
Genome-Wide Expression Profiling and Networking Reveals an Imperative Role of IMF-Associated Novel CircRNAs as ceRNA in Pigs

Abstract
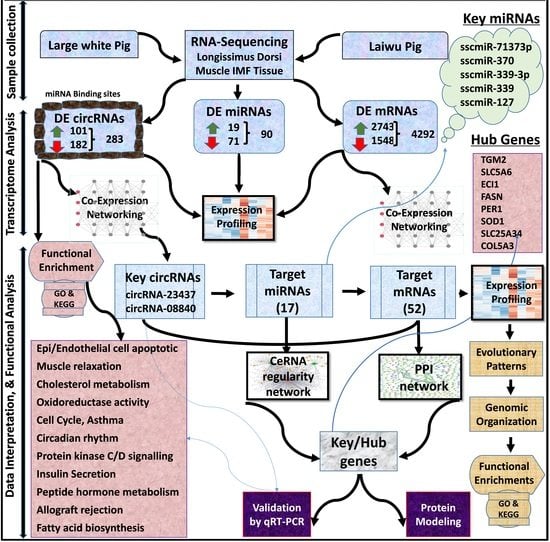
:
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Animals and Sample Collection
2.3. RNA Isolation and Quality Control
2.4. RNA Library Construction
2.5. Identification and Characterization of circRNAs
2.6. Identification and Characterization of miRNAs
2.7. Differential Expression Analysis and ceRNA Regulatory Networking
2.8. Functional Enrichment Analysis of DEC Host and Target Genes
2.9. Phylogenesis and Genomic Structures of Target Genes
2.10. Expression Profiling and Subcellular Localization of Target Genes
2.11. Protein-Protein Interaction Network Analysis
2.12. Real-Time Fluorescence Quantitative PCR Verification
2.13. Protein Modeling for Structural Analysis
2.14. Statistical Analysis
3. Results
3.1. Circular RNA Library Construction and Quality Control
3.2. Identification, Characterization, and Expression Profiling of circRNAs
3.3. miRNA Library Construction and Quality Control
3.4. Identification, Characterization, and Expression Profiling of miRNAs
3.5. Co-Expression Network Analysis of circRNA-miRNA Interaction
3.6. Interaction of ncRNAs with mRNA as ceRNA in IMF Tissues
3.7. Functional Enrichment Analysis of DEC Host Genes
3.8. Functional Enrichment Analysis of DEC-Target Genes
3.9. Expression Profiling of Target DEGs, Gene Structure, and Subcellular Localization
3.10. Protein-Protein Interaction Network Analysis for DEGs
3.11. Structural-Functional Relationship of IMF-Associated Key Genes
3.12. qRT-PCR Verification
4. Discussion
4.1. Significance of Intramuscular Fat Deposition in Pork
4.2. Crosstalk of circRNAs with Their Target miRNAs Stimulating Fat Metabolism
4.3. Coherence of the ceRNA Regulatory Network with Differential Expression Datasets to Regulate Fat Metabolism
4.4. Significance of IMF-Associated Key/Potential Genes in Regulating Various Lipid Metabolic Syndromes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lonergan, S.; Topel, D.; Marple, D. Chapter 5—Fat and fat cells in domestic animals. In The Science of Animal Growth and Meat Technology, 2nd ed.; Academic Press: New York, NY, USA, 2019; pp. 51–69. [Google Scholar] [CrossRef]
- Luo, L.; Liu, M. Adipose tissue in control of metabolism. J. Endocrinol. 2016, 231, R77–R99. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Kamiya, M.; Higuchi, M. Fat depot-specific effects of body fat distribution and adipocyte size on intramuscular fat accumulation in Wagyu cattle. Anim. Sci. J. 2020, 91, e13449. [Google Scholar] [CrossRef]
- Yang, X.; Smith, U. Adipose tissue distribution and risk of metabolic disease: Does thiazolidinedione-induced adipose tissue redistribution provide a clue to the answer? Diabetologia 2007, 50, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, J.F.; Grace, D. The consequences of human actions on risks for infectious diseases: A review. Infect. Ecol. Epidemiol. 2015, 5, 30048. [Google Scholar] [CrossRef] [PubMed]
- Gardan, D.; Gondret, F.; Louveau, I. Lipid metabolism and secretory function of porcine intramuscular adipocytes compared with subcutaneous and perirenal adipocytes. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E372–E380. [Google Scholar] [CrossRef]
- Ayuso, D.; González, A.; Peña, F.; Hernández-García, F.I.; Izquierdo, M. Effect of fattening period length on intramuscular and subcutaneous fatty acid profiles in Iberian pigs finished in the montanera sustainable system. Sustainability 2020, 12, 7937. [Google Scholar] [CrossRef]
- Pérez-Palacios, T.; Ruiz, J.; Tejeda, J.F.; Antequera, T. Subcutaneous and intramuscular lipid traits as tools for classifying Iberian pigs as a function of their feeding background. Meat Sci. 2009, 81, 632–640. [Google Scholar] [CrossRef]
