
Macrophages Upregulate Estrogen Receptor Expression in the Model of Obesity-Associated Breast Carcinoma
 and
and
Abstract
:1. Introduction
2. Materials and Methods
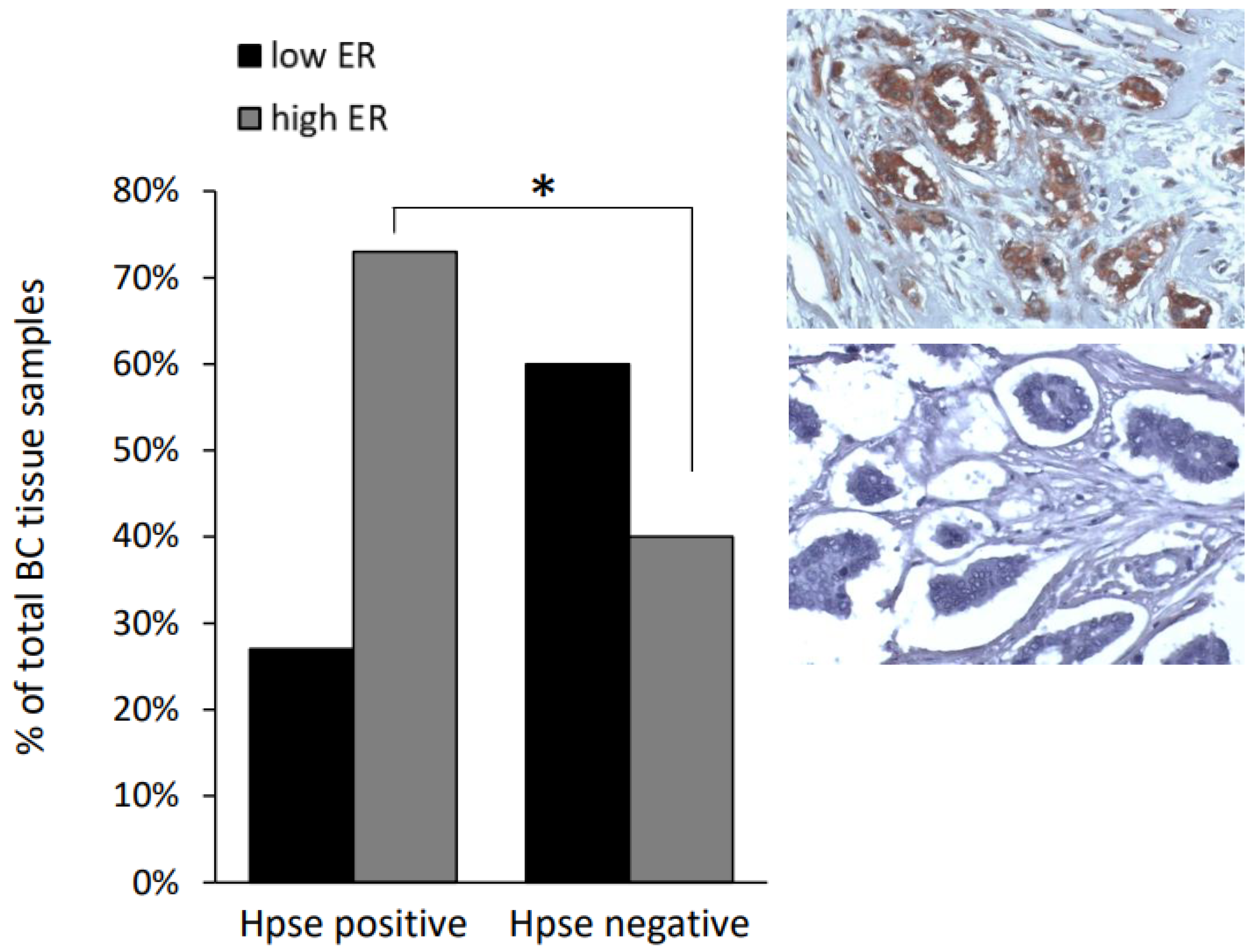
2.1. Clinical Data Analysis
2.2. Cell Culture
2.3. Macrophage Isolation and Treatment
2.4. MTS Assay
2.5. Orthotopic Immunocompetent Mouse Model of Obesity-Associated Breast Cancer
2.6. Immunoblotting
2.7. Antibodies
2.8. Immunofluorescence
2.9. Analysis of Gene Expression by Quantitative Real-Time PCR (qRT-PCR)
2.10. Statistical Analysis
3. Results
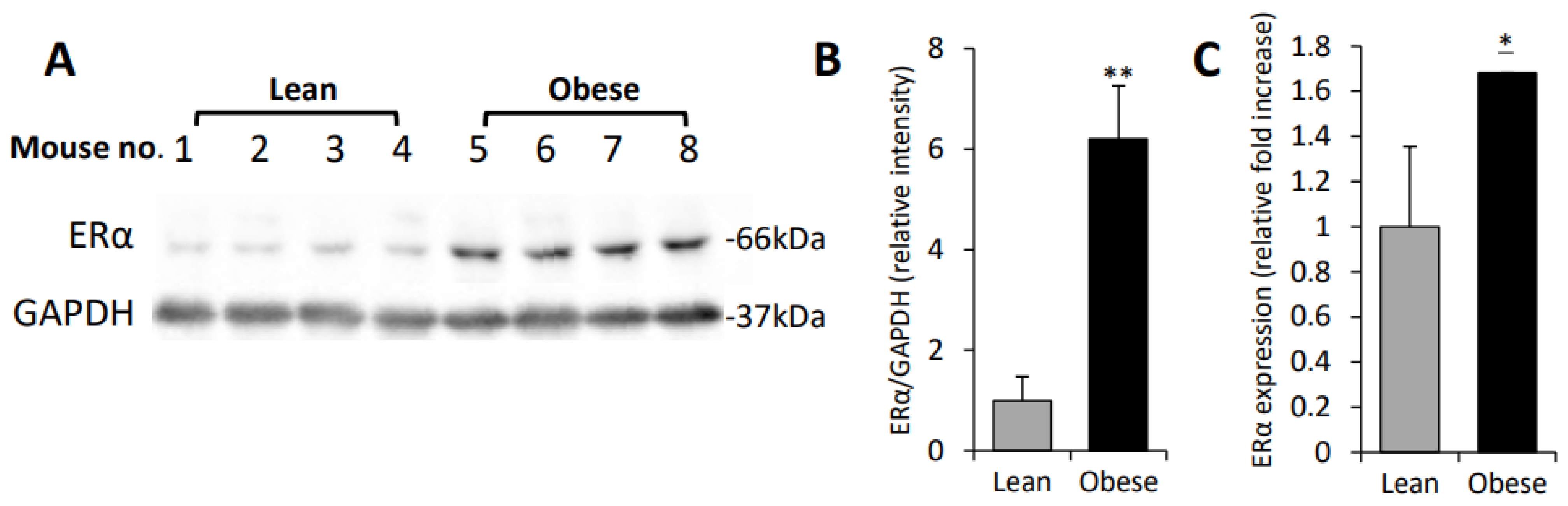
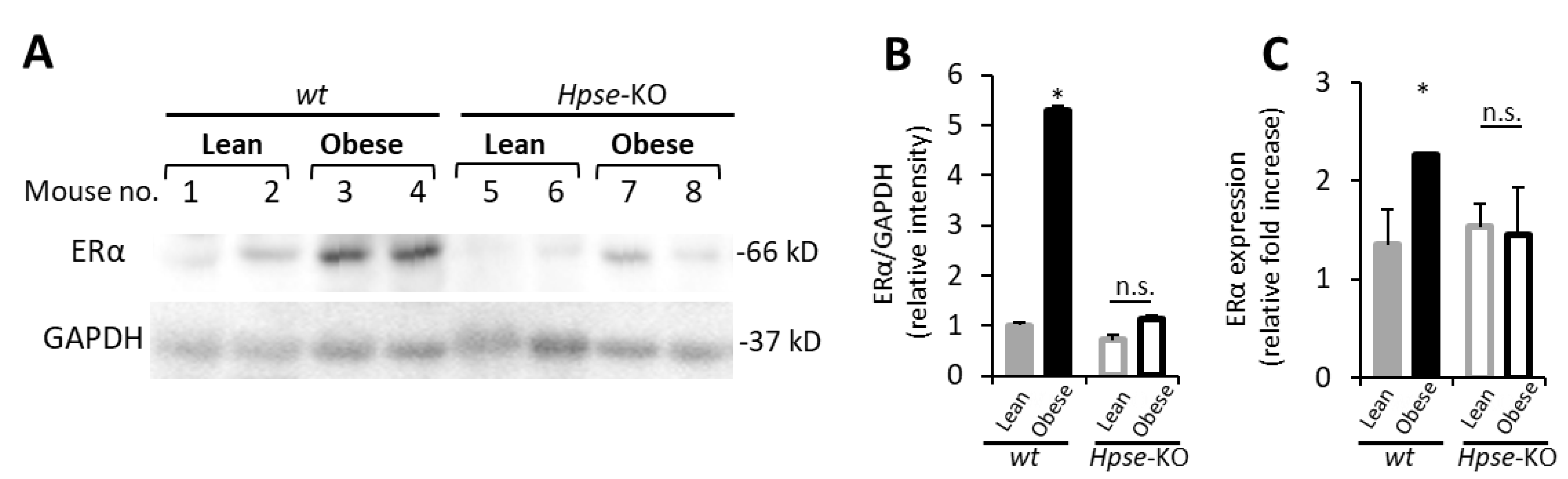
3.1. Increased ERα Expression Levels in Obesity-Associated E0771 Murine Breast Carcinoma
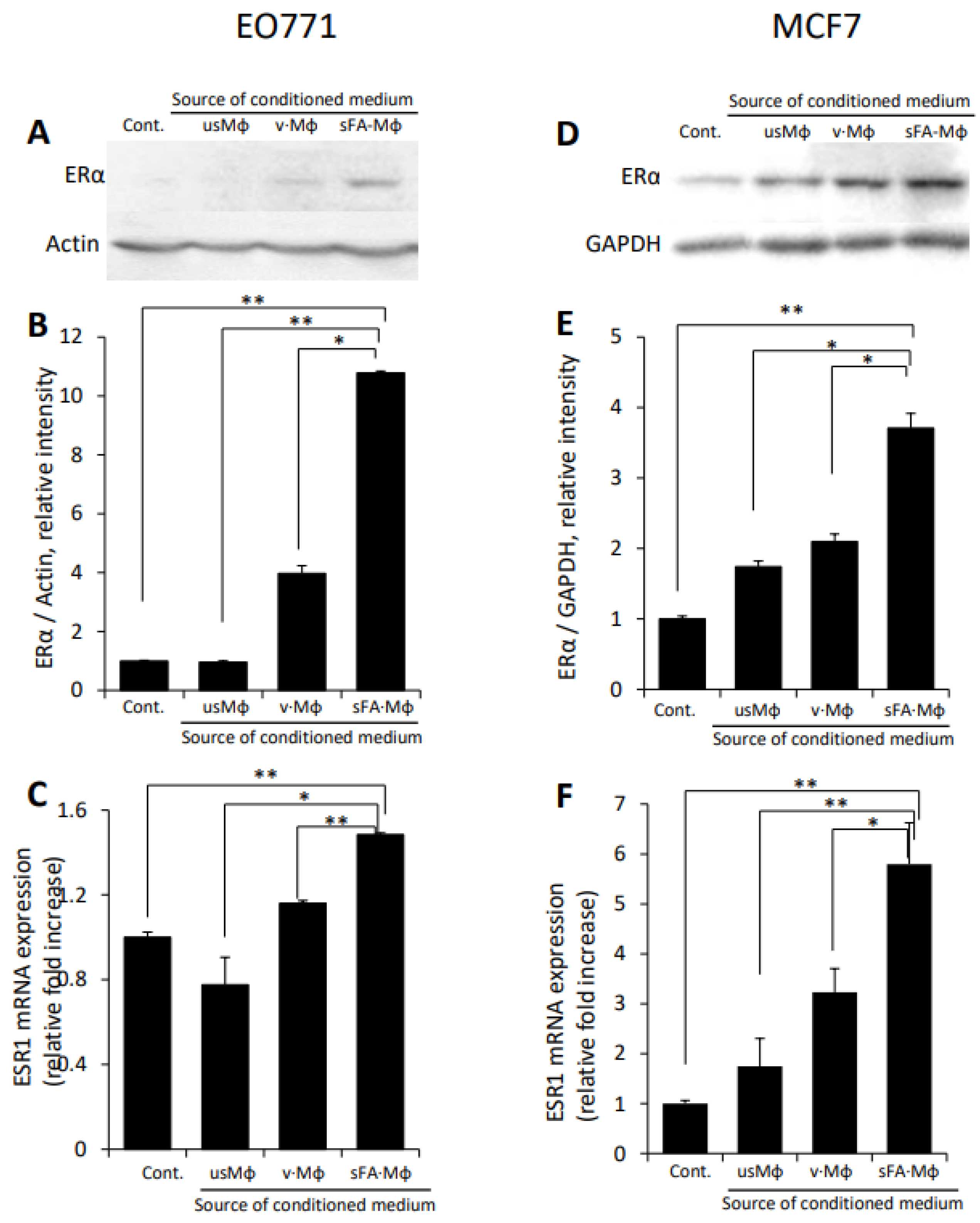
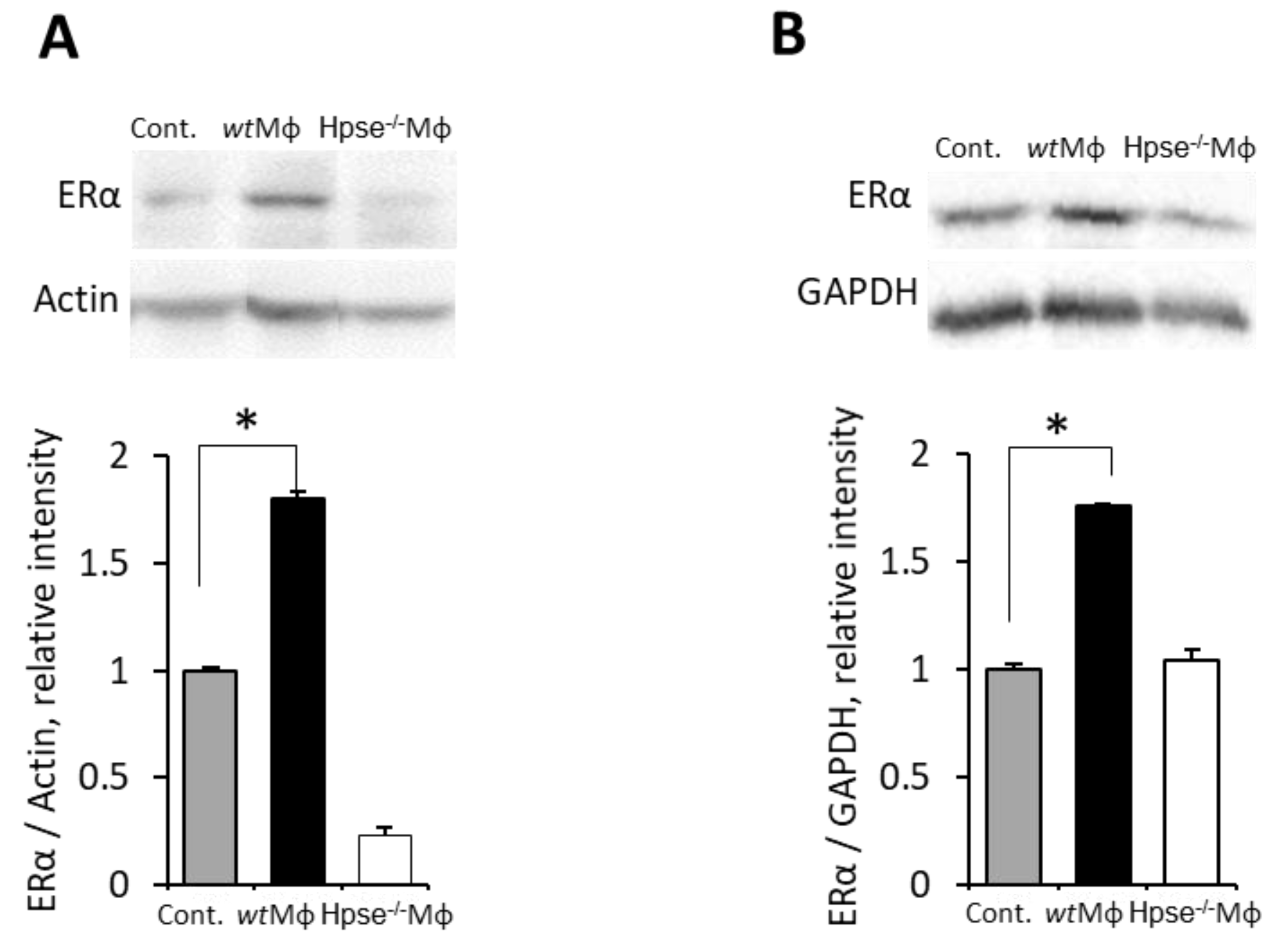
3.2. Macrophages Stimulated by Obese Milieu Components Mediate Upregulation of ERα in BC Cells
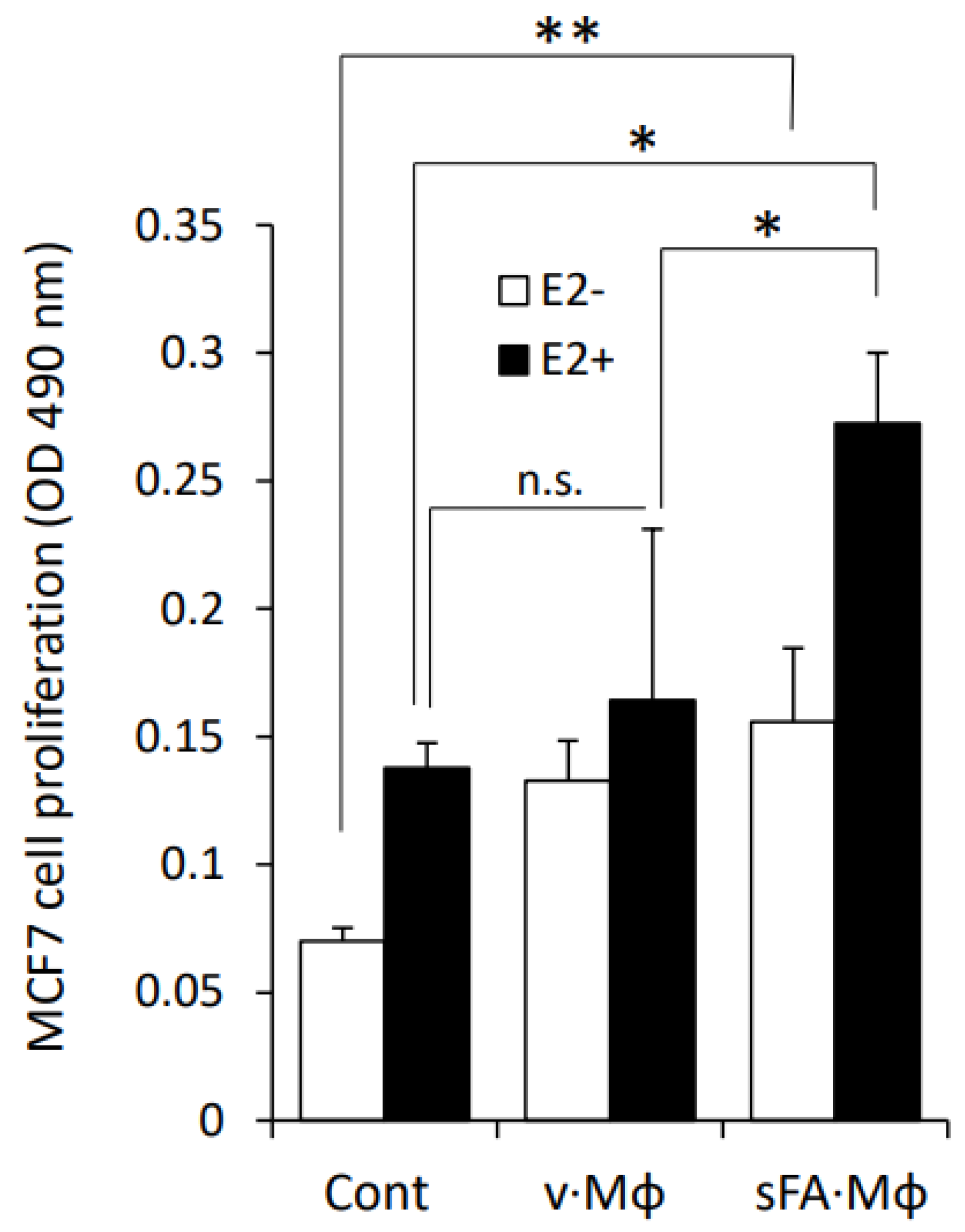
3.3. Macrophages Stimulated by SFA Increase the Sensitivity of BC Cells to Estrogen
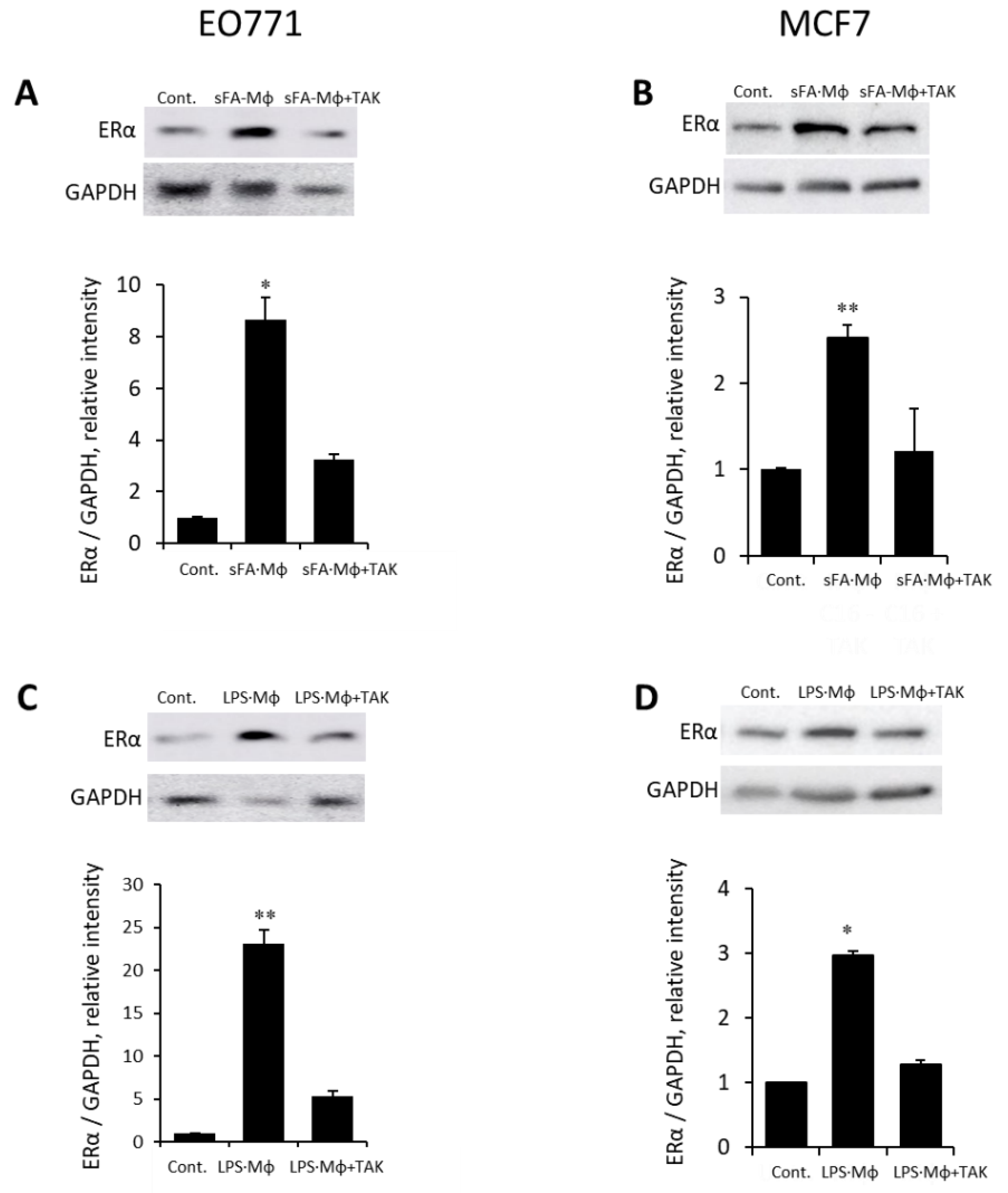
3.4. Mϕ-Mediated Augmentation of ER Expression in BC Is Dependent on Heparanase, the Endoglycosidase Enzyme Essential for Mϕ Reactivity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Prat, A.; Pineda, E.; Adamo, B.; Galvan, P.; Fernandez, A.; Gaba, L.; Diez, M.; Viladot, M.; Arance, A.; Munoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. S2), S26–S35. