
Demand Coupling Drives Neurodegeneration: A Model of Age-Related Cognitive Decline and Dementia

Abstract
:1. Introduction—Framing the Approach
2. Inadequacy of Current Model of Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
| Early-Onset (Classical) Alzheimer’s Disease | Age-Related Dementia | |
|---|---|---|
| Age of onset | 30–60 years | >60 years |
| Genetics | Monogenic/familial: PSEN1, PSEN2, APP. ~40–50% of mutations are sporadic. | Polygenic. Most significant risk gene (ApoE) contributes ~5% of total risk and is variably penetrant based on context and environment. |
| Clinical course | Homogeneous | Heterogeneous |
| Effect of environment | Minimal, or poorly described. | Substantial. Education, diet, exercise, cardiovascular and metabolic health, history of trauma, sleep, stress, pollutants, smoking, and alcohol all have a documented role. |
| Prevalence | ~1% of all AD cases and decreasing (previously estimated to represent 5% of all AD) [23]. | ~10% of individuals >65 and increasing (projected to double by 2050) [24]. ~99% of all AD cases [23]. |
3. Hierarchies of Explanation and Observations That the Model Must Account for
4. Population-Level Observations and Evolutionary Theory Derive the Demand-Driven Model of Age-Related Cognitive Decline
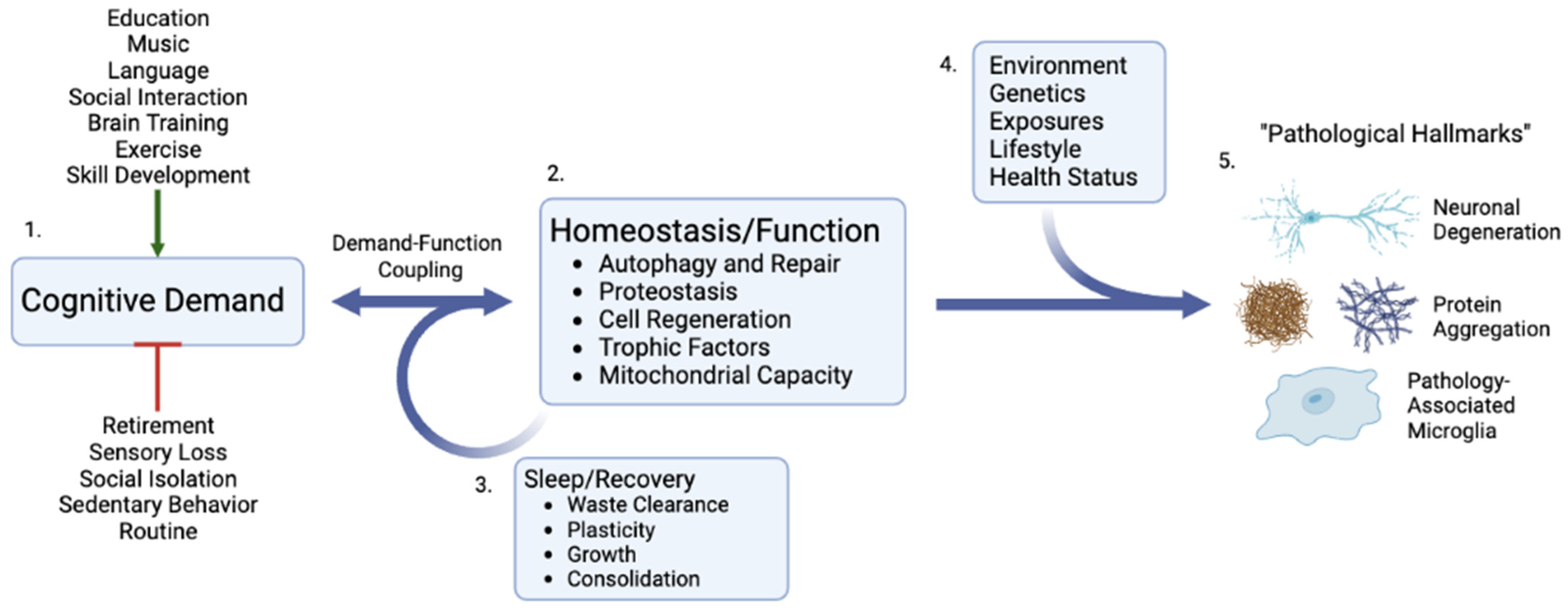
5. Demand as a Critical Stimulus and Initiator of Repair
6. Interventions—Stimuli with Known Benefits on Cognitive Headroom/Brain Aging
| Intervention/Exposure | Evidence |
|---|---|
| Exercise | |
| Language | |
| Music |
|
| Brain Training | |
| Social Connection | |
| Sensory Input |
7. Cognitive Demands across the Lifespan
7.1. Genetic Forces
7.2. Neurological Forces
7.3. Cultural Forces
7.4. Summary of Forces Conspiring to Hasten Our Neurological Demise
8. Supporting Growth and Repair in Response to Cognitive Demands
9. Summary of Model, Observations, and Supportive Evidence
- Absence in Early Life. As mentioned previously, the exclusive onset of ARCD and ARD in mid-to-late life, as well as the stability of brain structure and function in early life could conceivably be explained by either the presence of harmful factors in the adult environment that are absent in the childhood environment, or the presence of protective factors in the childhood environment that are absent in the adult environment. To date, no factors that fit the first criterion have been identified. However, the disproportionately sustained and heightened demands of the childhood developmental environment, via demand coupling, is a suitable candidate for the second criterion. The consistently high demands in early life and related upregulation of restorative mechanisms foster the preservation of brain tissue that counteracts deterioration driven by environmental mismatch. A significant and progressive drop in cognitive demands, as is currently typical of mid-life and beyond, would be expected to lead to progressive cognitive deterioration, including to degrees that are maladaptive and pathological.
- Heterogeneity of Pathology and Clinical Course. The heterogeneity in pathological findings and clinical course in ARDs and ARCD can also be entirely accounted for in this model. Both the accumulation of environmental contributors over time and the cognitive demands over the lifespan will significantly modify tissue structure and function. Together, these factors would produce wide variations in the clinical course, as is observed. Generally, individuals leading lifestyles of low mismatch that include expected physiological inputs (e.g., sleep, movement, social connection) and high cognitive demands throughout adulthood would be expected to display the lowest rates of deterioration. On the other end of the spectrum, those with lifestyles of high mismatch (e.g., sleep deprived, sedentary, isolated) and low cognitive demands throughout adulthood would exhibit the most rapid deterioration.
- Lack of Unique Environmental Exposures or Risk Factors. To date, no single genetic or environmental factor unique to the diseased population has been identified, indicating that the disease state is multifactorial in origin; the result of accelerated structural deterioration over time brought about by normal life sustaining functions in a range of environmental conditions. As stated, in this model, the differential rates of deterioration are largely explained by differing degrees of environmental mismatch and cognitive demands.