- Mourot, J.; Kouba, M.; Bonneau, M. Comparative study of in vitro lipogenesis in various adipose tissues in the growing Meishan pig: Comparison with the Large White pig (Sus domesticus). Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 1996, 115, 383–388. [Google Scholar] [CrossRef]
- Zhang, S.; Song, G.; Yuan, J.; Qiao, S.; Xu, S.; Si, Z.; Yang, Y.; Xu, X.; Wang, A. Circular RNA circ_0003204 inhibits proliferation, migration and tube formation of endothelial cell in atherosclerosis via miR-370-3p/TGFβR2/phosph-SMAD3 axis. J. Biomed. Sci. 2020, 27, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Ye, Z.; Stanton, R. Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. RNA 2020, 26, 903–909. [Google Scholar] [CrossRef]
- Li, M.; Zhu, M.; Chai, W.; Wang, Y.; Song, Y.; Liu, B.; Cai, C.; Song, Y.; Sun, X.; Xue, P.; et al. Determination of the heterogeneity of intramuscular fat and visceral adipose tissue from Dezhou donkey by lipidomics and transcriptomics profiling. Front. Nutr. 2021, 8, 746684. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.; Ball, A.; Hughes, J.; Krishnamurthy, R.; Piyasiri, U.; Stark, J.; Watkins, P.; Warner, R. Sensory and flavor chemistry characteristics of Australian beef: Influence of intramuscular fat, feed, and breed. J. Agric. Food Chem. 2016, 64, 4299–4311. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.L.; Wang, Q.; Zhang, Z.M.; Xu, Y.L.; Yan, H.C.; Li, H.C.; Gao, C.Q.; Wang, X.Q. Dietary supplementation with pioglitazone hydrochloride and chromium methionine improves growth performance, meat quality, and antioxidant ability in finishing pigs. J. Agric. Food Chem. 2018, 66, 4345–4351. [Google Scholar] [CrossRef] [PubMed]
- Tyra, M.; Ropka-Molik, K.; Terman, A.; Piórkowska, K.; Oczkowicz, M.; Bereta, A. Association between subcutaneous and intramuscular fat content in porcine ham and loin depending on age, breed and FABP3 and LEPR genes transcript abundance. Mol. Biol. Rep. 2013, 40, 2301–2308. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.Q.; Wang, G.L.; Wang, C.F.; Wei, S.D.; Wu, Y.; Wang, L.Y.; Wang, H.; Yang, H.L. Genetic variation of H-FABP gene and association with intramuscular fat content in Laiwu Black and four western pig breeds. Asian Australas. J. Anim. Sci. 2005, 18, 13–16. [Google Scholar] [CrossRef]
- Spurlock, M.E.; Gabler, N.K. The development of porcine models of obesity and the metabolic syndrome. J. Nutr. 2008, 138, 397–402. [Google Scholar] [CrossRef]
- Chen, Q.; Zeng, Y.; Wang, H.; Yang, L.; Yang, Y.; Zhu, H.; Shi, Y.; Chen, W.; Hu, Y. Molecular characterization and expression analysis of NDUFS4 gene in m. longissimus dorsi of Laiwu pig (Sus scrofa). Mol. Biol. Rep. 2013, 40, 1599–1608. [Google Scholar] [CrossRef]
- Grzes, M.; Sadkowski, S.; Rzewuska, K.; Szydlowski, M.; Switonski, M. Pig fatness in relation to FASN and INSIG2 genes polymorphism and their transcript level. Mol. Biol. Rep. 2016, 43, 381–389. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, X.; Li, A.; Xie, L.; Miao, X. Genome-wide analysis of mRNAs and lncRNAs of intramuscular fat related to lipid metabolism in two pig breeds. Cell. Physiol. Biochem. 2018, 50, 2406–2422. [Google Scholar] [CrossRef]
- Li, A.; Huang, W.; Zhang, X.; Xie, L.; Miao, X. Identification and characterization of CircRNAs of two pig breeds as a new biomarker in metabolism-related diseases. Cell. Physiol. Biochem. 2018, 47, 2458–2470. [Google Scholar] [CrossRef]
