
Ablation of Sphingosine Kinase 1 Protects Cornea from Neovascularization in a Mouse Corneal Injury Model
 , and
, and 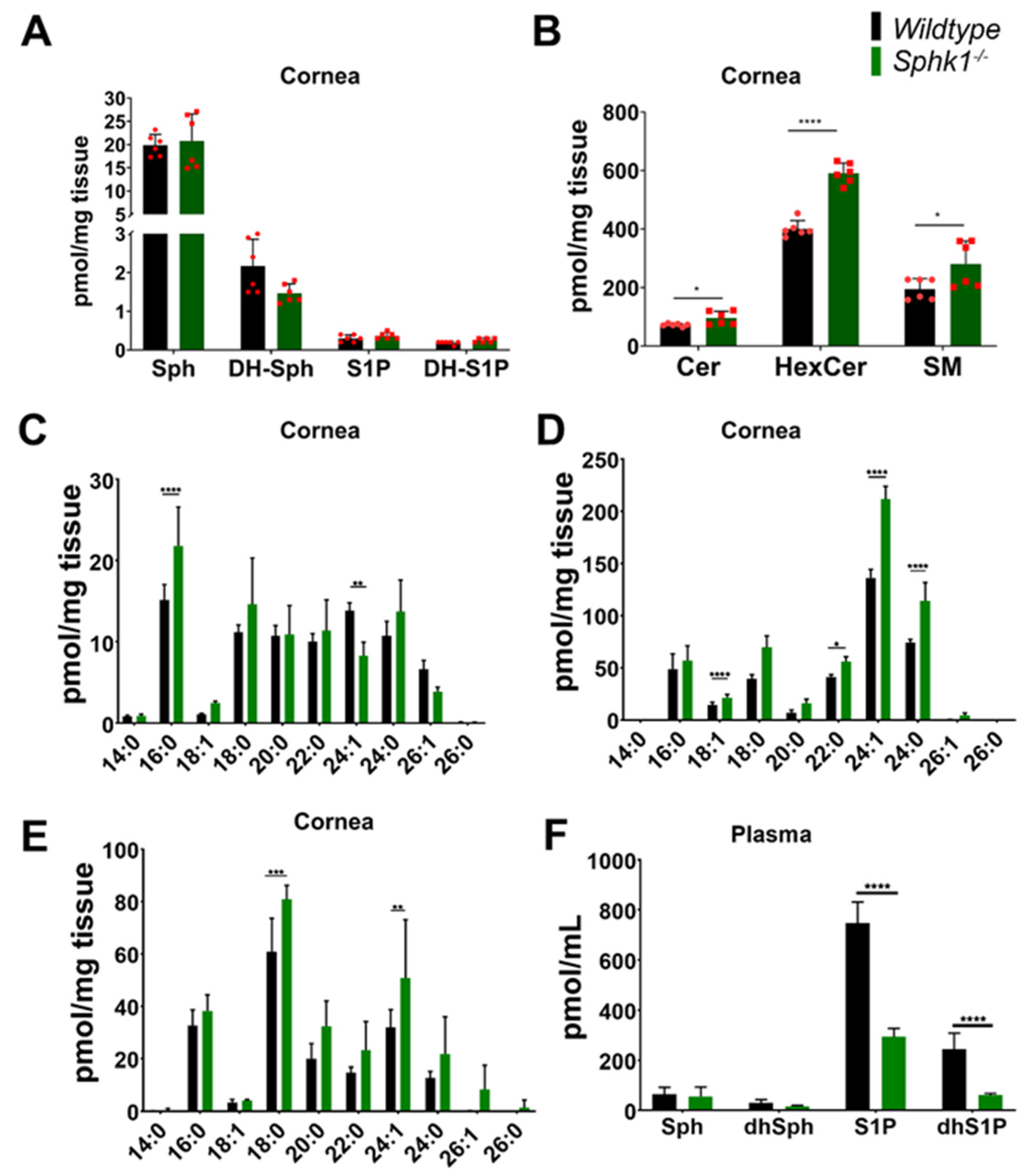{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animal Care
2.3. Blood/Plasma Collection
2.4. Sphingolipid Analysis
2.5. Cornea Alkali Burn
2.6. Ethynyl Deoxy Uridine (EdU) Pulse
2.7. Immunohistochemistry
2.8. Multiplex Assay
2.9. Statistics
3. Results
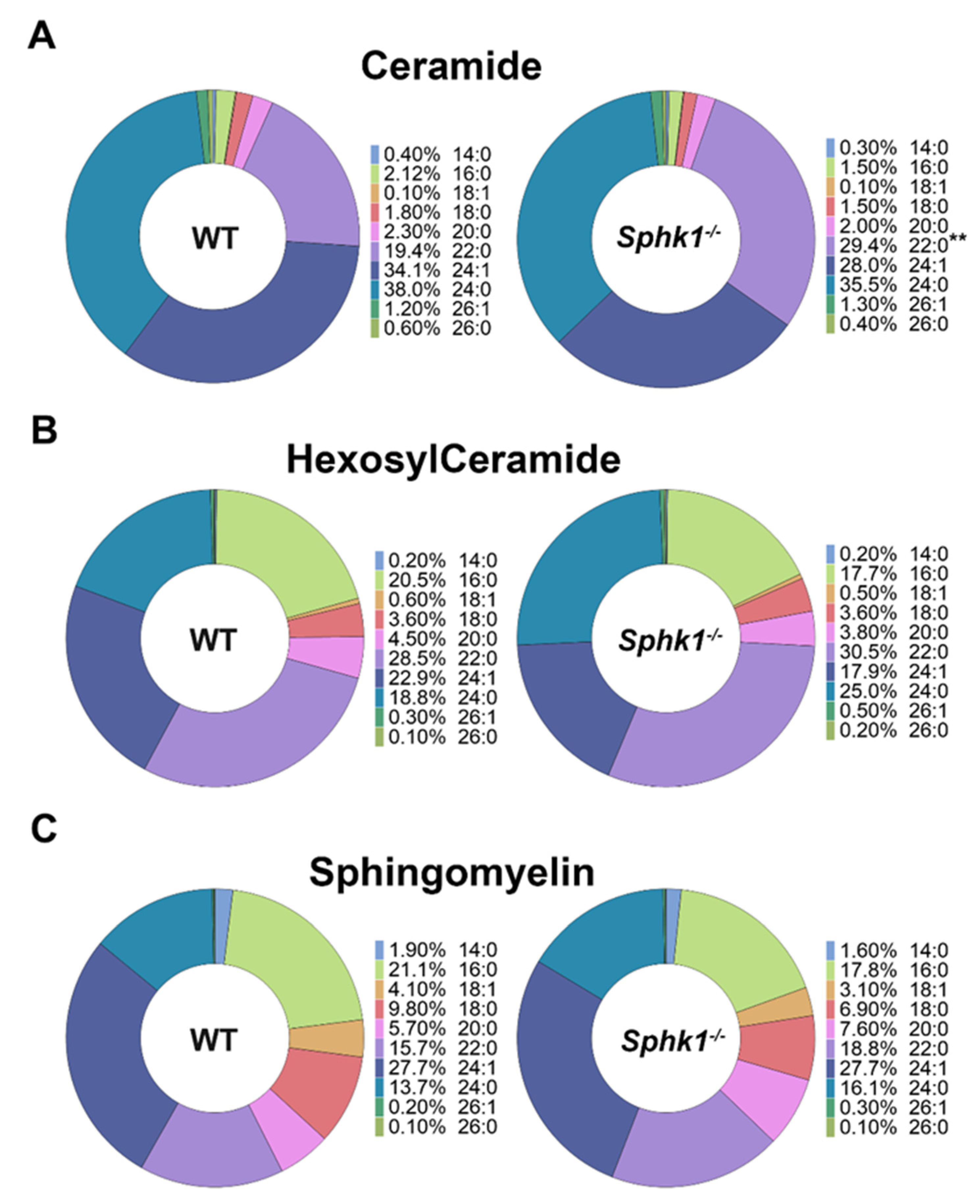
3.1. Sphingolipid Profile Is Altered in the Cornea and Plasma of Sphk1−/− Mice
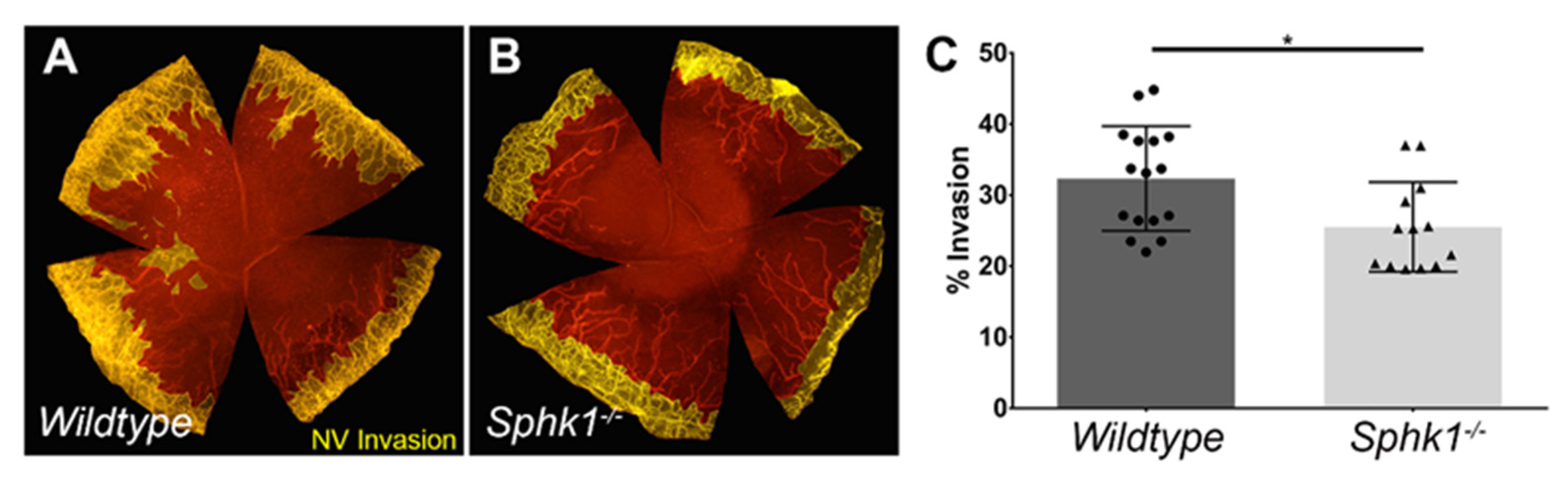
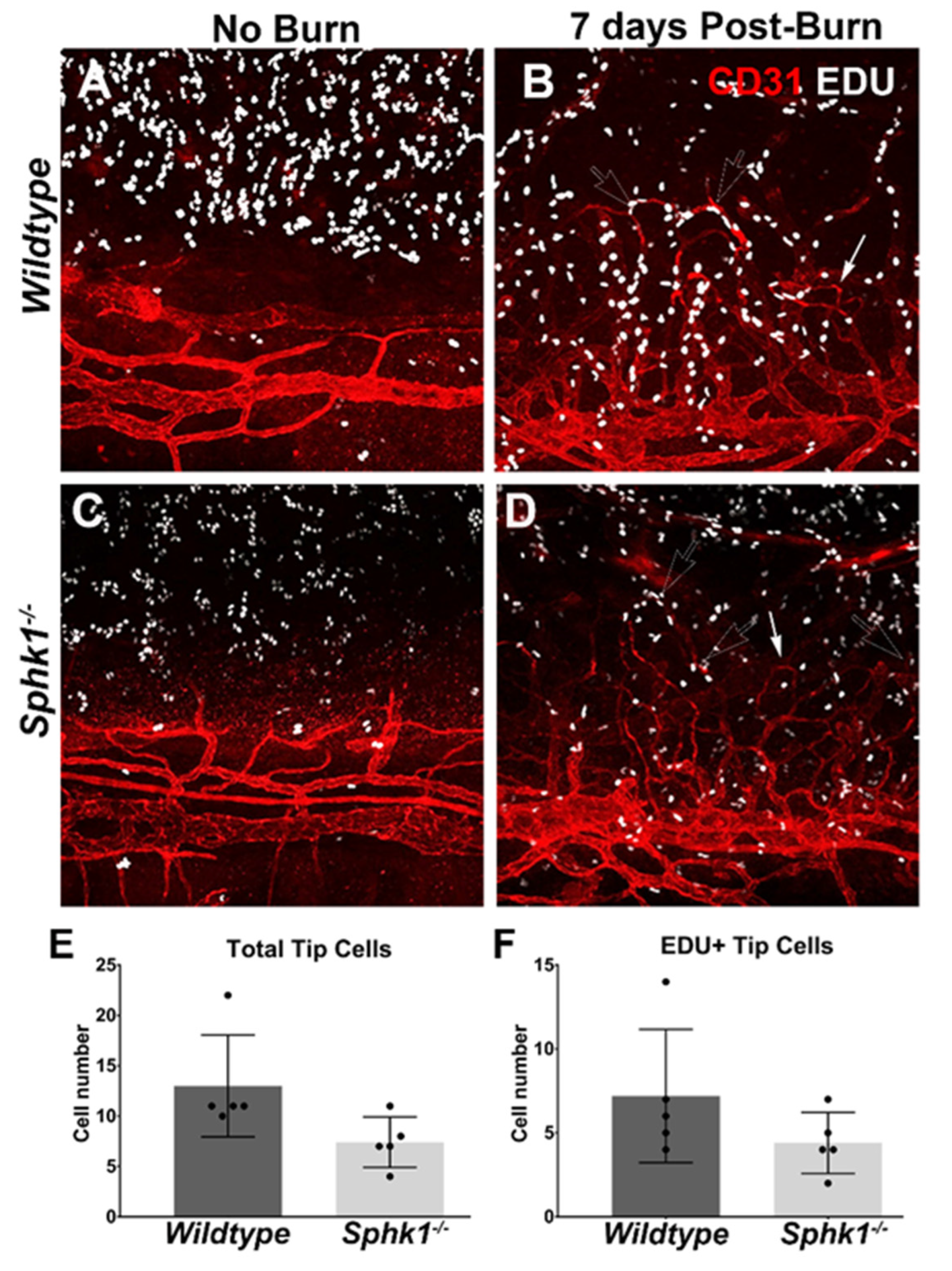
3.2. Sphk1−/− Mice Have Reduced Corneal Neovascularization following Injury
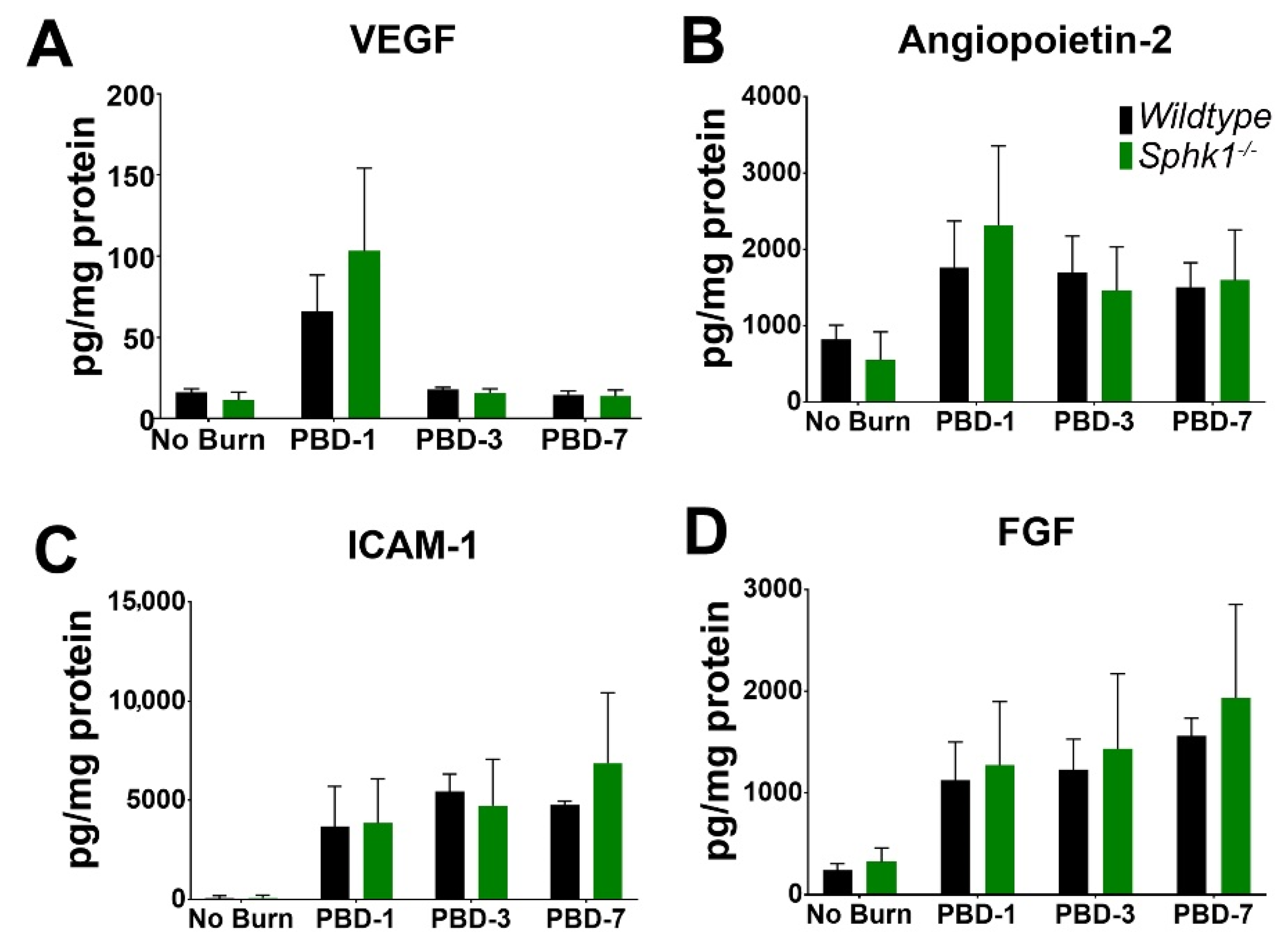
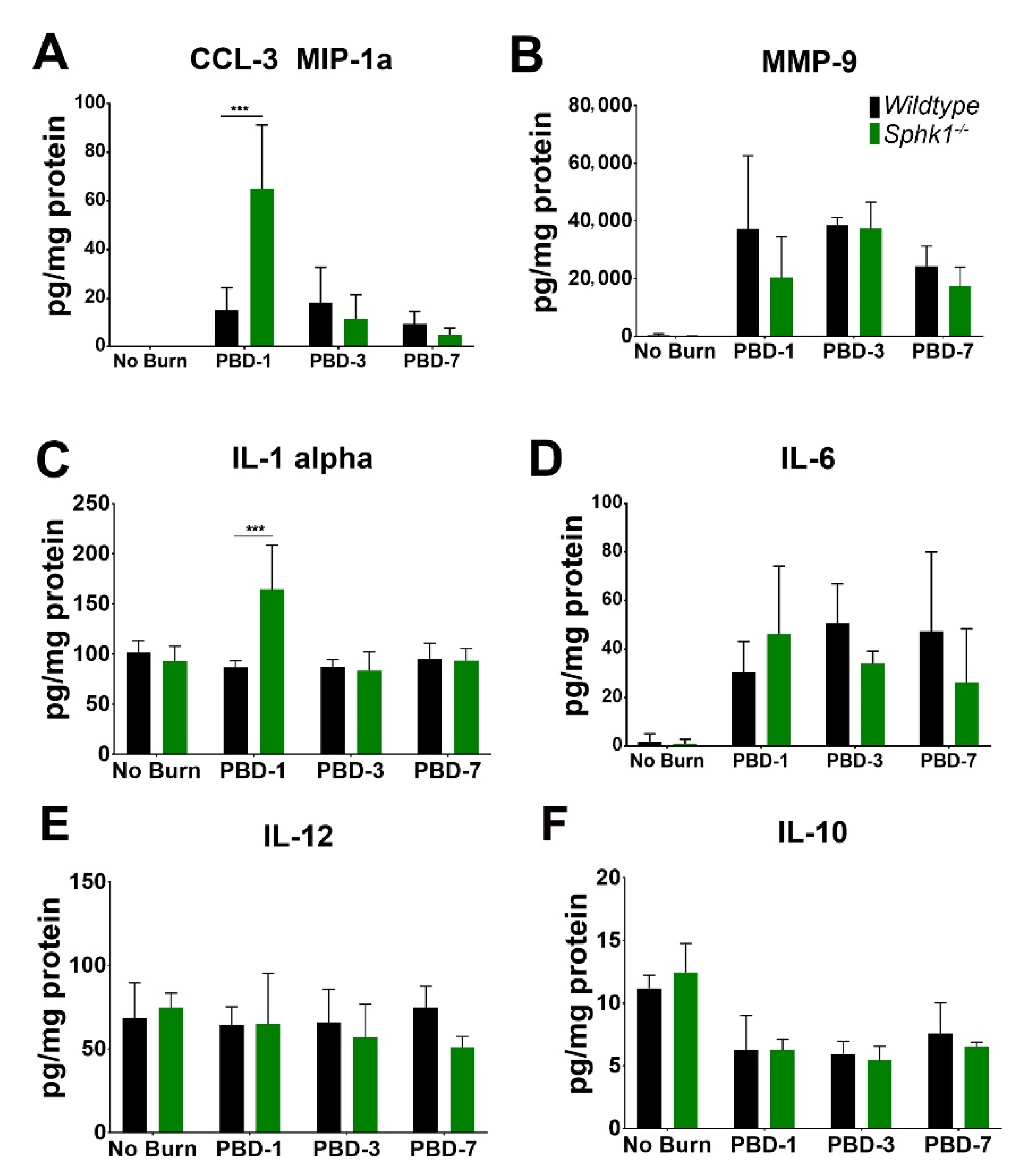
3.3. Alkali Burn Alters Angiogenic and Inflammatory Cytokine Markers in the Cornea
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Piatigorsky, J. Enigma of the abundant water-soluble cytoplasmic proteins of the cornea: The “refracton” hypothesis. Cornea 2001, 20, 853–858. [Google Scholar] [CrossRef]
- Pandolfi, A.; Holzapfel, G.A. Three-dimensional modeling and computational analysis of the human cornea considering distributed collagen fibril orientations. J. Biomech. Eng. 2008, 130, 061006. [Google Scholar] [CrossRef]
- Ambati, B.K.; Nozaki, M.; Singh, N.; Takeda, A.; Jani, P.D.; Suthar, T.; Albuquerque, R.J.; Richter, E.; Sakurai, E.; Newcomb, M.T.; et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature 2006, 443, 993–997. [Google Scholar] [CrossRef]
- Cheng, S.F.; Dastjerdi, M.H.; Ferrari, G.; Okanobo, A.; Bower, K.S.; Ryan, D.S.; Amparo, F.; Stevenson, W.; Hamrah, P.; Nallasamy, N.; et al. Short-term topical bevacizumab in the treatment of stable corneal neovascularization. Am. J. Ophthalmol. 2012, 154, 940–948.e1. [Google Scholar] [CrossRef]
- Bachmann, B.; Taylor, R.S.; Cursiefen, C. The association between corneal neovascularization and visual acuity: A systematic review. Acta Ophthalmol. 2013, 91, 12–19. [Google Scholar] [CrossRef]
- Qazi, Y.; Maddula, S.; Ambati, B.K. Mediators of ocular angiogenesis. J. Genet. 2009, 88, 495–515. [Google Scholar] [CrossRef]
- McKay, T.B.; Hutcheon, A.E.K.; Zieske, J.D. Biology of corneal fibrosis: Soluble mediators, integrins, and extracellular vesicles. Eye 2020, 34, 271–278. [Google Scholar] [CrossRef]