- Absence in Indigenous Populations, Explosive Growth With Industrialization. The relative absence of these conditions in indigenous populations and the explosive rise in prevalence during the industrial age is explained by the impact of this era on the two primary driving factors. On an evolutionary time scale, the industrial and information ages have ushered in a rapid and dramatic shift in every major facet of human life and a broad array of environmental changes that strain homeostatic capabilities. Not surprisingly, this has resulted in a rapid and dramatic increase in environmentally driven chronic diseases, including ARCD and ARDs [149]. The agricultural, industrial, and information ages have also dramatically altered the typical profile of cognitive activity across the human lifespan. The continued technological advances and globalization that characterize recent human history have led to an overriding trend of increasing specialization in the kinds of tasks humans perform in both the home and work environment. From a cognitive perspective, this has resulted in increasing reliance on a restricted set of automated cognitive capabilities to fulfill daily obligations. The net result of these recent major shifts in human life has been an unprecedented decrease in the cognitive demands placed on aging individuals at the population level. Left unaddressed, this reduction in cognitive demands over the course of adult life, alongside an increasing lifespan as medical and societal advances reduce both communicable and other non-communicable causes of disease, is likely to compound into an ever-increasing prevalence of ARCD and ARDs, as has been widely projected [150].
- Mitigation by Cognitive Activity. It is now well established that a cognitively active lifestyle is the single most protective factor associated with a reduction in risk of cognitive decline [10]. However, this association has been most commonly explained as cognitive activity compensating for deterioration brought on by the pathological condition, rather than as a primary cause of the disease state. In this model, cognitive activity is a central regulator of tissue maintenance. The nature and scope of cognitive activity must therefore be considered a primary driver of the disease state.
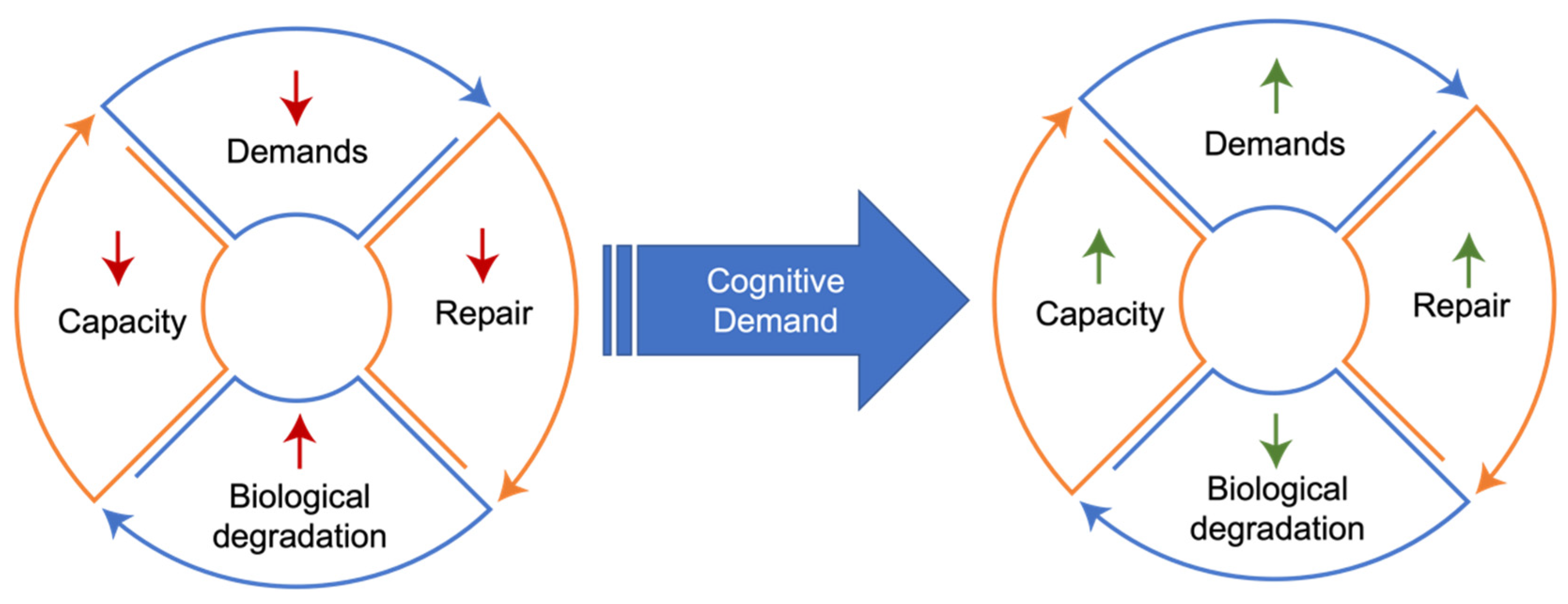
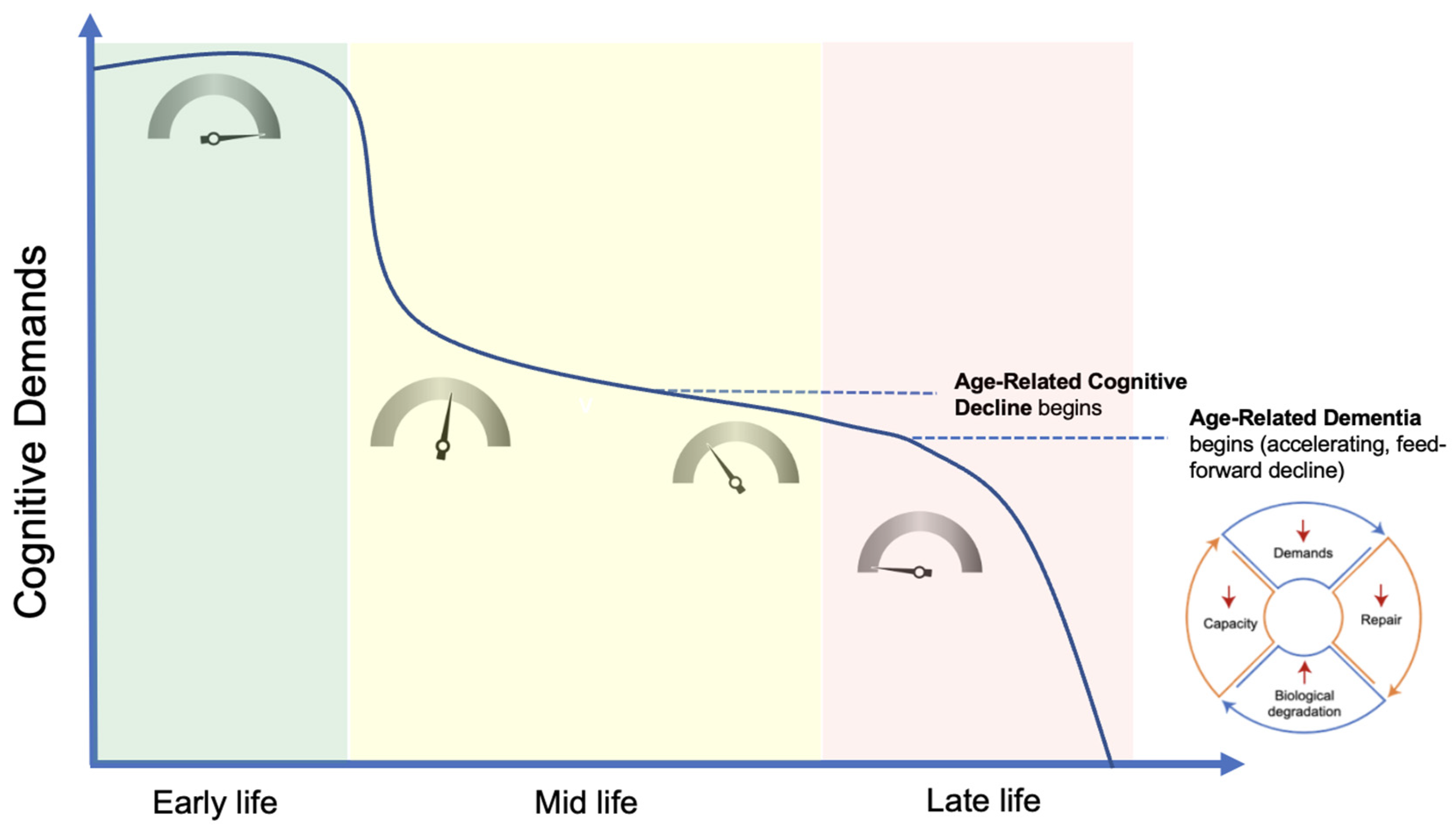
- Nonlinear Progression. The phenomenon of demand coupling would also be expected to produce a nonlinear, accelerating decline after cognitive function deteriorates beneath a certain threshold. As cognitive demands drop precipitously in mid-life, the downregulation of restorative mechanisms leads to greater deterioration and dysfunction, manifested by the beginnings of pathological accumulations and declining cognitive function. This process is then moderated by environmental exposures and the degree of mismatch. Once this process of deterioration and dysfunction progresses to the point of cognitive frailty, the range of potential cognitive challenges is constrained. Like physical frailty, cognitive frailty constrains the scope of cognitive activities that can be performed independently. This reduction in the scope of cognitive activities reduces cognitive demands, leading to further reduction in tissue restoration, and a positive feedback cycle is created, producing a nonlinear, accelerating decline (Figure 3).
10. Future Directions, Therapeutic Implications, and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rocca, W.A.; Petersen, R.C.; Knopman, D.S.; Hebert, L.E.; Evans, D.A.; Hall, K.S.; Gao, S.; Unverzagt, F.W.; Langa, K.M.; Larson, E.B.; et al. Trends in the incidence and prevalence of Alzheimer’s disease, dementia, and cognitive impairment in the United States. Alzheimers Dement. J. Alzheimers Assoc. 2011, 7, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.W.; Victor, M.B.; Tsai, L.-H. Dissecting the complexities of Alzheimer disease with in vitro models of the human brain. Nat. Rev. Neurol. 2022, 18, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Altomare, D.; Thal, D.R.; Ribaldi, F.; van der Kant, R.; Ossenkoppele, R.; Blennow, K.; Cummings, J.; van Duijn, C.; Nilsson, P.M.; et al. The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat. Rev. Neurol. 2022, 23, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Isla, T.; Frosch, M.P. Lesions without symptoms: Understanding resilience to Alzheimer disease neuropathological changes. Nat. Rev. Neurol. 2022, 18, 323–332. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Roda, A.R.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666–1674. [Google Scholar] [CrossRef]
- Se Thoe, E.; Fauzi, A.; Tang, Y.Q.; Chamyuang, S.; Chia, A.Y.Y. A review on advances of treatment modalities for Alzheimer’s disease. Life Sci. 2021, 276, 119129. [Google Scholar] [CrossRef]
- Yu, J.T.; Xu, W.; Tan, C.C.; Andrieu, S.; Suckling, J.; Evangelou, E.; Pan, A.; Zhang, C.; Jia, J.; Feng, L.; et al. Evidence-based prevention of Alzheimer’s disease: Systematic review and meta-analysis of 243 observational prospective studies and 153 randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1201–1209. [Google Scholar] [CrossRef]