- Lim, K.S.; Lee, K.T.; Park, J.E.; Chung, W.H.; Jang, G.W.; Choi, B.H.; Hong, K.C.; Kim, T.H. Identification of differentially expressed genes in longissimus muscle of pigs with high and low intramuscular fat content using RNA sequencing. Anim. Genet. 2017, 48, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.H.; Lu, J.X.; Chen, Y.; Zhao, Y.Q.; Guo, P.H.; Yang, J.T.; Zang, R.X. Comparison of the adipogenesis in intramuscular and subcutaneous adipocytes from Bamei and Landrace pigs. Biochem. Cell Biol. 2014, 92, 259–267. [Google Scholar] [CrossRef]
- Zhao, J.; Li, K.; Yang, Q.; Du, M.; Liu, X.; Cao, G. Enhanced adipogenesis in Mashen pigs compared with Large White pigs. Ital. J. Anim. Sci. 2017, 16, 217–225. [Google Scholar] [CrossRef]
- Ding, H.X.; Xu, Q.; Wang, B.G.; Lv, Z.; Yuan, Y. MetaDE-based analysis of circRNA expression profiles involved in gastric cancer. Dig. Dis. Sci. 2020, 65, 2884–2895. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Goff, L.A.; Trapnell, C.; Alexander, R.; Lo, K.A.; Hacisuleyman, E.; Sauvageau, M.; Tazon-Vega, B.; Kelley, D.R.; Hendrickson, D.G.; et al. Long noncoding RNAs regulate adipogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3387–3392. [Google Scholar] [CrossRef]
- Gorur, A.; Celik, A.; Yildirim, D.D.; Gundes, A.; Tamer, L. Investigation of possible effects of microRNAs involved in regulation of lipid metabolism in the pathogenesis of atherosclerosis. Mol. Biol. Rep. 2019, 46, 909–920. [Google Scholar] [CrossRef]
- Dube, U.; Del-Aguila, J.L.; Li, Z.; Budde, J.P.; Jiang, S.; Hsu, S.; Ibanez, L.; Fernandez, M.V.; Farias, F.; Norton, J.; et al. An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat. Neurosci. 2019, 22, 1903–1912. [Google Scholar] [CrossRef]
- Tang, Q.; Hann, S.S. Biological roles and mechanisms of circular RNA in human cancers. OncoTargets Ther. 2020, 13, 2067. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.-B.; Niu, Y.-L.; Huang, G.-X.; Lu, J.-J.; Chen, A.; Zhu, L. Silencing of circRNA. 2837 plays a protective role in sciatic nerve injury by sponging the miR-34 family via regulating neuronal autophagy. Mol. Ther. Nucleic Acids 2018, 12, 718–729. [Google Scholar] [CrossRef]
- Dong, W.; Dai, Z.H.; Liu, F.C.; Guo, X.G.; Ge, C.M.; Ding, J.; Liu, H.; Yang, F. The RNA-binding protein RBM3 promotes cell proliferation in hepatocellular carcinoma by regulating circular RNA SCD-circRNA 2 production. EBioMedicine 2019, 45, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Ragusa, M.; Barbagallo, D.; Chioccarelli, T.; Manfrevola, F.; Cobellis, G.; Di Pietro, C.; Brex, D.; Battaglia, R.; Fasano, S.; Ferraro, B.; et al. CircNAPEPLD is expressed in human and murine spermatozoa and physically interacts with oocyte miRNAs. RNA Biol. 2019, 16, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.-S.; Pan, F.; Mao, X.-D.; Liu, C.; Chen, Y.-J. Biological functions of circular RNAs and their roles in occurrence of reproduction and gynecological diseases. Am. J. Transl. Res. 2019, 11, 1–15. [Google Scholar] [PubMed]
- Steger, G.; Riesner, D. Viroid research and its significance for RNA technology and basic biochemistry. Nucleic Acids Res. 2018, 46, 10563–10576. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E. A 360 view of circular RNAs: From biogenesis to functions. Wiley Interdiscip. Rev. RNA 2018, 9, e1478. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.R.; Lee, J.H.; Sohn, K.C.; Lee, Y.; Seo, Y.J.; Kim, C.D.; Lee, J.H.; Hong, S.P.; Seo, S.J.; Kim, S.J.; et al. Adiponectin signaling regulates lipid production in human sebocytes. PLoS ONE 2017, 12, e0169824. [Google Scholar] [CrossRef]
- Song, W.W.; McLennan, S.V.; Tam, C.; Williams, P.F.; Baxter, R.C.; Twigg, S.M. CCN2 requires TGF-β signalling to regulate CCAAT/enhancer binding proteins and inhibit fat cell differentiation. J. Cell Commun. Signal. 2015, 9, 27–36. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Wang, X.P.; Luoreng, Z.M.; Zan, L.S.; Raza, S.H.; Li, F.; Li, N.; Liu, S. Expression patterns of miR-146a and miR-146b in mastitis infected dairy cattle. Mol. Cell. Probes 2016, 30, 342–344. [Google Scholar] [CrossRef]