- Dressler, K.A.; Mathias, S.; Kolesnick, R.N. Tumor necrosis factor-alpha activates the sphingomyelin signal transduction pathway in a cell-free system. Science 1992, 255, 1715–1718. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Loomis, C.R.; Merrill, A.H.; Bell, R.M. Sphingosine inhibition of protein kinase C activity and of phorbol dibutyrate binding in vitro and in human platelets. J. Biol. Chem. 1986, 261, 12604–12609. [Google Scholar] [CrossRef]
- Kolesnick, R.N. 1,2-Diacylglycerols but not phorbol esters stimulate sphingomyelin hydrolysis in GH3 pituitary cells. J. Biol. Chem. 1987, 262, 16759–16762. [Google Scholar] [CrossRef]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Cosot, O.A.; Gutkind, J.S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine- 1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- Gomez-Muñoz, A.; Duffy, P.A.; Martin, A.; O’Brien, L.; Byun, H.S.; Bittman, R.; Brindley, D.N. Short-chain ceramide-1-phosphates are novel stimulators of DNA synthesis and cell division: Antagonism by cell-permeable ceramides. Mol. Pharmacol. 1995, 47, 833–839. [Google Scholar]
- Gomez-Muñoz, A.; Frago, L.M.; Alvarez, L.; Varela-Nieto, I. Stimulation of DNA synthesis by natural ceramide 1-phosphate. Biochem. J. 1997, 325 Pt 2, 435–440. [Google Scholar] [CrossRef]
- Zhang, H.; Desai, N.N.; Olivera, A.; Seki, T.; Brooker, G.; Spiegel, S. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J. Cell Biol. 1991, 114, 155–167. [Google Scholar] [CrossRef]
- Olivera, A.; Spiegel, S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 1993, 365, 557–560. [Google Scholar] [CrossRef]
- Simon, M.V.; Basu, S.K.; Qaladize, B.; Grambergs, R.; Rotstein, N.P.; Mandal, N. Sphingolipids as critical players in retinal physiology and pathology. J. Lipid Res. 2021, 62, 100037. [Google Scholar] [CrossRef]
- Merrill, A.H., Jr. De novo sphingolipid biosynthesis: A necessary, but dangerous, pathway. J. Biol. Chem. 2002, 277, 25843–25846. [Google Scholar] [CrossRef]
- Marchesini, N.; Hannun, Y.A. Acid and neutral sphingomyelinases: Roles and mechanisms of regulation. Biochem. Cell Biol. 2004, 82, 27–44. [Google Scholar] [CrossRef]
- Le Stunff, H.; Giussani, P.; Maceyka, M.; Lepine, S.; Milstien, S.; Spiegel, S. Recycling of sphingosine is regulated by the concerted actions of sphingosine-1-phosphate phosphohydrolase 1 and sphingosine kinase 2. J. Biol. Chem. 2007, 282, 34372–34380. [Google Scholar] [CrossRef]
- Grassme, H.; Bock, J.; Kun, J.; Gulbins, E. Clustering of CD40 ligand is required to form a functional contact with CD40. J. Biol. Chem. 2002, 277, 30289–30299. [Google Scholar] [CrossRef] [Green Version]