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Caan, B.J.; Sweeney, C.; Habel, L.A.; Kwan, M.L.; Kroenke, C.H.; Weltzien, E.K.; Quesenberry, C.P., Jr.; Castillo, A.; Factor, R.E.; Kushi, L.H.; et al. Intrinsic subtypes from the PAM50 gene expression assay in a population-based breast cancer survivor cohort: Prognostication of short- and long-term outcomes. Cancer Epidemiol. Biomark. Prev. 2014, 23, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Piperigkou, Z.; Karamanos, N.K. Estrogen receptor-mediated targeting of the extracellular matrix network in cancer. Semin. Cancer Biol. 2020, 62, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Kong, Q.; Jiang, Y. Mechanisms for estrogen receptor expression in human cancer. Exp. Hematol. Oncol. 2018, 7, 24. [Google Scholar] [CrossRef]
- Thomas, C.; Gustafsson, J.A. The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef]
- Rong, C.; Meinert, E.; Hess, J. Estrogen Receptor Signaling in Radiotherapy: From Molecular Mechanisms to Clinical Studies. Int. J. Mol. Sci. 2018, 19, 713. [Google Scholar] [CrossRef]
- Siersbaek, R.; Kumar, S.; Carroll, J.S. Signaling pathways and steroid receptors modulating estrogen receptor alpha function in breast cancer. Genes Dev. 2018, 32, 1141–1154. [Google Scholar] [CrossRef]