- Miao, Z.; Wang, S.; Wang, Y.; Wei, P.; Khan, M.A.; Zhang, J.; Guo, L.; Liu, D. Comparison of microRNAs in the intramuscular adipose tissue from Jinhua and Landrace pigs. J. Cell. Biochem. 2019, 120, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S. Multiple roles for lipids in the Hedgehog signalling pathway. Nat. Rev. Mol. Cell Biol. 2008, 9, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D. MicroRNA targets in Drosophila. Genome Biol. 2003, 4, P8. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Drosatos, K.; Hiyama, Y.; Goldberg, I.J.; Zannis, V.I. MicroRNA-370 controls the expression of MicroRNA-122 and Cpt1α and affects lipid metabolism. J. Lipid Res. 2010, 51, 1513–1523. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Wang, J.; Wang, D.; Yao, A.; Li, Q. Prognostic and biological significance of microRNA-127 expression in human breast cancer. Dis. Markers 2014, 2014, 401986. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, J.; Sun, R.; He, Z.; Chen, Q.; Liu, W.; Wu, M.; Bao, J.; Liu, Z.; Wang, J.; et al. Comprehensive Construction of a Circular RNA-Associated Competing Endogenous RNA Network Identified Novel Circular RNAs in Hypertrophic Cardiomyopathy by Integrated Analysis. Front. Genet. 2020, 11, 764. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34 (Suppl. 2), W720–W724. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Li, B.; Feng, C.; Zhu, S.; Zhang, J.; Irwin, D.M.; Zhang, X.; Wang, Z.; Zhang, S. Identification of Candidate Circular RNAs Underlying Intramuscular Fat Content in the Donkey. Front. Genet. 2020, 11, 1580. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhang, Y.; Li, H.P.; Han, L.; Yan, X.M.; Li, H.B.; Du, W.; Zhang, J.S.; Yu, Q.L. Differential expression of mRNA-miRNAs related to intramuscular fat content in the longissimus dorsi in Xinjiang brown cattle. PLoS ONE 2018, 13, e0206757. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Yu, C.; Cui, S.; Wang, H.; Jin, H.; Wang, C.; Li, B.; Qin, M.; Yang, C.; He, J.; et al. circtp63 functions as a cerna to promote lung squamous cell carcinoma progression by upregulating foxm1. Nat. Commun. 2019, 10, 3200. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Wang, X.; Li, H.; Lu, Y.; Cheng, L. Regulatory Effects of Circular RNAs on Host Genes in Human Cancer. Front. Oncol. 2021, 10, 3477. [Google Scholar] [CrossRef] [PubMed]
- Malik, W.A.; Wang, X.; Wang, X.; Shu, N.; Cui, R.; Chen, X.; Wang, D.; Lu, X.; Yin, Z.; Wang, J.; et al. Genome-wide expression analysis suggests glutaredoxin genes response to various stresses in cotton. Int. J. Biol. Macromol. 2020, 153, 470–491. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, X.; Malik, W.A.; Chen, X.; Wang, J.; Wang, D.; Wang, S.; Chen, C.; Guo, L.; Ye, W. Differentially expressed bZIP transcription factors confer multi-tolerances in Gossypium hirsutum L. Int. J. Biol. Macromol. 2020, 146, 569–578. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, X.; Li, A.; Xie, L.; Miao, X. Differential regulation of mRNAs and lncRNAs related to lipid metabolism in two pig breeds. Oncotarget 2017, 8, 87539. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Huang, W.; Guo, Y.; Miao, X. An integrated analysis of microRNAs involved in fat deposition in different pig breeds. Oncotarget 2017, 10. [Google Scholar] [CrossRef]