- Sundström, A.; Rönnlund, M.; Josefsson, M. A nationwide Swedish study of age at retirement and dementia risk. Int. J. Geriatr. Psychiatry 2020, 35, 1243–1249. [Google Scholar] [CrossRef]
- Dufouil, C.; Pereira, E.; Chêne, G.; Glymour, M.M.; Alpérovitch, A.; Saubusse, E.; Risse-Fleury, M.; Heuls, B.; Salord, J.C.; Brieu, M.A.; et al. Older age at retirement is associated with decreased risk of dementia. Eur. J. Epidemiol. 2014, 29, 353–361. [Google Scholar] [CrossRef]
- Marioni, R.E.; van den Hout, A.; Valenzuela, M.J.; Brayne, C.; Matthews, F.E.; Function, M.R.C.C.; Ageing, S. Active cognitive lifestyle associates with cognitive recovery and a reduced risk of cognitive decline. J. Alzheimers Dis. 2012, 28, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Park, D.C.; Lodi-Smith, J.; Drew, L.; Haber, S.; Hebrank, A.; Bischof, G.N.; Aamodt, W. The impact of sustained engagement on cognitive function in older adults: The Synapse Project. Psychol. Sci. 2014, 25, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Friedland, R.P.; Fritsch, T.; Smyth, K.A.; Koss, E.; Lerner, A.J.; Chen, C.H.; Petot, G.J.; Debanne, S.M. Patients with Alzheimer’s disease have reduced activities in midlife compared with healthy control-group members. Proc. Natl. Acad. Sci. USA 2001, 98, 3440–3445. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Muller, U.; Winter, P.; Graeber, M.B. A presenilin 1 mutation in the first case of Alzheimer’s disease. Lancet Neurol. 2013, 12, 129–130. [Google Scholar] [CrossRef]
- Rupp, C.; Beyreuther, K.; Maurer, K.; Kins, S. A presenilin 1 mutation in the first case of Alzheimer’s disease: Revisited. Alzheimers Dement. 2014, 10, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.; Nance, E. Disease-directed engineering for physiology-driven treatment interventions in neurological disorders. APL Bioeng. 2019, 3, 040901. [Google Scholar] [CrossRef]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef]
- Herrup, K. Fallacies in Neuroscience: The Alzheimer’s Edition. eNeuro 2022, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Potter, R.R., III; Long, A.P.; Lichtenstein, M.L. Population prevalence of autosomal dominant Alzheimer’s disease: A systematic review. Alzheimers Dement. 2020, 16, e037129. [Google Scholar] [CrossRef]
- 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [CrossRef] [PubMed]
- Vogel, J.W.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; Iturria-Medina, Y.; et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med. 2021, 27, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C. How early can we diagnose Alzheimer disease (and is it sufficient)? Neurology 2018, 91, 395. [Google Scholar] [CrossRef]
- Fessel, J. Amyloid is essential but insufficient for Alzheimer causation: Addition of subcellular cofactors is required for dementia. Int. J. Geriatr. Psychiatry 2018, 33, e14–e21. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef]
- Ridge, P.G.; Mukherjee, S.; Crane, P.K.; Kauwe, J.S.K.; Alzheimer’s Disease Genetics, C. Alzheimer’s Disease: Analyzing the Missing Heritability. PLoS ONE 2013, 8, e79771. [Google Scholar] [CrossRef] [Green Version]
- Trumble, B.C.; Stieglitz, J.; Blackwell, A.D.; Allayee, H.; Beheim, B.; Finch, C.E.; Gurven, M.; Kaplan, H. Apolipoprotein E4 is associated with improved cognitive function in Amazonian forager-horticulturalists with a high parasite burden. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 1508–1515. [Google Scholar] [CrossRef]
- Suchy-Dicey, A.; Howard, B.; Longstreth, W.T., Jr.; Reiman, E.M.; Buchwald, D. APOE genotype, hippocampus, and cognitive markers of Alzheimer’s disease in American Indians: Data from the Strong Heart Study. Alzheimers Dement. J. Alzheimers Assoc. 2022. [Google Scholar] [CrossRef]
- Gureje, O.; Ogunniyi, A.; Baiyewu, O.; Price, B.; Unverzagt, F.W.; Evans, R.M.; Smith-Gamble, V.; Lane, K.A.; Gao, S.; Hall, K.S.; et al. APOE epsilon4 is not associated with Alzheimer’s disease in elderly Nigerians. Ann. Neurol. 2006, 59, 182–185. [Google Scholar] [CrossRef]
- Brooks, D.S.; Eronen, M.I. The significance of levels of organization for scientific research: A heuristic approach. Stud. Hist. Philos. Biol. Biomed. Sci. 2018, 68–69, 34–41. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.W.; Walker, D.H. Pathogenesis of Infectious Diseases and Mechanisms of Immunity. In Vaccinology; Wiley: Hoboken, NJ, USA, 2015; pp. 59–72. [Google Scholar] [CrossRef]
- Ko, Y.; Chye, S.M. Lifestyle intervention to prevent Alzheimer’s disease. Rev. Neurosci. 2020, 31, 817–824. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Li, Y.-Z.; Zhang, Y.-R.; Huang, Y.-Y.; Wu, B.-S.; Zhang, W.; Deng, Y.-T.; Chen, S.-D.; He, X.-Y.; Chen, S.-F.; et al. Sleep, physical activity, sedentary behavior, and risk of incident dementia: A prospective cohort study of 431,924 UK Biobank participants. Mol. Psychiatry 2022. [Google Scholar] [CrossRef]