- Elkin, M.; Cohen, I.; Zcharia, E.; Orgel, A.; Guatta-Rangini, Z.; Peretz, T.; Vlodavsky, I.; Kleinman, H.K. Regulation of heparanase gene expression by estrogen in breast cancer. Cancer Res. 2003, 63, 8821–8826. [Google Scholar]
- Elkin, M.; Orgel, A.; Kleinman, H.K. An angiogenic switch in breast cancer involves estrogen and soluble vascular endothelial growth factor receptor 1. J. Natl. Cancer Inst. 2004, 96, 875–878. [Google Scholar] [CrossRef] [Green Version]
- Lung, D.K.; Reese, R.M.; Alarid, E.T. Intrinsic and Extrinsic Factors Governing the Transcriptional Regulation of ESR1. Horm Cancer 2020, 11, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Blair, C.K.; Wiggins, C.L.; Nibbe, A.M.; Storlie, C.B.; Prossnitz, E.R.; Royce, M.; Lomo, L.C.; Hill, D.A. Obesity and survival among a cohort of breast cancer patients is partially mediated by tumor characteristics. NPJ Breast Cancer 2019, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; International Agency for Research on Cancer Handbook Working. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Stuttard, J.; Zhou, B.; Kontis, V.; Bentham, J.; Gunter, M.J.; Ezzati, M. Worldwide burden of cancer attributable to diabetes and high body-mass index: A comparative risk assessment. Lancet Diabetes Endocrinol. 2018, 6, e6–e15. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Baumgarten, S.C.; Frasor, J. Minireview: Inflammation: An instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol. Endocrinol. 2012, 26, 360–371. [Google Scholar] [CrossRef]
- Cleary, M.P.; Grossmann, M.E. Minireview: Obesity and breast cancer: The estrogen connection. Endocrinology 2009, 150, 2537–2542. [Google Scholar] [CrossRef]
- Lee, K.; Kruper, L.; Dieli-Conwright, C.M.; Mortimer, J.E. The Impact of Obesity on Breast Cancer Diagnosis and Treatment. Curr. Oncol. Rep. 2019, 21, 41. [Google Scholar] [CrossRef]
- Gerard, C.; Brown, K.A. Obesity and breast cancer—Role of estrogens and the molecular underpinnings of aromatase regulation in breast adipose tissue. Mol. Cell Endocrinol. 2018, 466, 15–30. [Google Scholar] [CrossRef]
- Santa-Maria, C.A.; Yan, J.; Xie, X.J.; Euhus, D.M. Aggressive estrogen-receptor-positive breast cancer arising in patients with elevated body mass index. Int. J. Clin. Oncol. 2015, 20, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.S.; Norat, T. Obesity and breast cancer: Not only a risk factor of the disease. Curr. Treat Options Oncol. 2015, 16, 22. [Google Scholar] [CrossRef]
- Sestak, I.; Distler, W.; Forbes, J.F.; Dowsett, M.; Howell, A.; Cuzick, J. Effect of body mass index on recurrences in tamoxifen and anastrozole treated women: An exploratory analysis from the ATAC trial. J. Clin. Oncol. 2010, 28, 3411–3415. [Google Scholar] [CrossRef]