- Grassme, H.; Schwarz, H.; Gulbins, E. Molecular mechanisms of ceramide-mediated CD95 clustering. Biochem. Biophys. Res. Commun. 2001, 284, 1016–1030. [Google Scholar] [CrossRef]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of Histone Acetylation in the Nucleus by Sphingosine-1-Phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef]
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-Out” Signaling of Sphingosine-1-Phosphate: Therapeutic Targets. Pharmacol. Rev. 2008, 60, 181–195. [Google Scholar] [CrossRef]
- Hla, T.; Lee, M.-J.; Ancellin, N.; Paik, J.H.; Kluk, M.J. Lysophospholipids-Receptor Revelations. Science 2001, 294, 1875–1878. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine 1-phosphate, a key cell signaling molecule. J. Biol. Chem. 2002, 277, 25851–25854. [Google Scholar] [CrossRef]
- Kunkel, G.T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 2013, 12, 688–702. [Google Scholar] [CrossRef]
- Kono, M.; Mi, Y.; Liu, Y.; Sasaki, T.; Allende, M.L.; Wu, Y.P.; Yamashita, T.; Proia, R.L. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J. Biol. Chem. 2004, 279, 29367–29373. [Google Scholar] [CrossRef]
- Tabasinezhad, M.; Samadi, N.; Ghanbari, P.; Mohseni, M.; Saei, A.A.; Sharifi, S.; Saeedi, N.; Pourhassan, A. Sphingosin 1-phosphate contributes in tumor progression. J. Cancer Res. Ther. 2013, 9, 556. [Google Scholar] [CrossRef]
- Hänel, P.; Andréani, P.; Gräler, M.H. Erythrocytes store and release sphingosine 1-phosphate in blood. FASEB J. 2007, 21, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, K.; Lee, Y.-M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular Endothelium as a Contributor of Plasma Sphingosine 1-Phosphate. Circ. Res. 2008, 102, 669–676. [Google Scholar] [CrossRef]
- Yanagida, K.; Hla, T. Vascular and Immunobiology of the Circulatory Sphingosine 1-Phosphate Gradient. Annu. Rev. Physiol. 2017, 79, 67–91. [Google Scholar] [CrossRef] [PubMed]
- Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G.F.; Spiegel, S.; Proia, R.L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 2005, 25, 11113–11121. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wada, R.; Yamashita, T.; Mi, Y.; Deng, C.X.; Hobson, J.P.; Rosenfeldt, H.M.; Nava, V.E.; Chae, S.S.; Lee, M.J.; et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Investig. 2000, 106, 951–961. [Google Scholar] [CrossRef]