- Fortin, A.; Robertson, W.; Tong, A. The eating quality of Canadian pork and its relationship with intramuscular fat. Meat Sci. 2005, 69, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, A.; Óvilo, C.; Núñez, Y.; Benítez, R.; López-Garcia, A.; García, F.; Félix, M.D.R.; Laranjo, M.; Charneca, R.; Martins, J.M. Transcriptomic Profiling of Skeletal Muscle Reveals Candidate Genes Influencing Muscle Growth and Associated Lipid Composition in Portuguese Local Pig Breeds. Animals 2021, 11, 1423. [Google Scholar] [CrossRef] [PubMed]
- Bergen, W.G.; Brandebourg, T.D. Regulation of lipid deposition in farm animals: Parallels between agriculture and human physiology. Exp. Biol. Med. 2016, 241, 1272–1280. [Google Scholar] [CrossRef]
- Micha, R.; Wallace, S.K.; Mozaffarian, D. Response to Letter Regarding Article,“Red and Processed Meat Consumption and Risk of Incident Coronary Heart Disease, Stroke, and Diabetes Mellitus: A Systematic Review and Meta-Analysis”. Circulation 2011, 123, e17. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Wu, Z.; Xiong, X.; Zhang, J.; Ma, J.; Xiao, S.; Huang, L.; Yang, B. Subcutaneous and intramuscular fat transcriptomes show large differences in network organization and associations with adipose traits in pigs. Sci. China Life Sci. 2021, 64, 1732–1746. [Google Scholar] [CrossRef]
- Qiu, Y.; Pu, C.; Li, Y.; Qi, B. Construction of a circRNA-miRNA-mRNA network based on competitive endogenous RNA reveals the function of circRNAs in osteosarcoma. Cancer Cell Int. 2020, 20, 48. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Scotece, M.; Gómez, R.; López, V.; Gómez-Reino, J.J.; Lago, F.; Gualillo, O. Adipokines: Biofactors from white adipose tissue. A complex hub among inflammation, metabolism, and immunity. Biofactors 2011, 37, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.M.; Mantzoros, C.S. Drug Insight: The role of leptin in human physiology and pathophysiology—Emerging clinical applications. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 318–327. [Google Scholar] [CrossRef]
- Han, M.H.; Kim, H.J.; Jeong, J.-W.; Park, C.; Kim, B.W.; Choi, Y.H. Inhibition of adipocyte differentiation by anthocyanins isolated from the fruit of Vitis coignetiae pulliat is associated with the activation of AMPK signaling pathway. Toxicol. Res. 2018, 34, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yasen, M.; Tang, D.; Ye, J.; Aisa, H.A.; Xin, X. Polyphenol-enriched extract of Rosa rugosa Thunb regulates lipid metabolism in diabetic rats by activation of AMPK pathway. Biomed. Pharmacother. 2018, 100, 29–35. [Google Scholar] [CrossRef]
- Aylon, Y.; Oren, M. The Hippo pathway, p53 and cholesterol. Cell Cycle 2016, 15, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 signaling and tissue fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [PubMed]
- Csapo, R.; Gumpenberger, M.; Wessner, B. Skeletal muscle extracellular matrix—What do we know about its composition, regulation, and physiological roles? A narrative review. Front. Physiol. 2020, 11, 253. [Google Scholar] [CrossRef]
- Hillege, M.M.; Galli Caro, R.A.; Offringa, C.; de Wit, G.M.; Jaspers, R.T.; Hoogaars, W.M. TGF-β regulates collagen type I expression in myoblasts and myotubes via transient Ctgf and Fgf-2 expression. Cells 2020, 9, 375. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xue, Y.; Wang, Y.; Ding, Y.; Zou, Q.; Pan, M.; Qiao, L.; Zhang, C.; Ge, Q.; Wang, T.; et al. CiRS-126 inhibits proliferation of ovarian granulosa cells through targeting the miR-21-PDCD4-ROS axis in a polycystic ovarian syndrome model. Cell Tissue Res. 2020, 381, 189–201. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Gao, W.; He, H.W.; Wang, Z.M.; Zhao, H.; Lian, X.Q.; Wang, Y.S.; Zhu, J.; Yan, J.J.; Zhang, D.G.; Yang, Z.J.; et al. Plasma levels of lipometabolism-related miR-122 and miR-370 are increased in patients with hyperlipidemia and associated with coronary artery disease. Lipids Health Dis. 2012, 11, 55. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.; Guo, Y.; Zhao, H.; Li, X.; Wang, X. Identification of the interaction between bta-miR-370 and OLR 1 gene in bovine adipocyte. Anim. Genet. 2017, 48, 455–458. [Google Scholar] [CrossRef]