- Park, Y.; Colditz, G.A. Diabetes and adiposity: A heavy load for cancer. Lancet Diabetes Endocrinol. 2018, 6, 82–83. [Google Scholar] [CrossRef]
- Cespedes Feliciano, E.M.; Kwan, M.L.; Kushi, L.H.; Chen, W.Y.; Weltzien, E.K.; Castillo, A.L.; Sweeney, C.; Bernard, P.S.; Caan, B.J. Body mass index, PAM50 subtype, recurrence, and survival among patients with nonmetastatic breast cancer. Cancer 2017, 123, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Neuhouser, M.L.; Aragaki, A.K.; Prentice, R.L.; Manson, J.E.; Chlebowski, R.; Carty, C.L.; Ochs-Balcom, H.M.; Thomson, C.A.; Caan, B.J.; Tinker, L.F.; et al. Overweight, Obesity, and Postmenopausal Invasive Breast Cancer Risk: A Secondary Analysis of the Women’s Health Initiative Randomized Clinical Trials. JAMA Oncol. 2015, 1, 611–621. [Google Scholar] [CrossRef]
- Nattenmuller, C.J.; Kriegsmann, M.; Sookthai, D.; Fortner, R.T.; Steffen, A.; Walter, B.; Johnson, T.; Kneisel, J.; Katzke, V.; Bergmann, M.; et al. Obesity as risk factor for subtypes of breast cancer: Results from a prospective cohort study. BMC Cancer 2018, 18, 616. [Google Scholar] [CrossRef] [PubMed]
- AACR. AACR Cancer Progress Report 2021. Available online: https://cancerprogressreport.aacr.org/progress/ (accessed on 1 June 2022).
