
Rule-Based Pruning and In Silico Identification of Essential Proteins in Yeast PPIN

 ,
,  ,
,  , , and
, , and
Abstract
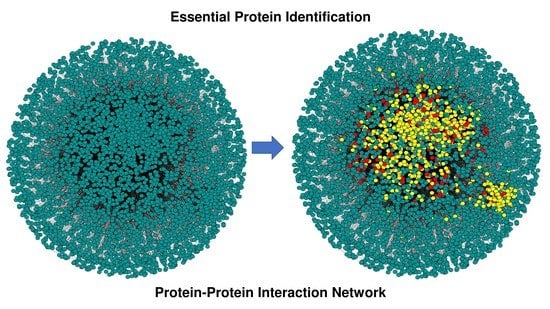
:
1. Introduction
2. Dataset
3. Methodology
| Algorithm1 (Essential Protein Prediction) |
| Input: PPIN of yeast Output: List of Essential and Non-essential Protein |
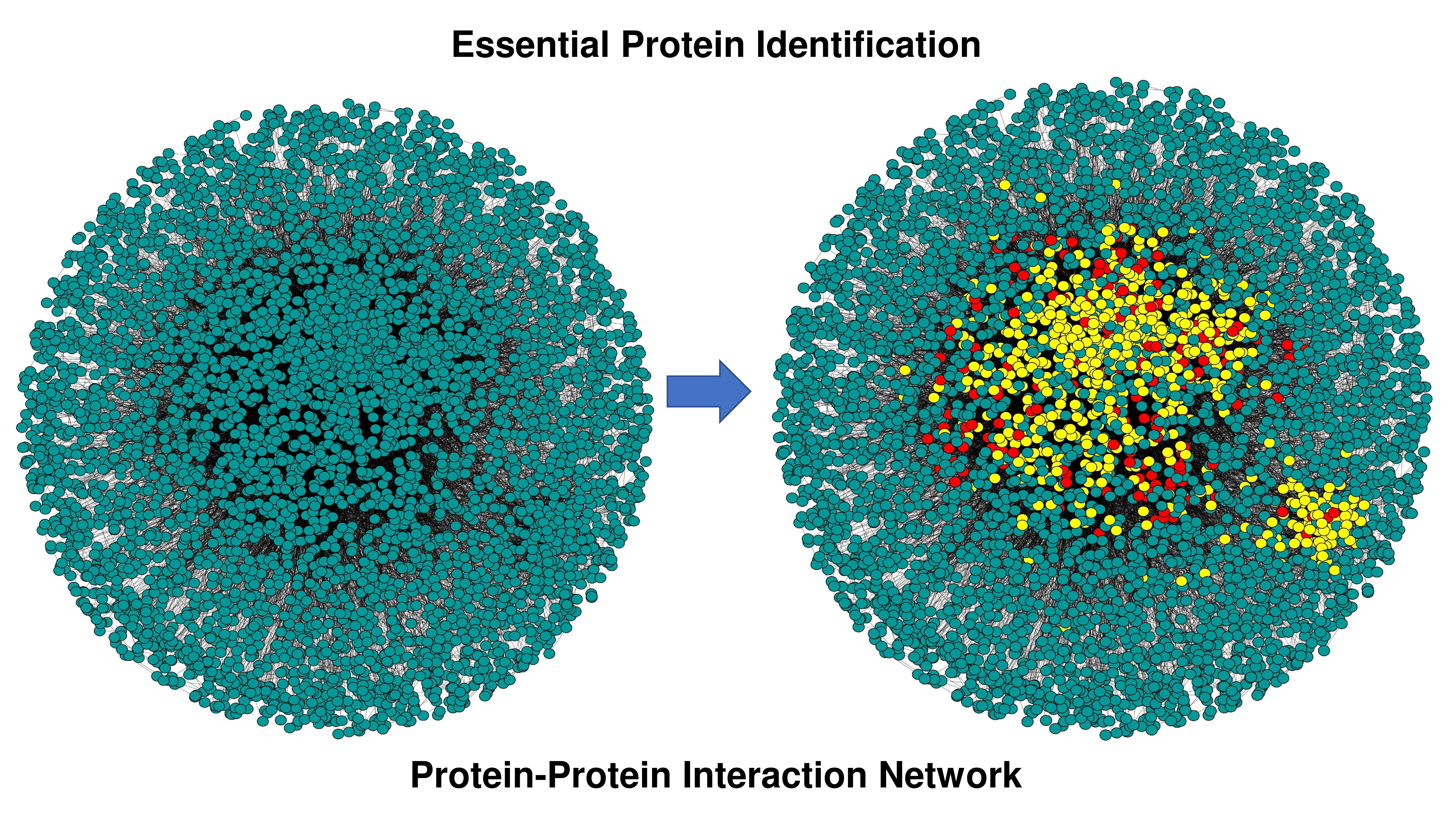
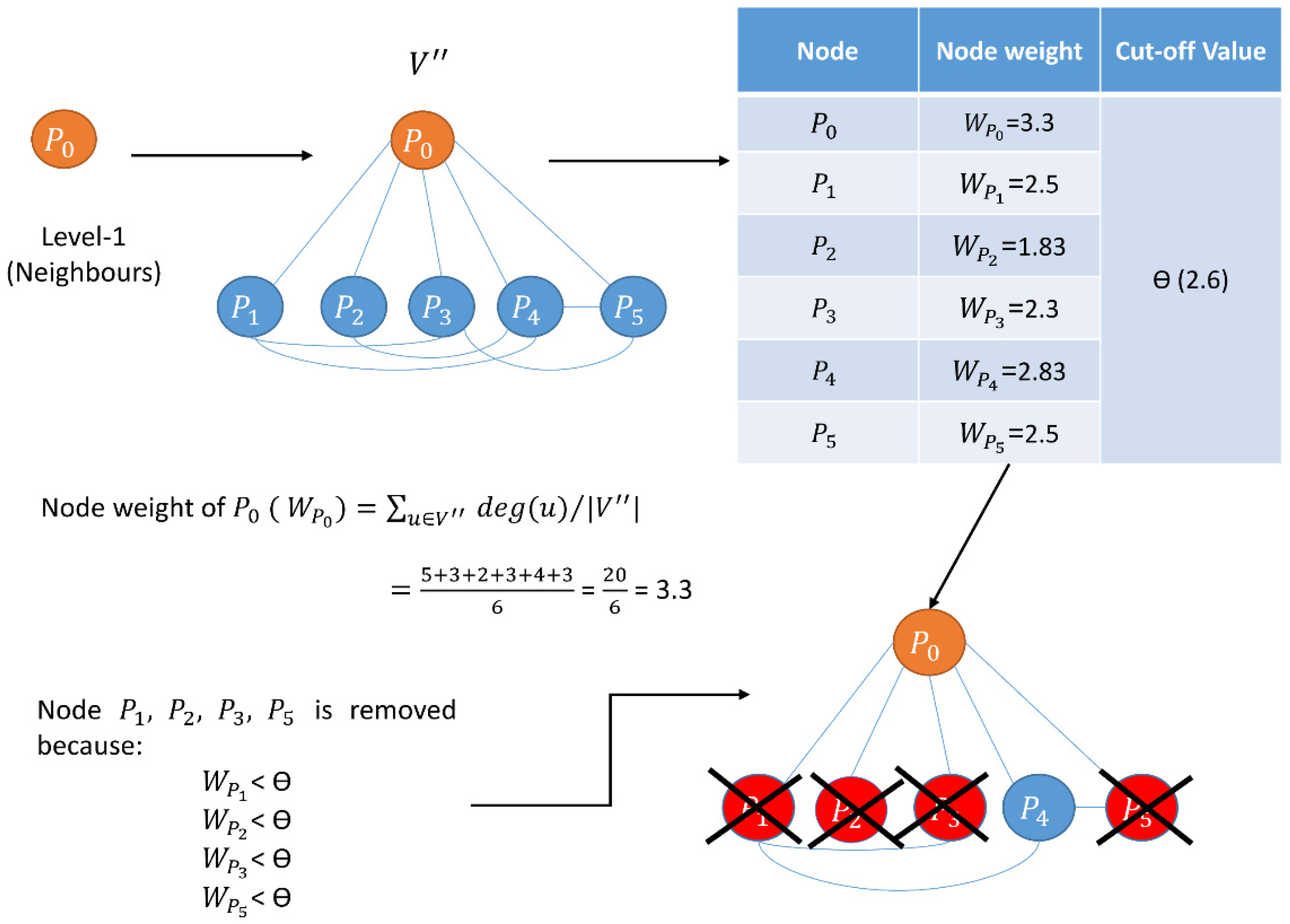
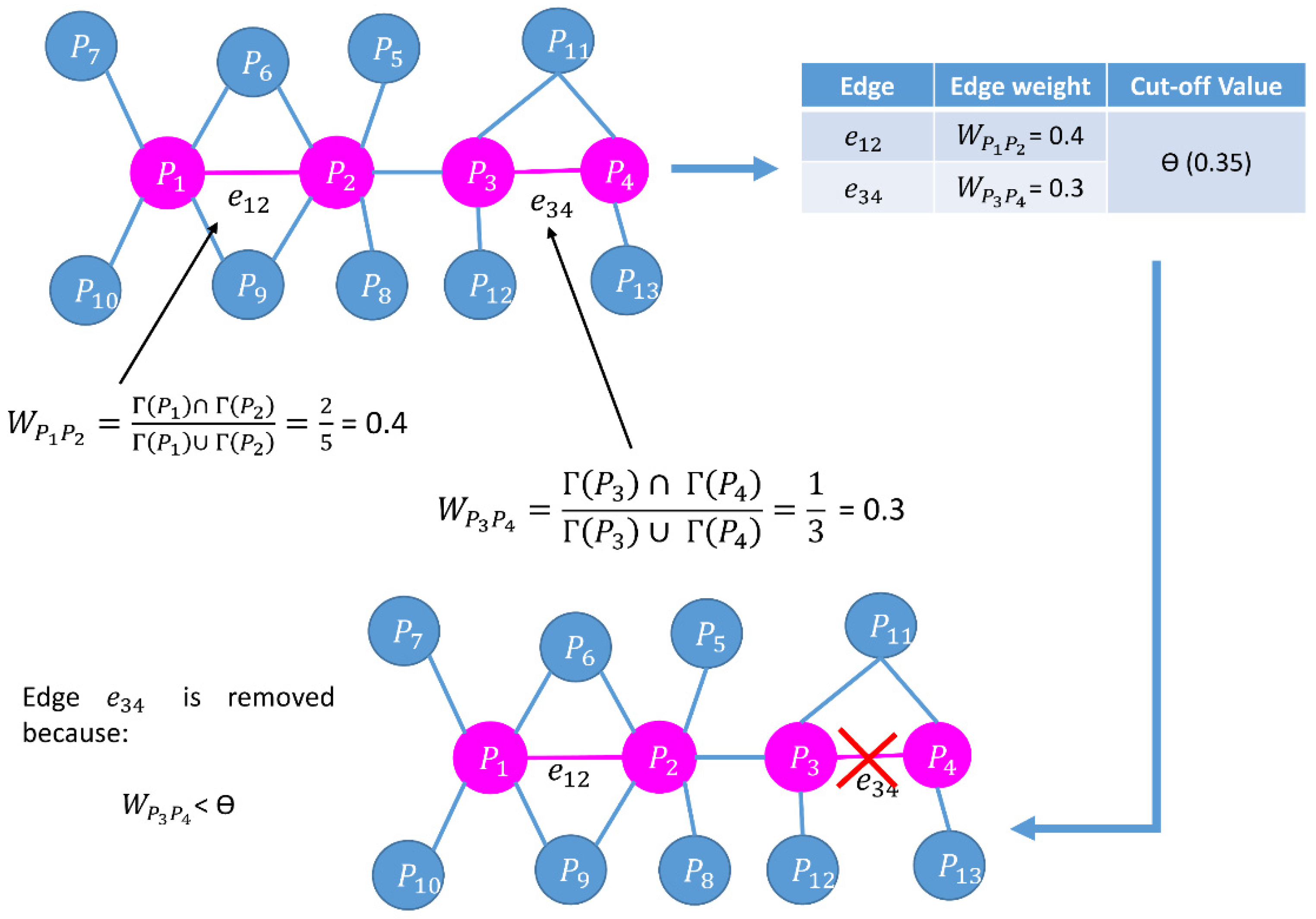
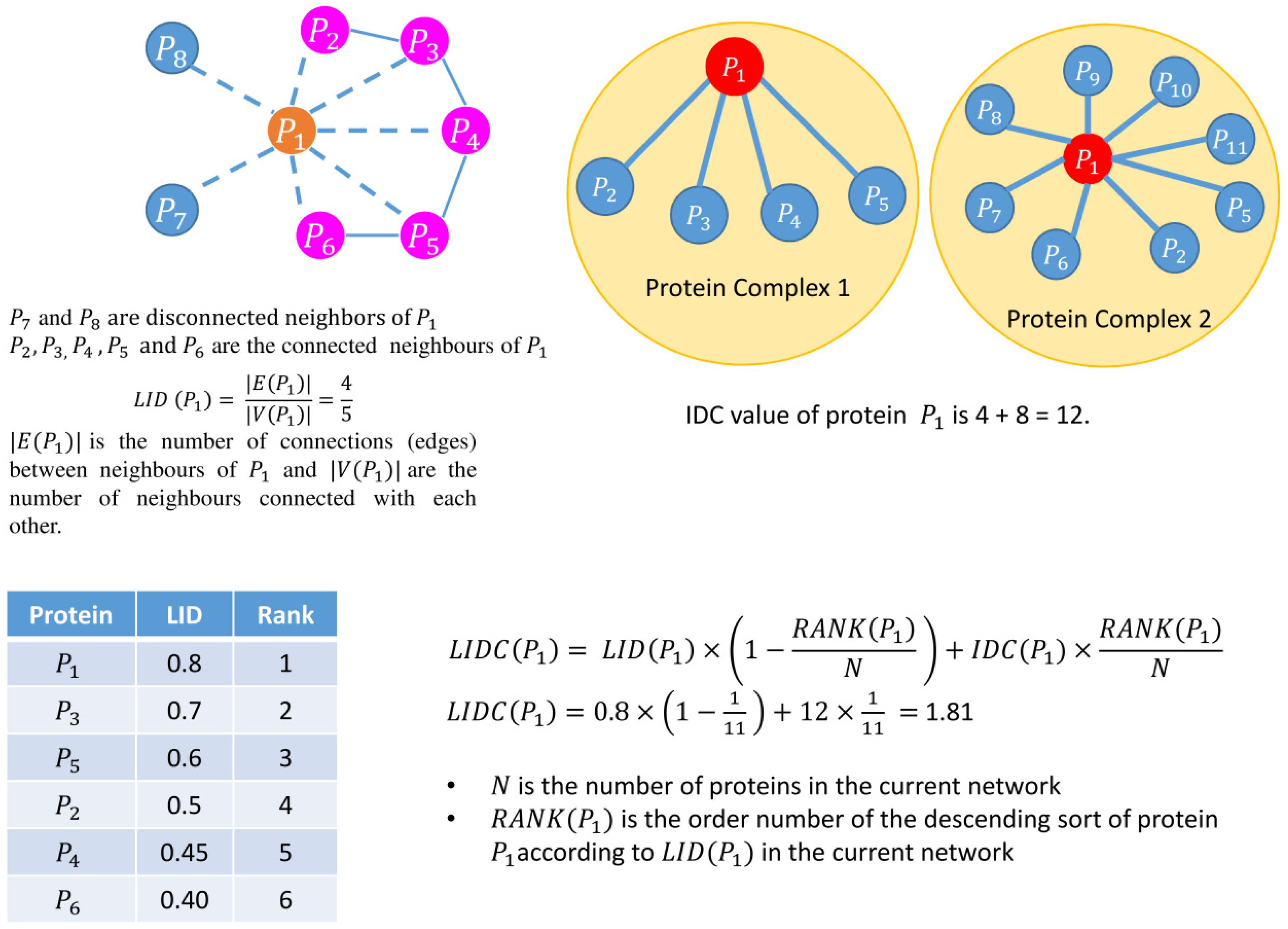
| Begin //calculating node weight for every node in the network Calculate the node weight, // is the set of neighbors of node , and is the number of proteins in // is the degree of a node //end of calculating node weight Compute // Cut-off calculation of node weight // is the mean of node weight, is the standard deviation of node weight, denotes three different //cut-offs, i.e., low, medium, and high, respectively. //reduction of network based on of node weights for every node in the network if node weight of remove from the network //end of reduction of network based on of node weights //edge weight calculation for every edge in the network Calculate edge weight, // and are the neighbors of and respectively //Γ(u) ∩ Γ(v) represents all common neighbors of u and v //Γ(u) ∪ Γ(v) represents all distinct neighbors of u and v //end of edge weight calculation Compute //Cut-off calculation of edge weight // is the mean of edge weight, is the standard deviation of edge weight, denotes three different //cut-offs, i.e., low, medium, and high, respectively. //reduction of network based on of edge weights for every edge in the network if edge weight of remove from the network //end of reduction of network based on of edge weights //calculate LIDC for low, medium, and high node edge weight //calculation of LIDC for every node in the pruned network, compute // is the number of connections (edges) between neighbors of , and are the number of neighbors //connected with each other //end of calculation of LID //) denotes a set of protein complexes that include protein // is the degree of protein in th protein complex that belongs to //end of calculation of IDC // is the value of the LID, is the value of IDC of the protein complex of protein , // is the number of proteins in the current network, // is the order number of the descending sort of protein according to in the current network //end of calculation of LIDC Choose proteins in six ranking ranges (top 100–600) as essential protein sets. End |
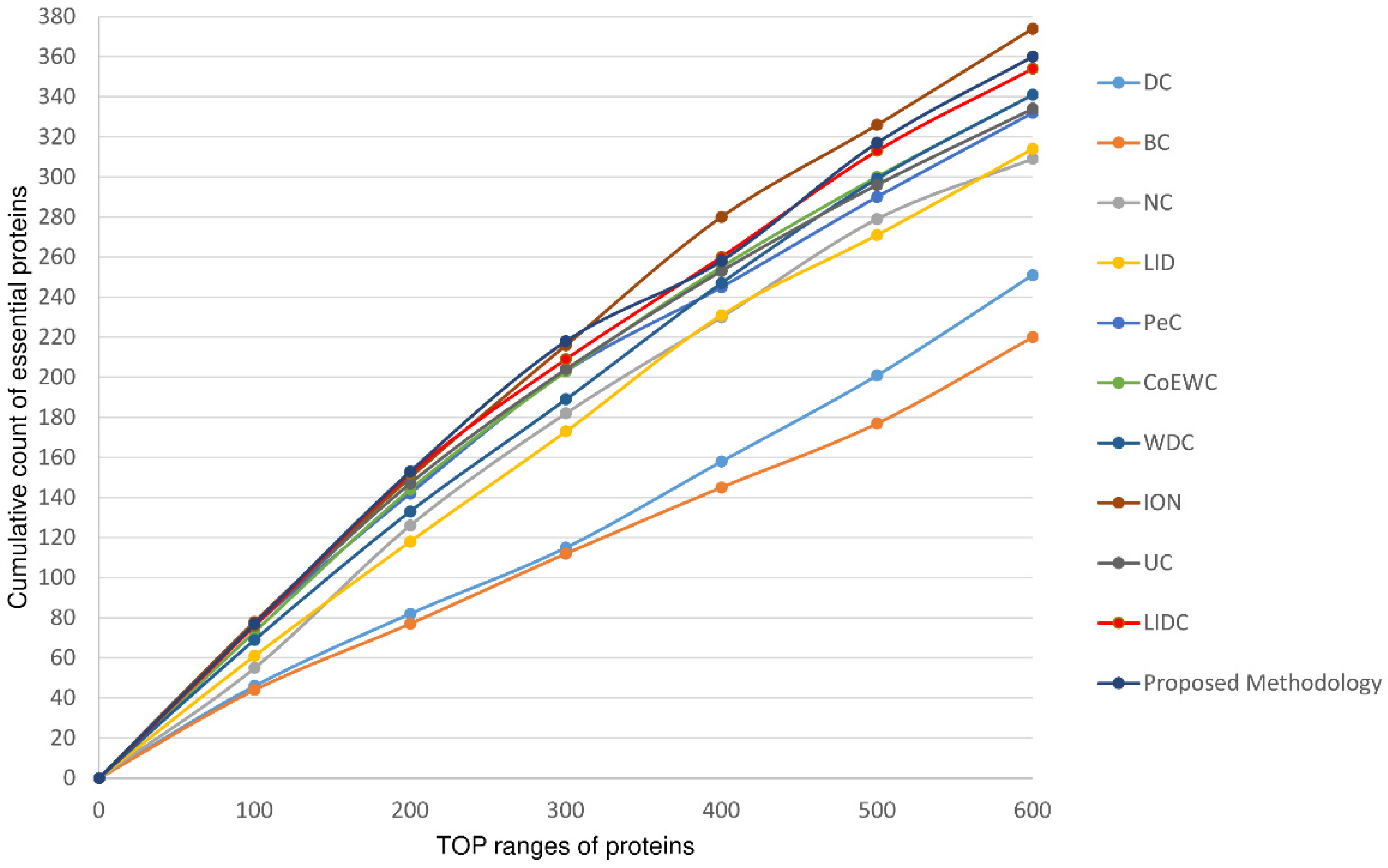