- Zhai, L.; Wu, R.; Han, W.; Zhang, Y.; Zhu, D. miR-127 enhances myogenic cell differentiation by targeting S1PR3. Cell Death Dis. 2017, 8, e2707. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.U.; Thirion, C.; Polesskaya, A.; Bauersachs, S.; Kaiser, S.; Krause, S.; Pfaffl, M.W. TNF-α and IGF1 modify the microRNA signature in skeletal muscle cell differentiation. Cell Commun. Signal. 2015, 13, 4. [Google Scholar] [CrossRef]
- Bi, R.; Ding, F.; He, Y.; Jiang, L.; Jiang, Z.; Mei, J.; Liu, H. miR-503 inhibits platelet-derived growth factor-induced human aortic vascular smooth muscle cell proliferation and migration through targeting the insulin receptor. Biomed. Pharmacother. 2016, 84, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Deng, L.; Zhao, D.; Chen, L.; Yao, Z.; Guo, X.; Liu, X.; Lv, L.; Leng, B.; Xu, W.; et al. Micro RNA-503 promotes angiotensin II-induced cardiac fibrosis by targeting Apelin-13. J. Cell. Mol. Med. 2016, 20, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Luo, E.C.; Chang, Y.C.; Sher, Y.P.; Huang, W.Y.; Chuang, L.L.; Chiu, Y.C.; Tsai, M.H.; Chuang, E.Y.; Lai, L.C. MicroRNA-769-3p down-regulates NDRG1 and enhances apoptosis in MCF-7 cells during reoxygenation. Sci. Rep. 2014, 4, 5908. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E.; Ercan, B.; Morin, K.M.; Liu, C.T.; Tamer, L.; Ayaz, L.; Kanadasi, M.; Cicek, D.; Seyhan, A.I.; Akilli, R.E.; et al. The distribution of circulating microRNA and their relation to coronary disease. FResearch 2012, 1, 50. [Google Scholar] [CrossRef]
- Hammarström, P.; Nyström, S. Porcine prion protein amyloid. Prion 2015, 9, 266–277. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Lepore, E.; Martini, M.; Barberi, L.; Nunn, A.; Scicchitano, B.M.; Musarò, A. Metabolic changes associated with muscle expression of SOD1G93A. Front. Physiol. 2018, 9, 831. [Google Scholar] [CrossRef]
- Ghosal, A.; Lambrecht, N.; Subramanya, S.B.; Kapadia, R.; Said, H.M. Conditional knockout of the Slc5a6 gene in mouse intestine impairs biotin absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G64–G71. [Google Scholar] [CrossRef]
- Albuquerque, A.; Neves, J.A.; Redondeiro, M.; Laranjo, M.; Félix, M.R.; Freitas, A.; Tirapicos, J.L.; Martins, J.M. Long term betaine supplementation regulates genes involved in lipid and cholesterol metabolism of two muscles from an obese pig breed. Meat Sci. 2017, 124, 25–33. [Google Scholar] [CrossRef]
- van Weeghel, M.; Brinke, H.T.; van Lenthe, H.; Kulik, W.; Minkler, P.E.; Stoll, M.S.; Sass, J.O.; Janssen, U.; Stoffel, W.; Schwab, K.O.; et al. Functional redundancy of mitochondrial enoyl-CoA isomerases in the oxidation of unsaturated fatty acids. FASEB J. 2012, 26, 4316–4326. [Google Scholar] [CrossRef]
- Janssen, U.; Stoffel, W. Disruption of mitochondrial β-oxidation of unsaturated fatty acids in the 3, 2-trans-enoyl-CoA isomerase-deficient mouse. J. Biol. Chem. 2002, 277, 19579–19584. [Google Scholar] [CrossRef]
- Schwantje, M.; de Sain-van der Velden, M.; Jans, J.; van Gassen, K.; Dorrepaal, C.; Koop, K.; Visser, G. Genetic defect of the sodium-dependent multivitamin transporter: A treatable disease, mimicking biotinidase deficiency. JIMD Rep. 2019, 48, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Almannai, M.; El-Hattab, A.W. Inborn errors of metabolism with seizures: Defects of glycine and serine metabolism and cofactor-related disorders. Pediatric Clin. 2018, 65, 279–299. [Google Scholar] [CrossRef]
