Alterations in Occupational North Indian Suburban Population. Hum. Exp. Toxicol. 2013, 32, 1213–1227. [Google Scholar] [CrossRef]
- Shi, R.; Liu, Z.; Liu, T. The Antagonistic Effect of Bisphenol A and Nonylphenol on Liver and Kidney Injury in Rats. Immunopharmacol. Immunotoxicol. 2021, 43, 527–535. [Google Scholar] [CrossRef]
- Park, K.S.; Sohn, D.H.; Veech, R.L.; Song, B.J. Translational Activation of Ethanol-Inducible Cytochrome P450 (CYP2E1) by Isoniazid. Eur. J. Pharmacol. Environ. Toxicol. Pharmacol. 1993, 248, 7–14. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, J.-J.; Wang, X.; Bu, X.-Y.; Lou, Y.-Q.; Zhang, G.-L. Effect of Berberine on Hepatocyte Proliferation, Inducible Nitric Oxide Synthase Expression, Cytochrome P450 2E1 and 1A2 Activities in Diethylnitrosamine- and Phenobarbital-Treated Rats. Biomed. Pharmacother. 2008, 62, 567–572. [Google Scholar] [CrossRef]
- Shen, C.; Meng, Q.; Zhang, G.; Hu, W. Rifampicin Exacerbates Isoniazid-Induced Toxicity in Human but Not in Rat Hepatocytes in Tissue-like Cultures: Rifampicin on Isoniazid-Induced Hepatotoxicity in Vitro. Br. J. Pharmacol. 2008, 153, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Miao, M.; Zhong, Z.; Xu, P.; Chen, Y.; Liu, X. Chronic Administration of Caderofloxacin, a New Fluoroquinolone, Increases Hepatic CYP2E1 Expression and Activity in Rats. Acta Pharmacol. Sin. 2016, 37, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, J.P.; Lipscomb, J.C.; Wesselkamper, S.C. Putative Mechanisms of Environmental Chemical–Induced Steatosis. Int. J. Toxicol. 2012, 31, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Fromenty, B. Inhibition of Mitochondrial Fatty Acid Oxidation in Drug-Induced Hepatic Steatosis. Liver Res. 2019, 3, 157–169. [Google Scholar] [CrossRef]
- Correia, M.A.; Kwon, D. Why Hepatic CYP2E1-Elevation by Itself Is Insufficient for Inciting NAFLD/NASH: Inferences from Two Genetic Knockout Mouse Models. Biology 2020, 9, 419. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective Hepatic Mitochondrial Respiratory Chain in Patients with Nonalcoholic Steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Michaut, A.; Le Guillou, D.; Moreau, C.; Bucher, S.; McGill, M.R.; Martinais, S.; Gicquel, T.; Morel, I.; Robin, M.-A.; Jaeschke, H.; et al. A Cellular Model to Study Drug-Induced Liver Injury in Nonalcoholic Fatty Liver Disease: Application to Acetaminophen. Toxicol. Appl. Pharmacol. 2016, 292, 40–55. [Google Scholar] [CrossRef] [Green Version]
- Bucher, S.; Le Guillou, D.; Allard, J.; Pinon, G.; Begriche, K.; Tête, A.; Sergent, O.; Lagadic-Gossmann, D.; Fromenty, B. Possible Involvement of Mitochondrial Dysfunction and Oxidative Stress in a Cellular Model of NAFLD Progression Induced by Benzo[a]Pyrene/Ethanol CoExposure. Oxid. Med. Cell. Longev. 2018, 2018, 4396403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Henry, L. Epidemiology of Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma. JHEP Rep. 2021, 3, 100305. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Dalbeni, A. Treatments for NAFLD: State of Art. Int. J. Mol. Sci. 2021, 22, 2350. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massart, J.; Begriche, K.; Hartman, J.H.; Fromenty, B. Role of Mitochondrial Cytochrome P450 2E1 in Healthy and Diseased Liver. Cells 2022, 11, 288. https://doi.org/10.3390/cells11020288
Massart J, Begriche K, Hartman JH, Fromenty B. Role of Mitochondrial Cytochrome P450 2E1 in Healthy and Diseased Liver. Cells. 2022; 11(2):288. https://doi.org/10.3390/cells11020288
Chicago/Turabian StyleMassart, Julie, Karima Begriche, Jessica H. Hartman, and Bernard Fromenty. 2022. "Role of Mitochondrial Cytochrome P450 2E1 in Healthy and Diseased Liver" Cells 11, no. 2: 288. https://doi.org/10.3390/cells11020288