- Liang, Y.; Ngandu, T.; Laatikainen, T.; Soininen, H.; Tuomilehto, J.; Kivipelto, M.; Qiu, C. Cardiovascular health metrics from mid- to late-life and risk of dementia: A population-based cohort study in Finland. PLoS Med. 2020, 17, e1003474. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Sindi, S.; Kåreholt, I.; Ngandu, T.; Rosenberg, A.; Kulmala, J.; Johansson, L.; Wetterberg, H.; Skoog, J.; Sjöberg, L.; Wang, H.-X.; et al. Sex differences in dementia and response to a lifestyle intervention: Evidence from Nordic population-based studies and a prevention trial. Alzheimers Dement. 2021, 17, 1166–1178. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.; Yang, P.-S.; Jin, M.-N.; Yu, H.T.; Kim, T.-H.; Jang, E.; Uhm, J.-S.; Pak, H.-N.; Lee, M.-H.; Joung, B. Association of Physical Activity Level With Risk of Dementia in a Nationwide Cohort in Korea. JAMA Netw. Open 2021, 4, e2138526. [Google Scholar] [CrossRef]
- Gallardo-Gómez, D.; del Pozo-Cruz, J.; Noetel, M.; Álvarez-Barbosa, F.; Alfonso-Rosa, R.M.; del Pozo Cruz, B. Optimal dose and type of exercise to improve cognitive function in older adults: A systematic review and bayesian model-based network meta-analysis of RCTs. Ageing Res. Rev. 2022, 76, 101591. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Fu, W.; Wang, C.; Mao, J.; Liu, B.; Zou, L.; Lv, C. Association between sedentary behavior and the risk of dementia: A systematic review and meta-analysis. Transl. Psychiatry 2020, 10, 112. [Google Scholar] [CrossRef]
- Bubu, O.M.; Brannick, M.; Mortimer, J.; Umasabor-Bubu, O.; Sebastiao, Y.V.; Wen, Y.; Schwartz, S.; Borenstein, A.R.; Wu, Y.; Morgan, D.; et al. Sleep, Cognitive impairment, and Alzheimer’s disease: A Systematic Review and Meta-Analysis. Sleep 2017, 40, zsw032. [Google Scholar] [CrossRef] [PubMed]
- Sabia, S.; Fayosse, A.; Dumurgier, J.; van Hees, V.T.; Paquet, C.; Sommerlad, A.; Kivimäki, M.; Dugravot, A.; Singh-Manoux, A. Association of sleep duration in middle and old age with incidence of dementia. Nat. Commun. 2021, 12, 2289. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Qiu, C.; Winblad, B.; Fratiglioni, L. The Effect of Borderline Diabetes on the Risk of Dementia and Alzheimer’s Disease. Diabetes 2007, 56, 211–216. [Google Scholar] [CrossRef]
- Zheng, F.; Yan, L.; Yang, Z.; Zhong, B.; Xie, W. HbA1c, diabetes and cognitive decline: The English Longitudinal Study of Ageing. Diabetologia 2018, 61, 839–848. [Google Scholar] [CrossRef]
- Jernerén, F.; Elshorbagy, A.K.; Oulhaj, A.; Smith, S.M.; Refsum, H.; Smith, A.D. Brain atrophy in cognitively impaired elderly: The importance of long-chain ω-3 fatty acids and B vitamin status in a randomized controlled trial. Am. J. Clin. Nutr. 2015, 102, 215–221. [Google Scholar] [CrossRef]
- Klinedinst, B.S.; Pappas, C.; Le, S.; Yu, S.; Wang, Q.; Wang, L.; Allenspach-Jorn, K.; Mochel, J.P.; Willette, A.A. Aging-related changes in fluid intelligence, muscle and adipose mass, and sex-specific immunologic mediation: A longitudinal UK Biobank study. Brain Behav. Immun. 2019, 82, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, R.A.; Gustafson, D.R.; Barrett-Connor, E.; Haan, M.N.; Gunderson, E.P.; Yaffe, K. Central obesity and increased risk of dementia more than three decades later. Neurology 2008, 71, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Someya, Y.; Tamura, Y.; Kaga, H.; Sugimoto, D.; Kadowaki, S.; Suzuki, R.; Aoki, S.; Hattori, N.; Motoi, Y.; Shimada, K.; et al. Sarcopenic obesity is associated with cognitive impairment in community-dwelling older adults: The Bunkyo Health Study. Clin. Nutr. 2022, 41, 1046–1051. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Y.; Lv, Z.; Xiang, J. Sarcopenia and motoric cognitive risk syndrome: A moderated mediation model. BMC Geriatr. 2022, 22, 141. [Google Scholar] [CrossRef] [PubMed]
- Jett, S.; Schelbaum, E.; Jang, G.; Boneu Yepez, C.; Dyke, J.P.; Pahlajani, S.; Diaz Brinton, R.; Mosconi, L. Ovarian steroid hormones: A long overlooked but critical contributor to brain aging and Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 948219. [Google Scholar] [CrossRef]
- Penninkilampi, R.; Casey, A.N.; Singh, M.F.; Brodaty, H. The Association between Social Engagement, Loneliness, and Risk of Dementia: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2018, 66, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Toups, K.; Hathaway, A.; Gordon, D.; Chung, H.; Raji, C.; Boyd, A.; Hill, B.D.; Hausman-Cohen, S.; Attarha, M.; Chwa, W.J.; et al. Precision Medicine Approach to Alzheimer’s Disease: Successful Pilot Project. J. Alzheimers Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- LaPlume, A.A.; McKetton, L.; Levine, B.; Troyer, A.K.; Anderson, N.D. The adverse effect of modifiable dementia risk factors on cognition amplifies across the adult lifespan. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2022, 14, e12337. [Google Scholar] [CrossRef]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levälahti, E.; Ahtiluoto, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Eshraghi, M.; Ahmadi, M.; Afshar, S.; Lorzadeh, S.; Adlimoghaddam, A.; Rezvani Jalal, N.; West, R.; Dastghaib, S.; Igder, S.; Torshizi, S.R.N.; et al. Enhancing autophagy in Alzheimer’s disease through drug repositioning. Pharmacol. Ther. 2022, 237, 108171. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef]