4. Result and Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, H.; Jia, Y.; Zhang, Y. Protein structure prediction. Int. J. Mod. Phys. B 2018, 32, 1840009. [Google Scholar] [CrossRef] [PubMed]
- Krupa, P.; Mozolewska, M.A.; Joo, K.; Lee, J.; Czaplewski, C.; Liwo, A. Prediction of Protein Structure by Template-Based Modeling Combined with the UNRES Force Field. J. Chem. Inf. Model. 2015, 55, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Makrodimitris, S.; van Ham, R.C.H.J.; Reinders, M.J.T. Improving protein function prediction using protein sequence and GO-term similarities. Bioinformatics 2018, 35, 1116–1124. [Google Scholar] [CrossRef]
- Koskinen, P.; Törönen, P.; Nokso-Koivisto, J.; Holm, L. PANNZER: High-throughput functional annotation of uncharacterized proteins in an error-prone environment. Bioinformatics 2015, 31, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Orengo, C.A. Protein function annotation using protein domain family resources. Methods 2016, 93, 24–34. [Google Scholar] [CrossRef]
- Rentzsch, R.; Orengo, C.A. Protein function prediction using domain families. BMC Bioinform. 2013, 14, S5. [Google Scholar] [CrossRef]
- Saha, S.; Chatterjee, P.; Basu, S.; Nasipuri, M.; Plewczynski, D. FunPred 3.0: Improved protein function prediction using protein interaction network. PeerJ 2019, 7, e6830. [Google Scholar] [CrossRef]
- Saha, S.; Chatterjee, P.; Basu, S.; Kundu, M.; Nasipuri, M. FunPred-1: Protein function prediction from a protein interaction network using neighborhood analysis. Cell. Mol. Biol. Lett. 2014, 19, 675–691. [Google Scholar] [CrossRef]
- Basak, S.N.; Biswas, A.K.; Saha, S.; Chatterjee, P.; Basu, S.; Nasipuri, M. Target Protein Function Prediction by Identification of Essential Proteins in Protein-Protein Interaction Network. In Proceedings of the Computational Intelligence, Communications, and Business Analytics, Singapore, 26 June 2019; pp. 219–231. [Google Scholar]
- Saha, S.; Prasad, A.; Chatterjee, P.; Basu, S.; Nasipuri, M. Protein function prediction from protein–protein interaction network using gene ontology based neighborhood analysis and physico-chemical features. J. Bioinform. Comput. Biol. 2018, 16, 1850025. [Google Scholar] [CrossRef]
- Zhao, B.; Hu, S.; Li, X.; Zhang, F.; Tian, Q.; Ni, W. An efficient method for protein function annotation based on multilayer protein networks. Hum. Genom. 2016, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Savojardo, C.; Martelli Pier, L.; Fariselli, P.; Profiti, G.; Casadio, R. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018, 46, W459–W466. [Google Scholar] [CrossRef] [PubMed]