- Paik, J.H.; Skoura, A.; Chae, S.S.; Cowan, A.E.; Han, D.K.; Proia, R.L.; Hla, T. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev. 2004, 18, 2392–2403. [Google Scholar] [CrossRef]
- Jung, B.; Obinata, H.; Galvani, S.; Mendelson, K.; Ding, B.S.; Skoura, A.; Kinzel, B.; Brinkmann, V.; Rafii, S.; Evans, T.; et al. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev. Cell 2012, 23, 600–610. [Google Scholar] [CrossRef]
- Gaengel, K.; Niaudet, C.; Hagikura, K.; Lavina, B.; Muhl, L.; Hofmann, J.J.; Ebarasi, L.; Nystrom, S.; Rymo, S.; Chen, L.L.; et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Dev. Cell 2012, 23, 587–599. [Google Scholar] [CrossRef]
- Ancellin, N.; Colmont, C.; Su, J.; Li, Q.; Mittereder, N.; Chae, S.S.; Stefansson, S.; Liau, G.; Hla, T. Extracellular export of sphingosine kinase-1 enzyme. Sphingosine 1-phosphate generation and the induction of angiogenic vascular maturation. J. Biol. Chem. 2002, 277, 6667–6675. [Google Scholar] [CrossRef]
- Bode, C.; Sensken, S.C.; Peest, U.; Beutel, G.; Thol, F.; Levkau, B.; Li, Z.; Bittman, R.; Huang, T.; Tolle, M.; et al. Erythrocytes serve as a reservoir for cellular and extracellular sphingosine 1-phosphate. J. Cell. Biochem. 2010, 109, 1232–1243. [Google Scholar] [CrossRef]
- Pyne, N.J.; Dubois, G.; Pyne, S. Role of sphingosine 1-phosphate and lysophosphatidic acid in fibrosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2013, 1831, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine-1-phosphate: A Janus-faced mediator of fibrotic diseases. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2013, 1831, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; He, X.; Zeng, M. The Role of S1P and the Related Signaling Pathway in the Development of Tissue Fibrosis. Front. Pharmacol. 2019, 9, 1504. [Google Scholar] [CrossRef] [PubMed]
- Gellings Lowe, N.; Swaney, J.S.; Moreno, K.M.; Sabbadini, R.A. Sphingosine-1-phosphate and sphingosine kinase are critical for transforming growth factor-β-stimulated collagen production by cardiac fibroblasts. Cardiovasc. Res. 2009, 82, 303–312. [Google Scholar] [CrossRef]
- Nicholas, S.E.; Rowsey, T.G.; Priyadarsini, S.; Mandal, N.A.; Karamichos, D. Unravelling the interplay of sphingolipids and TGF-β signaling in the human corneal stroma. PLoS ONE 2017, 12, e0182390. [Google Scholar] [CrossRef]
- Wilkerson, J.L.; Mandal, N.A. Angiogenesis Model of Cornea to Understand the Role of Sphingosine 1-Phosphate. Methods Mol. Biol. 2017, 1609, 267–276. [Google Scholar] [CrossRef]