- McGlory, C.; Devries, M.C.; Phillips, S.M. Skeletal muscle and resistance exercise training; the role of protein synthesis in recovery and remodeling. J. Appl. Physiol. 2017, 122, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Rebelo-Marques, A.; De Sousa Lages, A.; Andrade, R.; Ribeiro, C.F.; Mota-Pinto, A.; Carrilho, F.; Espregueira-Mendes, J. Aging Hallmarks: The Benefits of Physical Exercise. Front. Endocrinol. 2018, 9, 258. [Google Scholar] [CrossRef]
- Farrell, C.; Turgeon, D.R. Normal Versus Chronic Adaptations to Aerobic Exercise. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rocchi, A.; He, C. Regulation of Exercise-Induced Autophagy in Skeletal Muscle. Curr. Pathobiol. Rep. 2017, 5, 177–186. [Google Scholar] [CrossRef]
- Sanchez, A.M.J.; Bernardi, H.; Py, G.; Candau, R.B. Autophagy is essential to support skeletal muscle plasticity in response to endurance exercise. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R956–R969. [Google Scholar] [CrossRef]
- Sebastián, D.; Zorzano, A. Self-Eating for Muscle Fitness: Autophagy in the Control of Energy Metabolism. Dev. Cell 2020, 54, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.-H.; Dong, L.-L.; Zhang, Y.-J.; Zhao, X.-M.; He, H.-Y. Enriched environment boosts the post-stroke recovery of neurological function by promoting autophagy. Neural Regen. Res. 2021, 16, 813–819. [Google Scholar] [CrossRef]
- Zhang, Q.Q.; Luo, L.; Liu, M.X.; Wang, C.J.; Wu, Y.; Yu, K.W. Enriched Environment-Induced Neuroprotection against Cerebral Ischemia-Reperfusion Injury Might Be Mediated via Enhancing Autophagy Flux and Mitophagy Flux. Mediat. Inflamm. 2022, 2022, 2396487. [Google Scholar] [CrossRef]
- Lazarov, O.; Robinson, J.; Tang, Y.P.; Hairston, I.S.; Korade-Mirnics, Z.; Lee, V.M.; Hersh, L.B.; Sapolsky, R.M.; Mirnics, K.; Sisodia, S.S. Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell 2005, 120, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Shimizu, K.; Shimazaki, K.; Toda, H.; Nibuya, M. Environmental enrichment enhances autophagy signaling in the rat hippocampus. Brain Res. 2014, 1592, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Qu, C.; Qu, C.; Shen, J.; Song, H.; Li, Y.; Li, T.; Zheng, J.; Zhang, J. Improvement of autophagy dysfunction as a potential mechanism for environmental enrichment to protect blood-brain barrier in rats with vascular cognitive impairment. Neurosci. Lett. 2020, 739, 135437. [Google Scholar] [CrossRef]
- Schwalm, C.; Jamart, C.; Benoit, N.; Naslain, D.; Prémont, C.; Prévet, J.; Van Thienen, R.; Deldicque, L.; Francaux, M. Activation of autophagy in human skeletal muscle is dependent on exercise intensity and AMPK activation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 3515–3526. [Google Scholar] [CrossRef]
- Brandt, N.; Gunnarsson, T.P.; Bangsbo, J.; Pilegaard, H. Exercise and exercise training-induced increase in autophagy markers in human skeletal muscle. Physiol. Rep. 2018, 6, e13651. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Sumpter, R., Jr.; Levine, B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012, 8, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.K.; Lee, S.; Yang, M.J.; Leeuwenburgh, C.; Kim, J.S. Exercise-Induced Autophagy in Fatty Liver Disease. Exerc. Sport Sci. Rev. 2017, 45, 181–186. [Google Scholar] [CrossRef]
- Møller, A.B.; Vendelbo, M.H.; Christensen, B.; Clasen, B.F.; Bak, A.M.; Jørgensen, J.O.; Møller, N.; Jessen, N. Physical exercise increases autophagic signaling through ULK1 in human skeletal muscle. J. Appl. Physiol. 2015, 118, 971–979. [Google Scholar] [CrossRef]
- Vendelbo, M.H.; Møller, A.B.; Christensen, B.; Nellemann, B.; Clasen, B.F.; Nair, K.S.; Jørgensen, J.O.; Jessen, N.; Møller, N. Fasting increases human skeletal muscle net phenylalanine release and this is associated with decreased mTOR signaling. PLoS ONE 2014, 9, e102031. [Google Scholar] [CrossRef]
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.-M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Woollett, K.; Maguire, E.A. Acquiring “the Knowledge” of London’s layout drives structural brain changes. Curr. Biol. CB 2011, 21, 2109–2114. [Google Scholar] [CrossRef] [Green Version]
- Suo, C.; Singh, M.F.; Gates, N.; Wen, W.; Sachdev, P.; Brodaty, H.; Saigal, N.; Wilson, G.C.; Meiklejohn, J.; Singh, N.; et al. Therapeutically relevant structural and functional mechanisms triggered by physical and cognitive exercise. Mol. Psychiatry 2016, 21, 1633–1642. [Google Scholar] [CrossRef]
- Parker, W.D.; Parks, J.K. Cytochrome C Oxidase in Alzheimer’s Disease Brain. Purif. Charact. 1995, 45, 482–486. [Google Scholar] [CrossRef]
- Gonzalez-Lima, F.; Barksdale, B.R.; Rojas, J.C. Mitochondrial respiration as a target for neuroprotection and cognitive enhancement. Biochem. Pharmacol. 2014, 88, 584–593. [Google Scholar] [CrossRef]
- Gonzalez-Lima, F.; Valla, J.; Jorandby, L. Cytochrome Oxidase Inhibition in Alzheimer’s Disease; Springer: Boston, MA, USA, 1998. [Google Scholar]
- Valla, J.; Berndt, J.D.; Gonzalez-Lima, F. Energy Hypometabolism in Posterior Cingulate Cortex of Alzheimer’s Patients: Superficial Laminar Cytochrome Oxidase Associated with Disease Duration. J. Neurosci. 2001, 21, 4923–4930. [Google Scholar] [CrossRef] [PubMed]
- Wong-Riley, M.T. Cytochrome oxidase: An endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989, 12, 94–101. [Google Scholar] [CrossRef]