- Nebenführ, A. Identifying Subcellular Protein Localization with Fluorescent Protein Fusions After Transient Expression in Onion Epidermal Cells. In Plant Cell Morphogenesis: Methods and Protocols; Žárský, V., Cvrčková, F., Eds.; Humana Press: Totowa, NJ, USA, 2014; pp. 77–85. [Google Scholar]
- Jianxin Wang, M.L.H.W.Y.P.; Min, L.; Huan, W.; Yi, P.; Wang, J.; Li, M.; Wang, H.; Pan, Y.; Jianxin, W.; Min, L.; et al. Identification of essential proteins based on edge clustering coefficient. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lu, Y.; Wang, J.; Wu, F.; Pan, Y. A Topology Potential-Based Method for Identifying Essential Proteins from PPI Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, J.; Wang, H.; Pan, Y. Essential Proteins Discovery from Weighted Protein Interaction Networks. In Proceedings of the Bioinformatics Research and Applications, Berlin, Germany, 26 June 2010; pp. 89–100. [Google Scholar]
- Jeong, H.; Mason, S.P.; Barabási, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, G.; Childs, B.; Valle, D. Human disease genes. Nature 2001, 409, 853–855. [Google Scholar] [CrossRef]
- Gill, N.; Singh, S.; Aseri, T.C. Computational Disease Gene Prioritization: An Appraisal. J. Comput. Biol. 2014, 21, 456–465. [Google Scholar] [CrossRef]
- Zhu, C.; Wu, C.; Aronow, B.J.; Jegga, A.G. Computational approaches for human disease gene prediction and ranking. Adv. Exp. Med. Biol. 2014, 799, 69–84. [Google Scholar] [CrossRef]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Véronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; André, B.; et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef]
- Yuan, Z.; Chong, W. Identification of Essential Proteins Using Improved Node and Edge Clustering Coefficient. In Proceedings of the 2018 37th Chinese Control Conference (CCC), Wuhan, China, 25–27 July 2018; pp. 3258–3262. [Google Scholar]
- Luo, J.; Qi, Y. Identification of Essential Proteins Based on a New Combination of Local Interaction Density and Protein Complexes. PLoS ONE 2015, 10, e0131418. [Google Scholar]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.-X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Bio Syst. 2015, 127, 67–72. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joy, M.P.; Brock, A.; Ingber, D.E.; Huang, S. High-Betweenness Proteins in the Yeast Protein Interaction Network. J. Biomed. Biotechnol. 2005, 2005, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Wuchty, S.; Stadler, P.F. Centers of complex networks. J. Theor. Biol. 2003, 223, 45–53. [Google Scholar] [CrossRef]
- Bonacich, P. Power and Centrality: A Family of Measures. Am. J. Sociol. 1987, 92, 1170–1182. [Google Scholar] [CrossRef]