- Qi, H.; Priyadarsini, S.; Nicholas, S.E.; Sarker-Nag, A.; Allegood, J.; Chalfant, C.E.; Mandal, N.A.; Karamichos, D. Analysis of sphingolipids in human corneal fibroblasts from normal and keratoconus patients. J. Lipid Res. 2017, 58, 636–648. [Google Scholar] [CrossRef]
- Paranjpe, V.; Tan, J.; Nguyen, J.; Lee, J.; Allegood, J.; Galor, A.; Mandal, N. Clinical signs of meibomian gland dysfunction (MGD) are associated with changes in meibum sphingolipid composition. Ocul. Surf. 2019, 17, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Sugano, E.; Edwards, G.; Saha, S.; Wilmott, L.A.; Grambergs, R.C.; Mondal, K.; Qi, H.; Stiles, M.; Tomita, H.; Mandal, N. Overexpression of acid ceramidase (ASAH1) protects retinal cells (ARPE19) from oxidative stress. J. Lipid Res. 2019, 60, 30–43. [Google Scholar] [CrossRef]
- Allende, M.L.; Sasaki, T.; Kawai, H.; Olivera, A.; Mi, Y.; van Echten-Deckert, G.; Hajdu, R.; Rosenbach, M.; Keohane, C.A.; Mandala, S.; et al. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J. Biol. Chem. 2004, 279, 52487–52492. [Google Scholar] [CrossRef]
- Dallinga, M.G.; Boas, S.E.; Klaassen, I.; Merks, R.H.; van Noorden, C.J.; Schlingemann, R.O. Tip Cells in Angiogenesis. In eLS; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2015; pp. 1–10. [Google Scholar]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.-Y.; Bocci, F.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Levchenko, A. Pericytes enable effective angiogenesis in the presence of proinflammatory signals. Proc. Natl. Acad. Sci. USA 2019, 116, 23551–23561. [Google Scholar] [CrossRef] [PubMed]
- Stapor, P.C.; Sweat, R.S.; Dashti, D.C.; Betancourt, A.M.; Murfee, W.L. Pericyte Dynamics during Angiogenesis: New Insights from New Identities. J. Vasc. Res. 2014, 51, 163–174. [Google Scholar] [CrossRef]
- Laskin, D.L.; Sunil, V.R.; Gardner, C.R.; Laskin, J.D. Macrophages and tissue injury: Agents of defense or destruction? Annu. Rev. Pharm. Toxicol. 2011, 51, 267–288. [Google Scholar] [CrossRef]
- Gurevich, D.B.; Severn, C.E.; Twomey, C.; Greenhough, A.; Cash, J.; Toye, A.M.; Mellor, H.; Martin, P. Live imaging of wound angiogenesis reveals macrophage orchestrated vessel sprouting and regression. EMBO J. 2018, 37, e97786. [Google Scholar] [CrossRef]
- Kang, H.; Li, X.; Xiong, K.; Song, Z.; Tian, J.; Wen, Y.; Sun, A.; Deng, X. The Entry and Egress of Monocytes in Atherosclerosis: A Biochemical and Biomechanical Driven Process. Cardiovasc. Ther. 2021, 2021, 6642927. [Google Scholar] [CrossRef]