- Rojas, J.C.; Bruchey, A.K.; Gonzalez-Lima, F. Neurometabolic mechanisms for memory enhancement and neuroprotection of methylene blue. Prog. Neurobiol. 2012, 96, 32–45. [Google Scholar] [CrossRef]
- Anton, S.C.; Potts, R.; Aiello, L.C. Human evolution. Evolution of early Homo: An integrated biological perspective. Science 2014, 345, 1236828. [Google Scholar] [CrossRef] [PubMed]
- Medic, G.; Wille, M.; Hemels, M.E. Short- and long-term health consequences of sleep disruption. Nat. Sci. Sleep 2017, 9, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.K.; Teasell, R. Complications of immobilization and bed rest. Part 1: Musculoskeletal and cardiovascular complications. Can. Fam. Physician 1993, 39, 1428–1432, 1435–1437. [Google Scholar] [PubMed]
- Krumholz, H.M. Post-hospital syndrome--an acquired, transient condition of generalized risk. N. Engl. J. Med. 2013, 368, 100–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meessen, J.M.; Pisani, S.; Gambino, M.L.; Bonarrigo, D.; van Schoor, N.M.; Fozzato, S.; Cherubino, P.; Surace, M.F. Assessment of mortality risk in elderly patients after proximal femoral fracture. Orthopedics 2014, 37, e194–e200. [Google Scholar] [CrossRef]
- CDC. Physiologic Responses and Long-Term Adaptations to Exercise. Physical Activity and Health: A Report of the Surgeon General. 1999. Available online: https://www.cdc.gov/nccdphp/sgr/pdf/chap3.pdf (accessed on 5 July 2022).
- Lee, C.S.; Gibbons, L.E.; Lee, A.Y.; Yanagihara, R.T.; Blazes, M.S.; Lee, M.L.; McCurry, S.M.; Bowen, J.D.; McCormick, W.C.; Crane, P.K.; et al. Association Between Cataract Extraction and Development of Dementia. JAMA Intern. Med. 2022, 182, 134–141. [Google Scholar] [CrossRef]
- Liu, C.M.; Lee, C.T. Association of Hearing Loss With Dementia. JAMA Netw. Open 2019, 2, e198112. [Google Scholar] [CrossRef] [PubMed]
- Paciello, F.; Rinaudo, M.; Longo, V.; Cocco, S.; Conforto, G.; Pisani, A.; Podda, M.V.; Fetoni, A.R.; Paludetti, G.; Grassi, C. Auditory sensory deprivation induced by noise exposure exacerbates cognitive decline in a mouse model of Alzheimer’s disease. eLife 2021, 10, e70908. [Google Scholar] [CrossRef]
- Kurioka, T.; Mogi, S.; Yamashita, T. Decreasing auditory input induces neurogenesis impairment in the hippocampus. Sci. Rep. 2021, 11, 423. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.J.; Ricci, A.J. Effects of cochlear hair cell ablation on spatial learning/memory. Sci. Rep. 2020, 10, 20687. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, H.J.; Lee, S.; Lee, H.S.; Jeong, Y.J.; Son, Y.; Kim, J.M.; Lee, Y.J.; Park, M.H. Conductive Hearing Loss Aggravates Memory Decline in Alzheimer Model Mice. Front. Neurosci. 2020, 14, 843. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, M.J.; Kim, H.L.; Kim, D.K.; Yeo, S.W.; Park, S.N. Cognitive decline and increased hippocampal p-tau expression in mice with hearing loss. Behav. Brain Res. 2018, 342, 19–26. [Google Scholar] [CrossRef]
- Brenowitz, W.D.; Robbins, N.M.; Strotmeyer, E.S.; Yaffe, K. Associations of Lower Extremity Peripheral Nerve Impairment and Risk of Dementia in Black and White Older Adults. Neurology 2022, 98, e1837. [Google Scholar] [CrossRef]
- Lin, Y.J.; Kao, T.W.; Chen, W.L. Relationship between peripheral neuropathy and cognitive performance in the elderly population. Medicine 2021, 100, e26071. [Google Scholar] [CrossRef]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef]
- Manoharan-Valerio, M.A.; Colon, J.; Martinez, N.; Quiroz, E.; Noel, R. Astrocytes expressing Nef promote apoptosis and SMAD signaling in Neurons. FASEB J. 2013, 27, lb213. [Google Scholar] [CrossRef]
- Hiew, L.F.; Poon, C.H.; You, H.Z.; Lim, L.W. TGF-β/Smad Signalling in Neurogenesis: Implications for Neuropsychiatric Diseases. Cells 2021, 10, 1382. [Google Scholar] [CrossRef] [PubMed]
- Vilalta, A.; Brown, G.C. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018, 285, 3566–3575. [Google Scholar] [CrossRef]
- de Villers-Sidani, E.; Alzghoul, L.; Zhou, X.; Simpson, K.L.; Lin, R.C.; Merzenich, M.M. Recovery of functional and structural age-related changes in the rat primary auditory cortex with operant training. Proc. Natl. Acad. Sci. USA 2010, 107, 13900–13905. [Google Scholar] [CrossRef] [PubMed]
- Andrews, E.; Eierud, C.; Banks, D.; Harshbarger, T.; Michael, A.; Rammell, C. Effects of Lifelong Musicianship on White Matter Integrity and Cognitive Brain Res.erve. Brain Sci. 2021, 11, 67. [Google Scholar] [CrossRef]
- Meng, X.; D’Arcy, C. Education and dementia in the context of the cognitive reserve hypothesis: A systematic review with meta-analyses and qualitative analyses. PLoS ONE 2012, 7, e38268. [Google Scholar] [CrossRef]
- Riley, K.P.; Snowdon, D.A.; Desrosiers, M.F.; Markesbery, W.R. Early life linguistic ability, late life cognitive function, and neuropathology: Findings from the Nun Study. Neurobiol. Aging 2005, 26, 341–347. [Google Scholar] [CrossRef]