- Li, M.; Wang, J.; Chen, X.; Wang, H.; Pan, Y. A local average connectivity-based method for identifying essential proteins from the network level. Comput. Biol. Chem. 2011, 35, 143–150. [Google Scholar] [CrossRef]
- Estrada, E.; Rodríguez-Velázquez, J.A. Subgraph centrality in complex networks. Phys. Rev. E 2005, 71, 056103. [Google Scholar] [CrossRef]
- S Karen, M.Z.; Stephenson, K.; Zelen, M. Rethinking centrality: Methods and examples. Soc. Netw. 1989, 11, 1–37. [Google Scholar] [CrossRef]
- Hsing, M.; Byler, K.G.; Cherkasov, A. The use of Gene Ontology terms for predicting highly-connected ‘hub’ nodes in protein-protein interaction networks. BMC Syst. Biol. 2008, 2, 80. [Google Scholar] [CrossRef]
- Ren, J.; Wang, J.; Li, M.; Wang, H.; Liu, B. Prediction of Essential Proteins by Integration of PPI Network Topology and Protein Complexes Information. In Proceedings of the International Symposium on Bioinformatics Research and Applications, Changsha, China, 27–29 May 2011; pp. 12–24. [Google Scholar]
- Li, M.; Lu, Y.; Niu, Z.; Wu, F.-X. United Complex Centrality for Identification of Essential Proteins from PPI Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 370–380. [Google Scholar] [CrossRef]
- Peng, W.; Wang, J.; Wang, W.; Liu, Q.; Wu, F.-X.; Pan, Y. Iteration method for predicting essential proteins based on orthology and protein-protein interaction networks. BMC Syst. Biol. 2012, 6, 87. [Google Scholar] [CrossRef]
- Peng, X.; Wang, J.; Zhong, J.; Luo, J.; Pan, Y. An efficient method to identify essential proteins for different species by integrating protein subcellular localization information. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Washington, DC, USA, 9–12 November 2015; pp. 277–280. [Google Scholar]
- Li, M.; Zhang, H.; Wang, J.-X.; Pan, Y. A new essential protein discovery method based on the integration of protein-protein interaction and gene expression data. BMC Syst. Biol. 2012, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Wang, J.; Peng, X.; Wu, F.-x.; Pan, Y. Identifying essential proteins from active PPI networks constructed with dynamic gene expression. BMC Genom. 2015, 16 (Suppl. S3), S1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xiao, W.; Hu, X. Predicting essential proteins by integrating orthology, gene expressions, and PPI networks. PLoS ONE 2018, 13, e0195410. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Jin, Y.-T.; Hua, H.-L.; Wen, Q.-F.; Luo, S.; Zheng, W.-X.; Guo, F.-B. Comprehensive review of the identification of essential genes using computational methods: Focusing on feature implementation and assessment. Brief. Bioinform. 2020, 21, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wu, W.; Zhang, Y.; Li, X.; Jiang, X.; Wei, G.; Tao, S. A new computational strategy for predicting essential genes. BMC Genom. 2013, 14, 910. [Google Scholar] [CrossRef]