- Chang, C.-H.; Randolph, G.J. Sphingosine-1-Phosphate as the Lymphocyte’s Ticket to Ride and Survive. Dev. Cell 2017, 41, 576–578. [Google Scholar] [CrossRef]
- Mendoza, A.; Bréart, B.; Ramos-Perez, W.D.; Pitt, L.A.; Gobert, M.; Sunkara, M.; Lafaille, J.J.; Morris, A.J.; Schwab, S.R. The transporter Spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep. 2012, 2, 1104–1110. [Google Scholar] [CrossRef]
- Swaney, J.S.; Moreno, K.M.; Gentile, A.M.; Sabbadini, R.A.; Stoller, G.L. Sphingosine-1-phosphate (S1P) is a novel fibrotic mediator in the eye. Exp. Eye Res. 2008, 87, 367–375. [Google Scholar] [CrossRef]
- Mitra, P.; Oskeritzian, C.A.; Payne, S.G.; Beaven, M.A.; Milstien, S.; Spiegel, S. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16394–16399. [Google Scholar] [CrossRef]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Laviad, E.L.; Albee, L.; Pankova-Kholmyansky, I.; Epstein, S.; Park, H.; Merrill, A.H., Jr.; Futerman, A.H. Characterization of ceramide synthase 2: Tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 2008, 283, 5677–5684. [Google Scholar] [CrossRef]
- Zemann, B.; Urtz, N.; Reuschel, R.; Mechtcheriakova, D.; Bornancin, F.; Badegruber, R.; Baumruker, T.; Billich, A. Normal neutrophil functions in sphingosine kinase type 1 and 2 knockout mice. Immunol. Lett. 2007, 109, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Wadgaonkar, R.; Patel, V.; Grinkina, N.; Romano, C.; Liu, J.; Zhao, Y.; Sammani, S.; Garcia, J.G.; Natarajan, V. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L603–L613. [Google Scholar] [CrossRef] [PubMed]
- Porter, H.; Qi, H.; Prabhu, N.; Grambergs, R.; McRae, J.; Hopiavuori, B.; Mandal, N. Characterizing Sphingosine Kinases and Sphingosine 1-Phosphate Receptors in the Mammalian Eye and Retina. Int. J. Mol. Sci. 2018, 19, 3885. [Google Scholar] [CrossRef] [PubMed]
- French, K.J.; Schrecengost, R.S.; Lee, B.D.; Zhuang, Y.; Smith, S.N.; Eberly, J.L.; Yun, J.K.; Smith, C.D. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003, 63, 5962–5969. [Google Scholar]
- French, K.J.; Upson, J.J.; Keller, S.N.; Zhuang, Y.; Yun, J.K.; Smith, C.D. Antitumor activity of sphingosine kinase inhibitors. J. Pharm. Exp. 2006, 318, 596–603. [Google Scholar] [CrossRef]
- Nagahashi, M.; Ramachandran, S.; Kim, E.Y.; Allegood, J.C.; Rashid, O.M.; Yamada, A.; Zhao, R.; Milstien, S.; Zhou, H.; Spiegel, S.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012, 72, 726–735. [Google Scholar] [CrossRef]