- Bhardwaj, P.; Au, C.C.; Benito-Martin, A.; Ladumor, H.; Oshchepkova, S.; Moges, R.; Brown, K.A. Estrogens and breast cancer: Mechanisms involved in obesity-related development, growth and progression. J. Steroid. Biochem. Mol. Biol. 2019, 189, 161–170. [Google Scholar] [CrossRef]
- Hermano, E.; Goldberg, R.; Rubinstein, A.M.; Sonnenblick, A.; Maly, B.; Nahmias, D.; Li, J.P.; Bakker, M.A.H.; van der Vlag, J.; Vlodavsky, I.; et al. Heparanase Accelerates Obesity-Associated Breast Cancer Progression. Cancer Res. 2019, 79, 5342–5354. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Iyengar, N.M.; Zhou, X.K.; Gucalp, A.; Subbaramaiah, K.; Wang, H.; Giri, D.D.; Morrow, M.; Falcone, D.J.; Wendel, N.K.; et al. Menopause Is a Determinant of Breast Aromatase Expression and Its Associations With BMI, Inflammation, and Systemic Markers. J. Clin. Endocrinol. Metab. 2017, 102, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Zahid, H.; Simpson, E.R.; Brown, K.A. Inflammation, dysregulated metabolism and aromatase in obesity and breast cancer. Curr. Opin. Pharm. 2016, 31, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [PubMed]
- Incio, J.; Tam, J.; Rahbari, N.N.; Suboj, P.; McManus, D.T.; Chin, S.M.; Vardam, T.D.; Batista, A.; Babykutty, S.; Jung, K.; et al. PlGF/VEGFR-1 Signaling Promotes Macrophage Polarization and Accelerated Tumor Progression in Obesity. Clin. Cancer Res. 2016, 22, 2993–3004. [Google Scholar] [CrossRef]
- Trinchieri, G. Cancer and inflammation: An old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D.; Haymond, M.W.; Rizza, R.A.; Cryer, P.E.; Miles, J.M. Influence of body fat distribution on free fatty acid metabolism in obesity. J. Clin. Investig. 1989, 83, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Lager, S.; Gaccioli, F.; Ramirez, V.I.; Jones, H.N.; Jansson, T.; Powell, T.L. Oleic acid stimulates system A amino acid transport in primary human trophoblast cells mediated by toll-like receptor 4. J. Lipid Res. 2013, 54, 725–733. [Google Scholar] [CrossRef]
- Tsukumo, D.M.; Carvalho-Filho, M.A.; Carvalheira, J.B.; Prada, P.O.; Hirabara, S.M.; Schenka, A.A.; Araujo, E.P.; Vassallo, J.; Curi, R.; Velloso, L.A.; et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 2007, 56, 1986–1998. [Google Scholar] [CrossRef]
- Bigornia, S.J.; Farb, M.G.; Mott, M.M.; Hess, D.T.; Carmine, B.; Fiscale, A.; Joseph, L.; Apovian, C.M.; Gokce, N. Relation of depot-specific adipose inflammation to insulin resistance in human obesity. Nutr. Diabetes 2012, 2, e30. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Brown, K.A. Obese Adipose Tissue as a Driver of Breast Cancer Growth and Development: Update and Emerging Evidence. Front Oncol. 2021, 11, 638918. [Google Scholar] [CrossRef]
- Brown, K.A. Metabolic pathways in obesity-related breast cancer. Nat. Rev. Endocrinol. 2021, 17, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Ning, C.; Xie, B.; Zhang, L.; Li, C.; Shan, W.; Yang, B.; Luo, X.; Gu, C.; He, Q.; Jin, H.; et al. Infiltrating Macrophages Induce ERalpha Expression through an IL17A-mediated Epigenetic Mechanism to Sensitize Endometrial Cancer Cells to Estrogen. Cancer Res. 2016, 76, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, W.J.; Izumi, K.; Jiang, Q.; Lai, K.P.; Xu, D.; Fang, L.Y.; Lu, T.; Li, L.; Xia, S.; et al. Increased infiltrated macrophages in benign prostatic hyperplasia (BPH): Role of stromal androgen receptor in macrophage-induced prostate stromal cell proliferation. J. Biol. Chem. 2012, 287, 18376–18385. [Google Scholar] [CrossRef]
- Masola, V.; Zaza, G.; Bellin, G.; Dall’Olmo, L.; Granata, S.; Vischini, G.; Secchi, M.F.; Lupo, A.; Gambaro, G.; Onisto, M. Heparanase regulates the M1 polarization of renal macrophages and their crosstalk with renal epithelial tubular cells after ischemia/reperfusion injury. FASEB J. 2018, 32, 742–756. [Google Scholar] [CrossRef]
- Mayfosh, A.J.; Baschuk, N.; Hulett, M.D. Leukocyte Heparanase: A Double-Edged Sword in Tumor Progression. Front Oncol. 2019, 9, 331. [Google Scholar] [CrossRef] [PubMed]
- Gutter-Kapon, L.; Alishekevitz, D.; Shaked, Y.; Li, J.P.; Aronheim, A.; Ilan, N.; Vlodavsky, I. Heparanase is required for activation and function of macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, E7808–E7817. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.; Rubinstein, A.M.; Gil, N.; Hermano, E.; Li, J.P.; van der Vlag, J.; Atzmon, R.; Meirovitz, A.; Elkin, M. Role of heparanase-driven inflammatory cascade in pathogenesis of diabetic nephropathy. Diabetes 2014, 63, 4302–4313. [Google Scholar] [CrossRef]
- Hermano, E.; Carlotti, F.; Abecassis, A.; Meirovitz, A.; Rubinstein, A.M.; Li, J.P.; Vlodavsky, I.; Rabelink, T.J.; Elkin, M. Dichotomic role of heparanase in a murine model of metabolic syndrome. Cell Mol. Life Sci. 2021, 78, 2771–2780. [Google Scholar] [CrossRef] [PubMed]
- Hermano, E.; Meirovitz, A.; Meir, K.; Nussbaum, G.; Appelbaum, L.; Peretz, T.; Elkin, M. Macrophage polarization in pancreatic carcinoma: Role of heparanase enzyme. J. Natl. Cancer Inst. 2014, 106, dju332. [Google Scholar] [CrossRef]
- Lerner, I.; Hermano, E.; Zcharia, E.; Rodkin, D.; Bulvik, R.; Doviner, V.; Rubinstein, A.M.; Ishai-Michaeli, R.; Atzmon, R.; Sherman, Y.; et al. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J. Clin. Investig. 2011, 121, 1709–1721. [Google Scholar] [CrossRef]
- Zahavi, T.; Salmon-Divon, M.; Salgado, R.; Elkin, M.; Hermano, E.; Rubinstein, A.M.; Francis, P.A.; Di Leo, A.; Viale, G.; de Azambuja, E.; et al. Heparanase: A potential marker of worse prognosis in estrogen receptor-positive breast cancer. NPJ Breast Cancer 2021, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Maly, B.; Simon, I.; Meirovitz, A.; Pikarsky, E.; Zcharia, E.; Peretz, T.; Vlodavsky, I.; Elkin, M. Tamoxifen induces heparanase expression in estrogen receptor-positive breast cancer. Clin. Cancer Res. 2007, 13, 4069–4077. [Google Scholar] [CrossRef]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. Arch. Pathol. Lab. Med. 2010, 134, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.; Treaba, D.O.; Speers, C.H.; Olivotto, I.A.; Bajdik, C.D.; Chia, S.K.; Goldstein, L.C.; Gelmon, K.A.; Huntsman, D.; Gilks, C.B.; et al. Immunohistochemical detection using the new rabbit monoclonal antibody SP1 of estrogen receptor in breast cancer is superior to mouse monoclonal antibody 1D5 in predicting survival. J. Clin. Oncol. 2006, 24, 5637–5644. [Google Scholar] [CrossRef]