- Cheng, J.; Xu, Z.; Wu, W.; Zhao, L.; Li, X.; Liu, Y.; Tao, S. Training Set Selection for the Prediction of Essential Genes. PLoS ONE 2014, 9, e86805. [Google Scholar] [CrossRef]
- Sakharkar, K.R.; Sakharkar, M.K.; Chow, V.T. A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. Silico Biol. 2004, 4, 355–360. [Google Scholar]
- Song, J.H.; Ko, K.S.; Lee, J.Y.; Baek, J.Y.; Oh, W.S.; Yoon, H.S.; Jeong, J.Y.; Chun, J. Identification of essential genes in Streptococcus pneumoniae by allelic replacement mutagenesis. Mol. Cells 2005, 19, 365–374. [Google Scholar]
- Sarangi, A.N.; Lohani, M.; Aggarwal, R. Prediction of essential proteins in prokaryotes by incorporating various physico-chemical features into the general form of Chou’s pseudo amino acid composition. Protein Pept. Lett. 2013, 20, 781–795. [Google Scholar] [CrossRef]
- Ning, L.W.; Lin, H.; Ding, H.; Huang, J.; Rao, N.; Guo, F.B. Predicting bacterial essential genes using only sequence composition information. Genet. Mol. Res. GMR 2014, 13, 4564–4572. [Google Scholar] [CrossRef]
- Jeong, H.; Oltvai, Z.N.; Barabási, A.L. Prediction of Protein Essentiality Based on Genomic Data. Complexus 2003, 1, 19–28. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, D. Understanding protein dispensability through machine-learning analysis of high-throughput data. Bioinformatics 2005, 21, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Zotenko, E.; Mestre, J.; O’Leary, D.P.; Przytycka, T.M. Why do hubs in the yeast protein interaction network tend to be essential: Reexamining the connection between the network topology and essentiality. PLoS Comput. Biol. 2008, 4, e1000140. [Google Scholar] [CrossRef]
- Yu, H.; Braun, P.; Yildirim, M.A.; Lemmens, I.; Venkatesan, K.; Sahalie, J.; Hirozane-Kishikawa, T.; Gebreab, F.; Li, N.N.; Simonis, N.; et al. High-quality binary protein interaction map of the yeast interactome network. Science 2008, 322, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Krogan, N.J.; Cunningham, P.; Cagney, G. All or nothing: Protein complexes flip essentiality between distantly related eukaryotes. Genome Biol. Evol. 2013, 5, 1049–1059. [Google Scholar] [CrossRef]
- Wang, H.; Kakaradov, B.; Collins, S.R.; Karotki, L.; Fiedler, D.; Shales, M.; Shokat, K.M.; Walther, T.C.; Krogan, N.J.; Koller, D. A Complex-based Reconstruction of the Saccharomyces cerevisiae Interactome. Mol. Cell. Proteom. 2009, 8, 1361–1381. [Google Scholar] [CrossRef]