- Xiong, Y.; Yang, P.; Proia, R.L.; Hla, T. Erythrocyte-derived sphingosine 1-phosphate is essential for vascular development. J. Clin. Investig. 2014, 124, 4823–4828. [Google Scholar] [CrossRef]
- Ben Shoham, A.; Malkinson, G.; Krief, S.; Shwartz, Y.; Ely, Y.; Ferrara, N.; Yaniv, K.; Zelzer, E. S1P1 inhibits sprouting angiogenesis during vascular development. Development 2012, 139, 3859–3869. [Google Scholar] [CrossRef]
- Freed, J.K.; Beyer, A.M.; LoGiudice, J.A.; Hockenberry, J.C.; Gutterman, D.D. Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ. Res. 2014, 115, 525–532. [Google Scholar] [CrossRef]
- Moreno, L.; Moral-Sanz, J.; Morales-Cano, D.; Barreira, B.; Moreno, E.; Ferrarini, A.; Pandolfi, R.; Ruperez, F.J.; Cortijo, J.; Sanchez-Luna, M.; et al. Ceramide mediates acute oxygen sensing in vascular tissues. Antioxid. Redox Signal. 2014, 20, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Weil, B.R.; Canty, J.M., Jr. Ceramide signaling in the coronary microcirculation: A double-edged sword? Circ. Res. 2014, 115, 475–477. [Google Scholar] [CrossRef]
- Liao, L.; Zhou, Q.; Song, Y.; Wu, W.; Yu, H.; Wang, S.; Chen, Y.; Ye, M.; Lu, L. Ceramide mediates Ox-LDL-induced human vascular smooth muscle cell calcification via p38 mitogen-activated protein kinase signaling. PLoS ONE 2013, 8, e82379. [Google Scholar] [CrossRef]
- Chowdhury, A.; Sarkar, J.; Pramanik, P.K.; Chakraborti, T.; Chakraborti, S. Cross talk between MMP2-Spm-Cer-S1P and ERK1/2 in proliferation of pulmonary artery smooth muscle cells under angiotensin II stimulation. Arch. Biochem. Biophys. 2016, 603, 91–101. [Google Scholar] [CrossRef]
- Akwii, R.G.; Sajib, M.S.; Zahra, F.T.; Mikelis, C.M. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells 2019, 8, 471. [Google Scholar] [CrossRef]
- Wallach, D.; Kang, T.-B.; Kovalenko, A. Concepts of tissue injury and cell death in inflammation: A historical perspective. Nat. Rev. Immunol. 2014, 14, 51–59. [Google Scholar] [CrossRef]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The Master Regulator of Immunity to Infection. J. Immunol. 2008, 180, 5771. [Google Scholar] [CrossRef]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Lambert, V.; Munaut, C.; Jost, M.; Noël, A.; Werb, Z.; Foidart, J.M.; Rakic, J.M. Matrix metalloproteinase-9 contributes to choroidal neovascularization. Am. J. Pathol. 2002, 161, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yoshida, S.; Kubo, Y.; Kobayashi, Y.; Nakama, T.; Yamaguchi, M.; Ishikawa, K.; Nakao, S.; Ikeda, Y.; Ishibashi, T.; et al. Interleukin-12 inhibits pathological neovascularization in mouse model of oxygen-induced retinopathy. Sci. Rep. 2016, 6, 28140. [Google Scholar] [CrossRef]