- Yuasa, M.; Matsui, T.; Ando, S.; Ishii, Y.; Sawamura, H.; Ebara, S.; Watanabe, T. Consumption of a low-carbohydrate and high-fat diet (the ketogenic diet) exaggerates biotin deficiency in mice. Nutrition 2013, 29, 1266–1270. [Google Scholar] [CrossRef]
- Zempleni, J.; Mock, D. Biotin biochemistry and human requirements. J. Nutr. Biochem. 1999, 10, 128–138. [Google Scholar] [CrossRef]
- Agrawal, S.; Agrawal, A.; Said, H.M. Biotin deficiency enhances the inflammatory response of human dendritic cells. Am. J. Physiol. Cell Physiol. 2016, 311, C386–C391. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Zhang, Y.-M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef]
- Eligini, S.; Fiorelli, S.; Tremoli, E.; Colli, S. Inhibition of transglutaminase 2 reduces efferocytosis in human macrophages: Role of CD14 and SR-AI receptors. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Falasca, L.; Iadevaia, V.; Ciccosanti, F.; Melino, G.; Serafino, A.; Piacentini, M. Transglutaminase type II is a key element in the regulation of the anti-inflammatory response elicited by apoptotic cell engulfment. J. Immunol. 2005, 174, 7330–7340. [Google Scholar] [CrossRef]
- Haitina, T.; Lindblom, J.; Renström, T.; Fredriksson, R. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics 2006, 88, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflügers Arch. 2004, 447, 689–709. [Google Scholar] [CrossRef]
- Roy, N.; May, M.; Delgado, E.R.; Alencastro, F.; Wilkinson, P.D.; Smyers, M.; Reynolds, M.J.; Shiva, S.; Duncan, A.W. SLC25A34 regulates bioenergetic metabolism in the murine liver. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Maier, T.; Leibundgut, M.; Boehringer, D.; Ban, N. Structure and function of eukaryotic fatty acid synthases. Q. Rev. Biophys. 2010, 43, 373–422. [Google Scholar] [CrossRef]
- Wang, Q.; Yuan, Z.; Wu, H.; Liu, F.; Zhao, J. Molecular characterization of a manganese superoxide dismutase and copper/zinc superoxide dismutase from the mussel Mytilus galloprovincialis. Fish Shellfish. Immunol. 2013, 34, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qi, W.; Richardson, A.; Van Remmen, H.; Ikeno, Y.; Salmon, A.B. Oxidative damage associated with obesity is prevented by overexpression of CuZn-or Mn-superoxide dismutase. Biochem. Biophys. Res. Commun. 2013, 438, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Vettor, R.; Milan, G.; Franzin, C.; Sanna, M.; De Coppi, P.; Rizzuto, R.; Federspil, G. The origin of intermuscular adipose tissue and its pathophysiological implications. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E987–E998. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.C.; Pérez, V.I.; Song, W.; Lustgarten, M.S.; Salmon, A.B.; Mele, J.; Qi, W.; Liu, Y.; Liang, H.; Chaudhuri, A.; et al. Overexpression of Mn superoxide dismutase does not increase life span in mice. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2009, 64, 1114–1125. [Google Scholar] [CrossRef]
- Wu, L.; Guan, Y.; Wu, Z.; Yang, K.; Lv, J.; Converse, R.; Huang, Y.; Mao, J.; Zhao, Y.; Wang, Z.; et al. OsABCG15 encodes a membrane protein that plays an important role in anther cuticle and pollen exine formation in rice. Plant Cell Rep. 2014, 33, 1881–1899. [Google Scholar] [CrossRef]
- Gooley, J.J.; Chua, E.C.-P. Diurnal regulation of lipid metabolism and applications of circadian lipidomics. J. Genet. Genom. 2014, 41, 231–250. [Google Scholar] [CrossRef]
- Gooley, J.J. Circadian regulation of lipid metabolism. Proc. Nutr. Soc. 2016, 75, 440–450. [Google Scholar] [CrossRef]
- Garaulet, M.; Ordovás, J.M.; Gómez-Abellán, P.; Martínez, J.A.; Madrid, J.A. An approximation to the temporal order in endogenous circadian rhythms of genes implicated in human adipose tissue metabolism. J. Cell. Physiol. 2011, 226, 2075–2080. [Google Scholar] [CrossRef]