- He, X.; Zhang, J. Why do hubs tend to be essential in protein networks? PLoS Genet. 2006, 2, e88. [Google Scholar] [CrossRef]
- Lei, X.; Yang, X. A new method for predicting essential proteins based on participation degree in protein complex and subgraph density. PLoS ONE 2018, 13, e0198998. [Google Scholar] [CrossRef]
- Hart, G.T.; Lee, I.; Marcotte, E.R. A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality. BMC Bioinform. 2007, 8, 236. [Google Scholar] [CrossRef]
- Zhong, J.; Wang, J.; Peng, W.; Zhang, Z.; Pan, Y. Prediction of essential proteins based on gene expression programming. BMC Genom. 2013, 14, s4–s7. [Google Scholar] [CrossRef]
- Ercsey-Ravasz, M.; Lichtenwalter, R.N.; Chawla, N.V.; Toroczkai, Z. Range-limited centrality measures in complex networks. Phys. Rev. E 2012, 85, 066103. [Google Scholar] [CrossRef] [PubMed]
- Korn, A.; Schubert, A.; Telcs, A. Lobby index in networks. Phys. A Stat. Mech. Its Appl. 2009, 388, 2221–2226. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, J.; Xiao, W.X. A New Method for the Discovery of Essential Proteins. PLoS ONE 2013, 8, e58763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lü, L.; Zhang, Y.-C.; Yeung, C.H.; Zhou, T. Leaders in social networks, the Delicious case. PLoS ONE 2011, 6, e21202. [Google Scholar] [CrossRef]
- Tang, X.; Wang, J.; Zhong, J.; Pan, Y. Predicting essential proteins basedon weighted degree centrality. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 407–418. [Google Scholar] [CrossRef]
- Ghosh, R.; Lerman, K. Parameterized centrality metric for network analysis. Phys. Rev. E—Stat. Nonlinear Soft Matter Phys. 2011, 83, 066118. [Google Scholar] [CrossRef]
- Li, G.; Li, M.; Wang, J.; Li, Y.; Pan, Y. United neighborhood closeness centrality and orthology for predicting essential proteins. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 17, 1451–1458. [Google Scholar] [CrossRef]
- Wang, S.; Wu, F. Detecting overlapping protein complexes in PPI networks based on robustness. Proteome Sci. 2013, 11, S18. [Google Scholar] [CrossRef]
- Xenarios, I.; Rice, D.W.; Salwinski, L.; Baron, M.K.; Marcotte, E.M.; Eisenberg, D. DIP: The Database of Interacting Proteins. Nucleic Acids Res. 2000, 28, 289–291. [Google Scholar] [CrossRef]
- Xenarios, I. DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar] [CrossRef]
- Mewes, H.W. MIPS: Analysis and annotation of proteins from whole genomes in 2005. Nucleic Acids Res. 2005, 34, D169–D172. [Google Scholar] [CrossRef] [PubMed]