- Wang, J.; Tian, Y.; Phillips, K.L.; Chiverton, N.; Haddock, G.; Bunning, R.A.; Cross, A.K.; Shapiro, I.M.; Le Maitre, C.L.; Risbud, M.V. Tumor necrosis factor alpha- and interleukin-1beta-dependent induction of CCL3 expression by nucleus pulposus cells promotes macrophage migration through CCR1. Arthritis Rheum 2013, 65, 832–842. [Google Scholar] [CrossRef]
- Knapp, K.M.; English, B.K. Ceramide-mediated stimulation of inducible nitric oxide synthase (iNOS) and tumor necrosis factor (TNF) accumulation in murine macrophages requires tyrosine kinase activity. J. Leukoc. Biol. 2000, 67, 735–741. [Google Scholar] [CrossRef]
- Hammad, S.M.; Crellin, H.G.; Wu, B.X.; Melton, J.; Anelli, V.; Obeid, L.M. Dual and distinct roles for sphingosine kinase 1 and sphingosine 1 phosphate in the response to inflammatory stimuli in RAW macrophages. Prostaglandins Other Lipid Mediat. 2008, 85, 107–114. [Google Scholar] [CrossRef]
- Xiong, Y.; Lee, H.J.; Mariko, B.; Lu, Y.C.; Dannenberg, A.J.; Haka, A.S.; Maxfield, F.R.; Camerer, E.; Proia, R.L.; Hla, T. Sphingosine kinases are not required for inflammatory responses in macrophages. J. Biol. Chem. 2013, 288, 32563–32573. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilkerson, J.L.; Basu, S.K.; Stiles, M.A.; Prislovsky, A.; Grambergs, R.C.; Nicholas, S.E.; Karamichos, D.; Allegood, J.C.; Proia, R.L.; Mandal, N. Ablation of Sphingosine Kinase 1 Protects Cornea from Neovascularization in a Mouse Corneal Injury Model. Cells 2022, 11, 2914. https://doi.org/10.3390/cells11182914
Wilkerson JL, Basu SK, Stiles MA, Prislovsky A, Grambergs RC, Nicholas SE, Karamichos D, Allegood JC, Proia RL, Mandal N. Ablation of Sphingosine Kinase 1 Protects Cornea from Neovascularization in a Mouse Corneal Injury Model. Cells. 2022; 11(18):2914. https://doi.org/10.3390/cells11182914
Chicago/Turabian StyleWilkerson, Joseph L., Sandip K. Basu, Megan A. Stiles, Amanda Prislovsky, Richard C. Grambergs, Sarah E. Nicholas, Dimitrios Karamichos, Jeremy C. Allegood, Richard L. Proia, and Nawajes Mandal. 2022. "Ablation of Sphingosine Kinase 1 Protects Cornea from Neovascularization in a Mouse Corneal Injury Model" Cells 11, no. 18: 2914. https://doi.org/10.3390/cells11182914