- Zcharia, E.; Jia, J.; Zhang, X.; Baraz, L.; Lindahl, U.; Peretz, T.; Vlodavsky, I.; Li, J.P. Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS ONE 2009, 4, e5181. [Google Scholar] [CrossRef]
- Meirovitz, A.; Hermano, E.; Lerner, I.; Zcharia, E.; Pisano, C.; Peretz, T.; Elkin, M. Role of heparanase in radiation-enhanced invasiveness of pancreatic carcinoma. Cancer Res. 2011, 71, 2772–2780. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.M.; Arendt, L.M.; Alarid, E.T.; Beebe, D.J.; Johnson, B.P. Mammary adipose stromal cells derived from obese women reduce sensitivity to the aromatase inhibitor anastrazole in an organotypic breast model. FASEB J. 2019, 33, 8623–8633. [Google Scholar] [CrossRef] [PubMed]
- Maguire, O.A.; Ackerman, S.E.; Szwed, S.K.; Maganti, A.V.; Marchildon, F.; Huang, X.; Kramer, D.J.; Rosas-Villegas, A.; Gelfer, R.G.; Turner, L.E.; et al. Creatine-mediated crosstalk between adipocytes and cancer cells regulates obesity-driven breast cancer. Cell Metab. 2021, 33, 499–512.e496. [Google Scholar] [CrossRef]
- Le Naour, A.; Rossary, A.; Vasson, M.P. EO771, is it a well-characterized cell line for mouse mammary cancer model? Limit and uncertainty. Cancer Med. 2020, 9, 8074–8085. [Google Scholar] [CrossRef]
- Hiraga, T.; Ninomiya, T. Establishment and characterization of a C57BL/6 mouse model of bone metastasis of breast cancer. J. Bone Min. Metab. 2019, 37, 235–242. [Google Scholar] [CrossRef]
- Gu, J.W.; Young, E.; Busby, B.; Covington, J.; Johnson, J.W. Oral administration of pyrrolidine dithiocarbamate (PDTC) inhibits VEGF expression, tumor angiogenesis, and growth of breast cancer in female mice. Cancer Biol. Ther. 2009, 8, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, M.K.; Hallahan, N.L.; Brown, S.H.; Liu, M.; Mitchell, T.W.; Cooney, G.J.; Turner, N. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 2013, 56, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Kanfi, Y.; Peshti, V.; Gil, R.; Naiman, S.; Nahum, L.; Levin, E.; Kronfeld-Schor, N.; Cohen, H.Y. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 2010, 9, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Mentoor, I.; Nell, T.; Emjedi, Z.; van Jaarsveld, P.J.; de Jager, L.; Engelbrecht, A.M. Decreased Efficacy of Doxorubicin Corresponds with Modifications in Lipid Metabolism Markers and Fatty Acid Profiles in Breast Tumors From Obese vs. Lean Mice. Front. Oncol. 2020, 10, 306. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, U.S.; Walden, T.B.; Carlsson, P.O.; Jansson, L.; Phillipson, M. Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue. PLoS ONE 2012, 7, e46057. [Google Scholar] [CrossRef] [PubMed]
- Chantalat, E.; Boudou, F.; Laurell, H.; Palierne, G.; Houtman, R.; Melchers, D.; Rochaix, P.; Filleron, T.; Stella, A.; Burlet-Schiltz, O.; et al. The AF-1-deficient estrogen receptor ERalpha46 isoform is frequently expressed in human breast tumors. Breast Cancer Res. 2016, 18, 123. [Google Scholar] [CrossRef]
- Li, L.; Haynes, M.P.; Bender, J.R. Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4807–4812. [Google Scholar] [CrossRef]
- Majorini, M.T.; Cancila, V.; Rigoni, A.; Botti, L.; Dugo, M.; Triulzi, T.; De Cecco, L.; Fontanella, E.; Jachetti, E.; Tagliabue, E.; et al. Infiltrating Mast Cell-Mediated Stimulation of Estrogen Receptor Activity in Breast Cancer Cells Promotes the Luminal Phenotype. Cancer Res. 2020, 80, 2311–2324. [Google Scholar] [CrossRef]
- Koutsari, C.; Ali, A.H.; Mundi, M.S.; Jensen, M.D. Storage of circulating free fatty acid in adipose tissue of postabsorptive humans: Quantitative measures and implications for body fat distribution. Diabetes 2011, 60, 2032–2040. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Fu, J.; Zhang, L.; He, T.; Xiao, X.; Liu, X.; Wang, L.; Yang, L.; Yang, M.; Zhang, T.; Chen, R.; et al. TWIST represses estrogen receptor-alpha expression by recruiting the NuRD protein complex in breast cancer cells. Int. J. Biol. Sci. 2012, 8, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.W.; Graham, A.E.; Re, N.A.; Carr, I.M.; Robinson, J.I.; Mackie, S.L.; Morgan, A.W. Standardized protocols for differentiation of THP-1 cells to macrophages with distinct M(IFNgamma+LPS), M(IL-4) and M(IL-10) phenotypes. J. Immunol. Methods 2020, 478, 112721. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front Immunol. 2020, 11, 594150. [Google Scholar] [CrossRef]
- Cao, J.; Peng, J.; An, H.; He, Q.; Boronina, T.; Guo, S.; White, M.F.; Cole, P.A.; He, L. Endotoxemia-mediated activation of acetyltransferase P300 impairs insulin signaling in obesity. Nat. Commun. 2017, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferrieres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008, 87, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Troseid, M.; Nestvold, T.K.; Rudi, K.; Thoresen, H.; Nielsen, E.W.; Lappegard, K.T. Plasma lipopolysaccharide is closely associated with glycemic control and abdominal obesity: Evidence from bariatric surgery. Diabetes Care 2013, 36, 3627–3632. [Google Scholar] [CrossRef]
- Bergom, C.; Kelly, T.; Bedi, M.; Saeed, H.; Prior, P.; Rein, L.E.; Szabo, A.; Wilson, J.F.; Currey, A.D.; White, J. Association of Locoregional Control With High Body Mass Index in Women Undergoing Breast Conservation Therapy for Early-Stage Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 65–71. [Google Scholar] [CrossRef]
- Brunn, G.J.; Bungum, M.K.; Johnson, G.B.; Platt, J.L. Conditional signaling by Toll-like receptor 4. FASEB J. 2005, 19, 872–874. [Google Scholar] [CrossRef]
- Goodall, K.J.; Poon, I.K.; Phipps, S.; Hulett, M.D. Soluble Heparan Sulfate Fragments Generated by Heparanase Trigger the Release of Pro-Inflammatory Cytokines through TLR-4. PLoS ONE 2014, 9, e109596. [Google Scholar] [CrossRef]
- Johnson, G.B.; Brunn, G.J.; Kodaira, Y.; Platt, J.L. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J. Immunol. 2002, 168, 5233–5239. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Wang, L.; Chen, S. Endogenous toll-like receptor ligands and their biological significance. J. Cell Mol. Med. 2010, 14, 2592–2603. [Google Scholar] [CrossRef] [PubMed]
- Akbarshahi, H.; Axelsson, J.B.; Said, K.; Malmstrom, A.; Fischer, H.; Andersson, R. TLR4 dependent heparan sulphate-induced pancreatic inflammatory response is IRF3-mediated. J. Transl. Med. 2011, 9, 219. [Google Scholar] [CrossRef]
- WHO. Obesity and Overweight, World Health Organization Fact Sheet. Available online: http://www.who.int/mediacentre/factsheets/fs311/en/2018 (accessed on 25 May 2022).