- Friedel, C.C.; Krumsiek, J.; Zimmer, R. Bootstrapping the interactome: Unsupervised identification of protein complexes in yeast. In Research in Computational Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 3–16. [Google Scholar]
- Aloy, P.; Böttcher, B.; Ceulemans, H.; Leutwein, C.; Mellwig, C.; Fischer, S.; Gavin, A.C.; Bork, P.; Superti-Furga, G.; Serrano, L.; et al. Structure-Based Assembly of Protein Complexes in Yeast. Science 2004, 303, 2026–2029. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.M.; Adler, C.; Ball, C.; Chervitz, S.A.; Dwight, S.S.; Hester, E.T.; Jia, Y.; Juvik, G.; Roe, T.; Schroeder, M.; et al. SGD: Saccharomyces genome database. Nucleic Acids Res. 1998, 26, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Pu, S.; Wong, J.; Turner, B.; Cho, E.; Wodak, S.J. Up-to-date catalogues of yeast protein complexes. Nucleic Acids Res. 2009, 37, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Pu, S.; Vlasblom, J.; Emili, A.; Greenblatt, J.; Wodak, S.J. Identifying functional modules in the physical interactome of Saccharomyces cerevisiae. Proteomics 2007, 7, 944–960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, H.; Yang, Z.; Wang, J.; Liu, Y.; Sang, S. A method for predicting protein complex in dynamic PPI networks. BMC Bioinform. 2016, 17, 229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Lin, Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 2009, 37, D455–D458. [Google Scholar] [CrossRef]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef]
- Gurumayum, S.; Jiang, P.; Hao, X.; Campos, T.L.; Young, N.D.; Korhonen, P.K.; Gasser, R.B.; Bork, P.; Zhao, X.-M.; He, L.-j.; et al. OGEE v3: Online GEne Essentiality database with increased coverage of organisms and human cell lines. Nucleic Acids Res. 2021, 49, D998–D1003. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Utilized Features | Description | Database | References |
|---|---|---|---|
| Subcellular localization | An efficient method to identify essential proteins for different species by integrating protein subcellular localization information. | PPIN of Saccharomyces cerevisiae, Homo sapiens, Mus musculus and Drosophila melanogaster | [36] |
| Protein complex, degree, subgraph | A new method for predicting essential proteins based on participation degree in protein complex and subgraph Density. | PPIN of Saccharomyces cerevisiae | [54] |
| Orthology, gene expression, PPIN | Predicting essential proteins by integrating orthology, gene expressions, and PPIN. | PPIN of Saccharomyces cerevisiae | [39] |
| CC and orthology | United neighborhood closeness centrality and orthology for predicting essential proteins. | PPIN of Saccharomyces cerevisiae | [63] |