- Colangeli, S.; Boccia, M.; Verde, P.; Guariglia, P.; Bianchini, F.; Piccardi, L. Cognitive Reserve in Healthy Aging and Alzheimer’s Disease: A Meta-Analysis of fMRI Studies. Am, J. Alzheimers Dis. Other Demen 2016, 31, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.I.; Voss, M.W.; Prakash, R.S.; Basak, C.; Szabo, A.; Chaddock, L.; Kim, J.S.; Heo, S.; Alves, H.; White, S.M.; et al. Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujach, S.; Olek, R.A.; Byun, K.; Suwabe, K.; Sitek, E.J.; Ziemann, E.; Laskowski, R.; Soya, H. Acute Sprint Interval Exercise Increases Both Cognitive Functions and Peripheral Neurotrophic Factors in Humans: The Possible Involvement of Lactate. Front. Neurosci. 2019, 13, 1455. [Google Scholar] [CrossRef]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body beta-hydroxybutyrate. Elife 2016, 5, e15092. [Google Scholar] [CrossRef]
- Casaletto, K.; Ramos-Miguel, A.; VandeBunte, A.; Memel, M.; Buchman, A.; Bennett, D.; Honer, W. Late-life physical activity relates to brain tissue synaptic integrity markers in older adults. Alzheimers Dement. J. Alzheimers Assoc. 2022. [Google Scholar] [CrossRef] [PubMed]
- Casaletto, K.B.; Lindbergh, C.A.; VandeBunte, A.; Neuhaus, J.; Schneider, J.A.; Buchman, A.S.; Honer, W.G.; Bennett, D.A. Microglial Correlates of Late Life Physical Activity: Relationship with Synaptic and Cognitive Aging in Older Adults. J. Neurosci. 2022, 42, 288–298. [Google Scholar] [CrossRef]
- Slavich, G.M. Social Safety Theory: Understanding social stress, disease risk, resilience, and behavior during the COVID-19 pandemic and beyond. Curr. Opin. Psychol. 2022, 45, 101299. [Google Scholar] [CrossRef]
- Herold, F.; Törpel, A.; Schega, L.; Müller, N.G. Functional and/or structural brain changes in response to resistance exercises and resistance training lead to cognitive improvements—A systematic review. Eur. Rev. Aging Phys. Act. 2019, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Ludyga, S.; Gerber, M.; Pühse, U.; Looser, V.N.; Kamijo, K. Systematic review and meta-analysis investigating moderators of long-term effects of exercise on cognition in healthy individuals. Nat. Hum. Behav. 2020, 4, 603–612. [Google Scholar] [CrossRef]
- Rehfeld, K.; Müller, P.; Aye, N.; Schmicker, M.; Dordevic, M.; Kaufmann, J.; Hökelmann, A.; Müller, N.G. Dancing or Fitness Sport? The Effects of Two Training Programs on Hippocampal Plasticity and Balance Abilities in Healthy Seniors. Front. Hum. Neurosci. 2017, 11, 305. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, J.A.; Kates Rose, M.; Le, A.Y.; Spencer, K.A.; Goldstein, L.; Gubanova, A.; Lai, A.C.; Yossofzai, M.; Armstrong, S.E.M.; Bialystok, E. Improvement in executive function for older adults through smartphone apps: A randomized clinical trial comparing language learning and brain training. Aging Neuropsychol. Cogn. 2021, 1–22. [Google Scholar] [CrossRef]
- Bak, T.H.; Nissan, J.J.; Allerhand, M.M.; Deary, I.J. Does bilingualism influence cognitive aging? Ann. Neurol. 2014, 75, 959–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.A.E.; Hawrylewicz, K.; Grundy, J.G. Does bilingualism protect against dementia? A meta-analysis. Psychon. Bull. Rev. 2020, 27, 952–965. [Google Scholar] [CrossRef]
- Luk, G.; Bialystok, E.; Craik, F.I.; Grady, C.L. Lifelong bilingualism maintains white matter integrity in older adults. J. Neurosci. 2011, 31, 16808–16813. [Google Scholar] [CrossRef] [PubMed]
- Rogenmoser, L.; Kernbach, J.; Schlaug, G.; Gaser, C. Keeping brains young with making music. Brain Struct. Funct. 2018, 223, 297–305. [Google Scholar] [CrossRef]
- Verghese, J.; Lipton, R.B.; Katz, M.J.; Hall, C.B.; Derby, C.A.; Kuslansky, G.; Ambrose, A.F.; Sliwinski, M.; Buschke, H. Leisure activities and the risk of dementia in the elderly. N. Engl. J. Med. 2003, 348, 2508–2516. [Google Scholar] [CrossRef]
- Mansens, D.; Deeg, D.J.H.; Comijs, H.C. The association between singing and/or playing a musical instrument and cognitive functions in older adults. Aging Ment. Health 2018, 22, 970–977. [Google Scholar] [CrossRef]
- Edwards, J.D.; Xu, H.; Clark, D.O.; Guey, L.T.; Ross, L.A.; Unverzagt, F.W. Speed of processing training results in lower risk of dementia. Alzheimers Dement. Transl. Res. Clin. Interv. 2017, 3, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Shandera-Ochsner, A.L.; Chandler, M.J.; Locke, D.E.; Ball, C.T.; Crook, J.E.; Phatak, V.S.; Smith, G.E. Comparative Effects of Physical Exercise and Other Behavioral Interventions on Functional Status Outcomes in Mild Cognitive Impairment. J. Int. Neuropsychol. Soc. 2021, 27, 805–812. [Google Scholar] [CrossRef]
- Rosen, A.C.; Sugiura, L.; Kramer, J.H.; Whitfield-Gabrieli, S.; Gabrieli, J.D. Cognitive Training Changes Hippocampal Function in Mild Cognitive Impairment: A Pilot Study. J. Alzheimers Dis. 2011, 26, 349–357. [Google Scholar] [CrossRef]
- Barnes, D.E.; Yaffe, K.; Belfor, N.; Jagust, W.J.; DeCarli, C.; Reed, B.R.; Kramer, J.H. Computer-based Cognitive Training for Mild Cognitive Impairment: Results from a Pilot Randomized, Controlled Trial. Alzheimer Dis. Assoc. Disord. 2009, 23, 205–210. [Google Scholar] [CrossRef]
- Elovainio, M.; Lahti, J.; Pirinen, M.; Pulkki-Råback, L.; Malmberg, A.; Lipsanen, J.; Virtanen, M.; Kivimäki, M.; Hakulinen, C. Association of social isolation, loneliness and genetic risk with incidence of dementia: UK Biobank Cohort Study. BMJ Open 2022, 12, e053936. [Google Scholar] [CrossRef] [PubMed]