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- McTiernan, A.; Wu, L.; Chen, C.; Chlebowski, R.; Mossavar-Rahmani, Y.; Modugno, F.; Perri, M.G.; Stanczyk, F.Z.; Van Horn, L.; Wang, C.Y.; et al. Relation of BMI and physical activity to sex hormones in postmenopausal women. Obesity 2006, 14, 1662–1677. [Google Scholar] [CrossRef] [PubMed]
- Winuthayanon, W.; Lierz, S.L.; Delarosa, K.C.; Sampels, S.R.; Donoghue, L.J.; Hewitt, S.C.; Korach, K.S. Juxtacrine Activity of Estrogen Receptor alpha in Uterine Stromal Cells is Necessary for Estrogen-Induced Epithelial Cell Proliferation. Sci. Rep. 2017, 7, 8377. [Google Scholar] [CrossRef] [PubMed]
- Noordhoek, I.; de Groot, A.F.; Cohen, D.; Liefers, G.J.; Portielje, J.E.A.; Kroep, J.R. Higher ER load is not associated with better outcome in stage 1-3 breast cancer: A descriptive overview of quantitative HR analysis in operable breast cancer. Breast Cancer Res. Treat 2019, 176, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Pepe, G.; Locati, M.; Della Torre, S.; Mornata, F.; Cignarella, A.; Maggi, A.; Vegeto, E. The estrogen-macrophage interplay in the homeostasis of the female reproductive tract. Hum. Reprod. Update 2018, 24, 652–672. [Google Scholar] [CrossRef] [PubMed]
- Hillers-Ziemer, L.E.; McMahon, R.Q.; Hietpas, M.; Paderta, G.; LeBeau, J.; McCready, J.; Arendt, L.M. Obesity Promotes Cooperation of Cancer Stem-Like Cells and Macrophages to Enhance Mammary Tumor Angiogenesis. Cancers 2020, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Recht, L.; Strober, S. The Promise of Targeting Macrophages in Cancer Therapy. Clin. Cancer Res. 2017, 23, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Chamberlin, T.; Clack, M.; Silvers, C.; Kuziel, G.; Thompson, V.; Johnson, H.; Arendt, L.M. Targeting Obesity-Induced Macrophages during Preneoplastic Growth Promotes Mammary Epithelial Stem/Progenitor Activity, DNA Damage, and Tumor Formation. Cancer Res. 2020, 80, 4465–4475. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Schultze, J.L.; Murray, P.J.; Ochando, J.; Biswas, S.K. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 2016, 17, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, A.; Brown, E.J.; Garabedian, M.L.; Pena, S.; Sharma, M.; Lafaille, J.; Moore, K.J.; Fisher, E.A. Single-Cell RNA Sequencing of Visceral Adipose Tissue Leukocytes Reveals that Caloric Restriction Following Obesity Promotes the Accumulation of a Distinct Macrophage Population with Features of Phagocytic Cells. Immunometabolism 2019, 1, e190008. [Google Scholar] [CrossRef]
- Khanna, M.; Parish, C.R. Heparanase: Historical Aspects and Future Perspectives. Adv. Exp. Med. Biol. 2020, 1221, 71–96. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criteria | Range | No. Cases (%) |
|---|---|---|
| Tumor size (cm) | 0.5–10.5 | |
| <2 | 31 (25.2) | |
| 2–5 | 50 (40.7) | |
| >5 | 3 (2.4) | |
| Unknown | 39 (31.7) | |
| Tumor type | ||
| Ductal | 109 (88.6) | |
| Lobular | 5 (4.1) | |
| Others | 9 (7.3) | |
| Lymph node status | ||
| LN pos | 38 (30.9) | |
| LN neg | 37 (30.1) | |
| Unknown | 48 (39) | |
| Grade | ||
| 1 | 19 (15.5) | |
| 2 | 46 (37.4) | |
| 3 | 56 (45.5) | |
| Unknown | 2 (1.6) | |
| ER status | ||
| Negative | 40 (32.5) | |
| Positive | 78 (63.4) | |
| Unknown | 5 (4.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nahmias Blank, D.; Hermano, E.; Sonnenblick, A.; Maimon, O.; Rubinstein, A.M.; Drai, E.; Maly, B.; Vlodavsky, I.; Popovtzer, A.; Peretz, T.; et al. Macrophages Upregulate Estrogen Receptor Expression in the Model of Obesity-Associated Breast Carcinoma. Cells 2022, 11, 2844. https://doi.org/10.3390/cells11182844
Nahmias Blank D, Hermano E, Sonnenblick A, Maimon O, Rubinstein AM, Drai E, Maly B, Vlodavsky I, Popovtzer A, Peretz T, et al. Macrophages Upregulate Estrogen Receptor Expression in the Model of Obesity-Associated Breast Carcinoma. Cells. 2022; 11(18):2844. https://doi.org/10.3390/cells11182844
Chicago/Turabian StyleNahmias Blank, Daniela, Esther Hermano, Amir Sonnenblick, Ofra Maimon, Ariel M. Rubinstein, Emmy Drai, Bella Maly, Israel Vlodavsky, Aron Popovtzer, Tamar Peretz, and et al. 2022. "Macrophages Upregulate Estrogen Receptor Expression in the Model of Obesity-Associated Breast Carcinoma" Cells 11, no. 18: 2844. https://doi.org/10.3390/cells11182844