| Node, edge clustering coefficient | Identification of essential proteins using improved node and edge clustering coefficient. | PPIN of Saccharomyces cerevisiae and Drosophila melanogaster | [22] |
| Centrality scores | CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. | _ | [24] |
| Protein complex | Identification of essential proteins based on a new combination of local interaction density and protein complexes. | PPIN of Saccharomyces cerevisiae | [23] |
| PPIN, protein complex | Prediction of essential proteins by integration of PPI network topology and protein complex information. | PPIN of Saccharomyces cerevisiae | [33] |
| Methods | Precision | Recall | F-Score |
|---|---|---|---|
| DC (Jeong et al. 2001) | 0.41 | 0.35 | 0.38 |
| BC (Joy et al. 2005) | 0.35 | 0.31 | 0.33 |
| NC (Jianxin Wang et al. 2012) | 0.46 | 0.40 | 0.43 |
| LID (Luo and Qi 2015) | 0.45 | 0.39 | 0.42 |
| PeC (Li et al. 2012) | 0.46 | 0.40 | 0.43 |
| CoEWC (Zhang et al. 2013) | 0.47 | 0.41 | 0.44 |
| WDC (Xiwei et al. 2014) | 0.48 | 0.42 | 0.45 |
| ION (Peng et al. 2012) | 0.53 | 0.41 | 0.46 |
| UC (Li et al. 2017) | 0.48 | 0.42 | 0.45 |
| LIDC (Luo and Qi 2015) | 0.50 | 0.44 | 0.47 |
| Proposed Methodology | 0.77 | 0.44 | 0.56 |
| Cut-Off Levels | Proteins after Node Reduction | Interactions after Node Reduction | Proteins after Edge Reduction | Interactions after Node Reduction | Essential Protein | Non-Essential Protein |
|---|---|---|---|---|---|---|
| Low | 1393 | 14,063 | 985 | 3907 | 198 | 787 |
| Medium | 1374 | 13,924 | 969 | 3847 | 194 | 775 |
| High | 1340 | 13,714 | 931 | 3733 | 187 | 744 |
| Cut-Off Levels | Recall | Precision | F-Score |
|---|---|---|---|
| Low | 0.41 | 0.75 | 0.53 |
| Medium | 0.42 | 0.76 | 0.54 |
| High | 0.44 | 0.77 | 0.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banik, A.; Podder, S.; Saha, S.; Chatterjee, P.; Halder, A.K.; Nasipuri, M.; Basu, S.; Plewczynski, D. Rule-Based Pruning and In Silico Identification of Essential Proteins in Yeast PPIN. Cells 2022, 11, 2648. https://doi.org/10.3390/cells11172648
Banik A, Podder S, Saha S, Chatterjee P, Halder AK, Nasipuri M, Basu S, Plewczynski D. Rule-Based Pruning and In Silico Identification of Essential Proteins in Yeast PPIN. Cells. 2022; 11(17):2648. https://doi.org/10.3390/cells11172648
Chicago/Turabian StyleBanik, Anik, Souvik Podder, Sovan Saha, Piyali Chatterjee, Anup Kumar Halder, Mita Nasipuri, Subhadip Basu, and Dariusz Plewczynski. 2022. "Rule-Based Pruning and In Silico Identification of Essential Proteins in Yeast PPIN" Cells 11, no. 17: 2648. https://doi.org/10.3390/cells11172648