- Salinas, J.; Beiser, A.S.; Samra, J.K.; O’Donnell, A.; DeCarli, C.S.; Gonzales, M.M.; Aparicio, H.J.; Seshadri, S. Association of Loneliness With 10-Year Dementia Risk and Early Markers of Vulnerability for Neurocognitive Decline. Neurology 2022, 98, e1337–e1348. [Google Scholar] [CrossRef]
- Douaud, G.; Lee, S.; Alfaro-Almagro, F.; Arthofer, C.; Wang, C.; McCarthy, P.; Lange, F.; Andersson, J.L.R.; Griffanti, L.; Duff, E.; et al. SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature 2022, 604, 697–707. [Google Scholar] [CrossRef]
- Zamponi, H.P.; Juarez-Aguaysol, L.; Kukoc, G.; Dominguez, M.E.; Pini, B.; Padilla, E.G.; Calvó, M.; Molina-Rangeon, S.B.; Guerrero, G.; Figueredo-Aguiar, M.; et al. Olfactory dysfunction and chronic cognitive impairment following SARS-CoV-2 infection in a sample of older adults from the Andes mountains of Argentina. Alzheimers Dement. 2021, 17, e057897. [Google Scholar] [CrossRef]
- Hewitt, D. Age-Related Hearing Loss and Cognitive Decline: You Haven’t Heard the Half of It. Front. Aging Neurosci. 2017, 9, 112. [Google Scholar] [CrossRef]
- Lebel, C.; Walker, L.; Leemans, A.; Phillips, L.; Beaulieu, C. Microstructural maturation of the human brain from childhood to adulthood. Neuroimage 2008, 40, 1044–1055. [Google Scholar] [CrossRef]
- Poldrack, R.A.; Sabb, F.W.; Foerde, K.; Tom, S.M.; Asarnow, R.F.; Bookheimer, S.Y.; Knowlton, B.J. The neural correlates of motor skill automaticity. J. Neurosci. 2005, 25, 5356–5364. [Google Scholar] [CrossRef]
- Buettner, D. The Blue Zones: 9 Lessons for Living Longer from the People Who’ve Lived the Longest. 2012. Available online: https://www.bluezones.com/wp-content/uploads/backup/2012/02/BlueZonesStudyGuide.pdf (accessed on 5 July 2022).
- Hale, J.M.; Bijlsma, M.J.; Lorenti, A. Does postponing retirement affect cognitive function? A counterfactual experiment to disentangle life course risk factors. SSM—Popul. Health 2021, 15, 100855. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef]
- Shokri-Kojori, E.; Wang, G.-J.; Wiers, C.E.; Demiral, S.B.; Guo, M.; Kim, S.W.; Lindgren, E.; Ramirez, V.; Zehra, A.; Freeman, C.; et al. β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. USA 2018, 115, 4483. [Google Scholar] [CrossRef] [PubMed]
- Rasch, B.; Born, J. About sleep’s role in memory. Physiol. Rev. 2013, 93, 681–766. [Google Scholar] [CrossRef]
- Abel, T.; Havekes, R.; Saletin, J.M.; Walker, M.P. Sleep, plasticity and memory from molecules to whole-brain networks. Curr. Biol. 2013, 23, R774–R788. [Google Scholar] [CrossRef]
- Sexton, C.E.; Storsve, A.B.; Walhovd, K.B.; Johansen-Berg, H.; Fjell, A.M. Poor sleep quality is associated with increased cortical atrophy in community-dwelling adults. Neurology 2014, 83, 967–973. [Google Scholar] [CrossRef]
- Emamian, F.; Khazaie, H.; Tahmasian, M.; Leschziner, G.D.; Morrell, M.J.; Hsiung, G.Y.; Rosenzweig, I.; Sepehry, A.A. The Association Between Obstructive Sleep Apnea and Alzheimer’s Disease: A Meta-Analysis Perspective. Front. Aging Neurosci. 2016, 8, 78. [Google Scholar] [CrossRef]
- Leng, Y.; McEvoy, C.T.; Allen, I.E.; Yaffe, K. Association of Sleep-Disordered Breathing With Cognitive Function and Risk of Cognitive Impairment: A Systematic Review and Meta-analysis. JAMA Neurol. 2017, 74, 1237–1245. [Google Scholar] [CrossRef]
- Booth, F.W.; Gordon, S.E.; Carlson, C.J.; Hamilton, M.T. Waging war on modern chronic diseases: Primary prevention through exercise biology. J. Appl. Physiol. 2000, 88, 774–787. [Google Scholar] [CrossRef]
- 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. J. Alzheimers Assoc. 2021, 17, 327–406. [CrossRef] [PubMed]
- Lachman, M.E.; Lipsitz, L.; Lubben, J.; Castaneda-Sceppa, C.; Jette, A.M. When Adults Don’t Exercise: Behavioral Strategies to Increase Physical Activity in Sedentary Middle-Aged and Older Adults. Innov. Aging 2018, 2, igy007. [Google Scholar] [CrossRef]
- Almirall, D.; Nahum-Shani, I.; Sherwood, N.E.; Murphy, S.A. Introduction to SMART designs for the development of adaptive interventions: With application to weight loss research. Transl. Behav. Med. 2014, 4, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Hekler, E.; Tiro, J.A.; Hunter, C.M.; Nebeker, C. Precision Health: The Role of the Social and Behavioral Sciences in Advancing the Vision. Ann. Behav. Med. 2020, 54, 805–826. [Google Scholar] [CrossRef] [PubMed]
- Vinueza Veloz, A.F.; Carpio Arias, T.V.; Vargas Mejía, J.S.; Tapia Veloz, E.C.; Piedra Andrade, J.S.; Nicolalde Cifuentes, T.M.; Heredia Aguirre, S.I.; Vinueza Veloz, M.F. Cognitive function and vitamin B12 and D among community-dwelling elders: A cross-sectional study. Clin. Nutr. ESPEN 2022, 50, 270–276. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turknett, J.; Wood, T.R. Demand Coupling Drives Neurodegeneration: A Model of Age-Related Cognitive Decline and Dementia. Cells 2022, 11, 2789. https://doi.org/10.3390/cells11182789
Turknett J, Wood TR. Demand Coupling Drives Neurodegeneration: A Model of Age-Related Cognitive Decline and Dementia. Cells. 2022; 11(18):2789. https://doi.org/10.3390/cells11182789
Chicago/Turabian StyleTurknett, Josh, and Thomas R. Wood. 2022. "Demand Coupling Drives Neurodegeneration: A Model of Age-Related Cognitive Decline and Dementia" Cells 11, no. 18: 2789. https://doi.org/10.3390/cells11182789