- Vieira, E.; Ruano, E.G.; Figueroa, A.L.C.; Aranda, G.; Momblan, D.; Carmona, F.; Gomis, R.; Vidal, J.; Hanzu, F.A. Altered clock gene expression in obese visceral adipose tissue is associated with metabolic syndrome. PLoS ONE 2014, 9, e111678. [Google Scholar] [CrossRef] [PubMed]
- van der Kolk, B.W.; Goossens, G.H.; Jocken, J.W.; Blaak, E.E. Altered skeletal muscle fatty acid handling is associated with the degree of insulin resistance in overweight and obese humans. Diabetologia 2016, 59, 2686–2696. [Google Scholar] [CrossRef] [PubMed]
- Huang, G. Function of Collagens in Energy Metabolism and Metabolic Diseases. J. Cell Sci. Ther. 2014, 5, 1. [Google Scholar] [CrossRef]
- Huang, G.; Ge, G.; Wang, D.; Gopalakrishnan, B.; Butz, D.H.; Colman, R.J.; Nagy, A.; Greenspan, D.S. α3 (V) collagen is critical for glucose homeostasis in mice due to effects in pancreatic islets and peripheral tissues. J. Clin. Investig. 2011, 121, 769–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miR_Name | Mean (D) | Mean (L) | Log2 (Fold Change) (L/D) | p_Value | q_Value |
|---|---|---|---|---|---|
| ssc-miR-370 | 152.6247008 | 28.35987849 | −2.42807 | 8.04 × 10−78 | 1.10 × 10−76 |
| ssc-miR-127 | 3416.29341 | 954.4726318 | −1.83966 | 0 | 0 |
| ssc-miR-339-3p | 4.933988172 | 1.627206143 | −1.60036 | 0.011932 | 4.32 × 10−2 |
| ssc-miR-339 | 483.5308409 | 7.438656653 | −6.02242 | 0 | 0 |
| ssc-miR-7137-3p | 6.907583441 | 2.789496245 | −1.30818 | 0.010358 | 3.83 × 10−2 |
| ssc-miR-503 | 19.73595269 | 7.438656653 | −1.40771 | 4.39 × 1006 | 2.37 × 10−5 |
| ssc-miR-769-3p | 40.12977047 | 17.89926757 | −1.16477 | 1.67 × 1008 | 1.05 × 10−7 |
| miRNA | miRNA Expression | Transcript | circRNA Expression | Total Score | Total Energy | Max Score | Max Energy | miRNA Length | Transcript Length | Position |
|---|---|---|---|---|---|---|---|---|---|---|
| ssc-miR-127 | Down | circRNA_08840 (chr13:217685619_217779051_-) | Up | 158 | −31.47 | 158 | −31.47 | 22 | 93,433 | 7134 |
| ssc-miR-339-3p | Down | circRNA_08840 (chr13:217685619_217779051_-) | Up | 152 | −32.13 | 152 | −32.13 | 21 | 93,433 | 36837 |
| ssc-miR-339 | Down | circRNA_08840 (chr13:217685619_217779051_-) | Up | 309 | −60.47 | 156 | −30.36 | 21 | 93,433 | 63989 48747 |
| ssc-miR-370 | Down | circRNA_08840 (chr13:217685619_217779051_-) | Up | 476 | −105.8 | 166 | −40.11 | 22 | 93,433 | 10849 46,872 69308 |
| ssc-miR-7137-3p | Down | circRNA_08840 (chr13:217685619_217779051_-) | Up | 336 | −68.04 | 171 | −36.35 | 22 | 93,433 | 13332 48860 |
| ssc-miR-503 | Down | circRNA_23437 (chr7:24626858_24686216_-) | Up | 176 | −32.35 | 176 | −32.35 | 23 | 57,856 | 14200 |
| ssc-miR-769-3p | Down | circRNA_23437 (chr7:24626858_24686216_-) | Up | 176 | −36.55 | 176 | −36.55 | 23 | 57,856 | 45734 |
| ssc-miR-370 | Down | circRNA_23437 (chr7:24626858_24686216_-) | Up | 166 | −32.79 | 166 | −32.79 | 22 | 57,856 | 39180 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousuf, S.; Li, A.; Feng, H.; Lui, T.; Huang, W.; Zhang, X.; Xie, L.; Miao, X. Genome-Wide Expression Profiling and Networking Reveals an Imperative Role of IMF-Associated Novel CircRNAs as ceRNA in Pigs. Cells 2022, 11, 2638. https://doi.org/10.3390/cells11172638
Yousuf S, Li A, Feng H, Lui T, Huang W, Zhang X, Xie L, Miao X. Genome-Wide Expression Profiling and Networking Reveals an Imperative Role of IMF-Associated Novel CircRNAs as ceRNA in Pigs. Cells. 2022; 11(17):2638. https://doi.org/10.3390/cells11172638
Chicago/Turabian StyleYousuf, Salsabeel, Ai Li, Hui Feng, Tianyi Lui, Wanlong Huang, Xiuxiu Zhang, Lingli Xie, and Xiangyang Miao. 2022. "Genome-Wide Expression Profiling and Networking Reveals an Imperative Role of IMF-Associated Novel CircRNAs as ceRNA in Pigs" Cells 11, no. 17: 2638. https://doi.org/10.3390/cells11172638