
Identifying Disease Signatures in the Spinocerebellar Ataxia Type 1 Mouse Cortex

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Husbandry
2.2. Cell Culture
2.3. Protein Extraction and Western Blot Analysis
2.4. RNA Extraction and RT-qPCR
2.5. RNA Extraction and Bulk RNA Sequencing
2.6. RNA Sequencing Data Analysis
2.7. Immunohistochemistry
2.8. Fluorescent Image Quantification
2.9. Statistical Analysis
3. Results
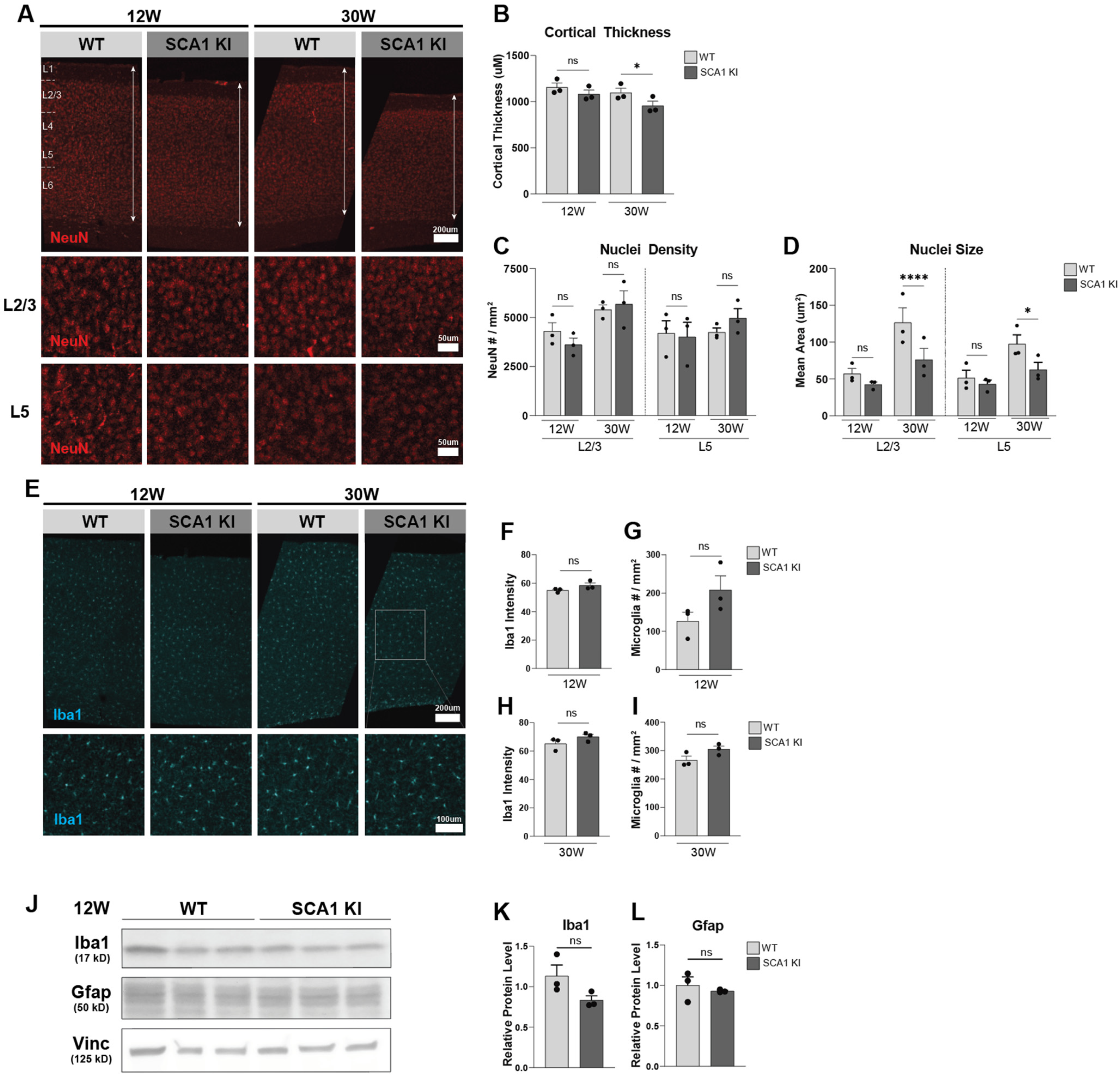
3.1. Progressive Cortical Thinning in the SCA1 KI Mouse Model
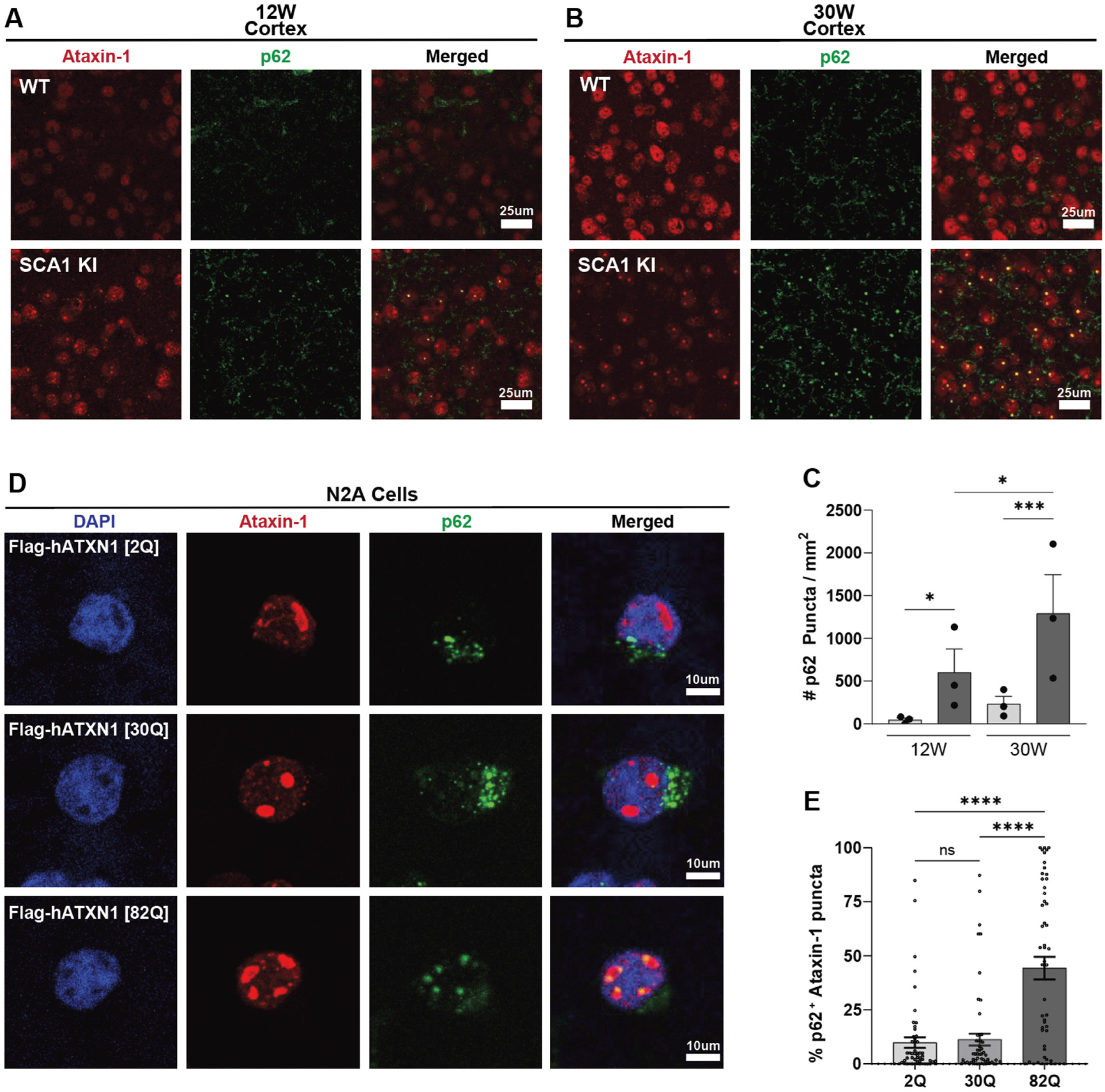
3.2. Progressive, Region-Specific, PolyQ-Dependent Colocalization of p62 with Mutant Ataxin-1 Nuclear Inclusions
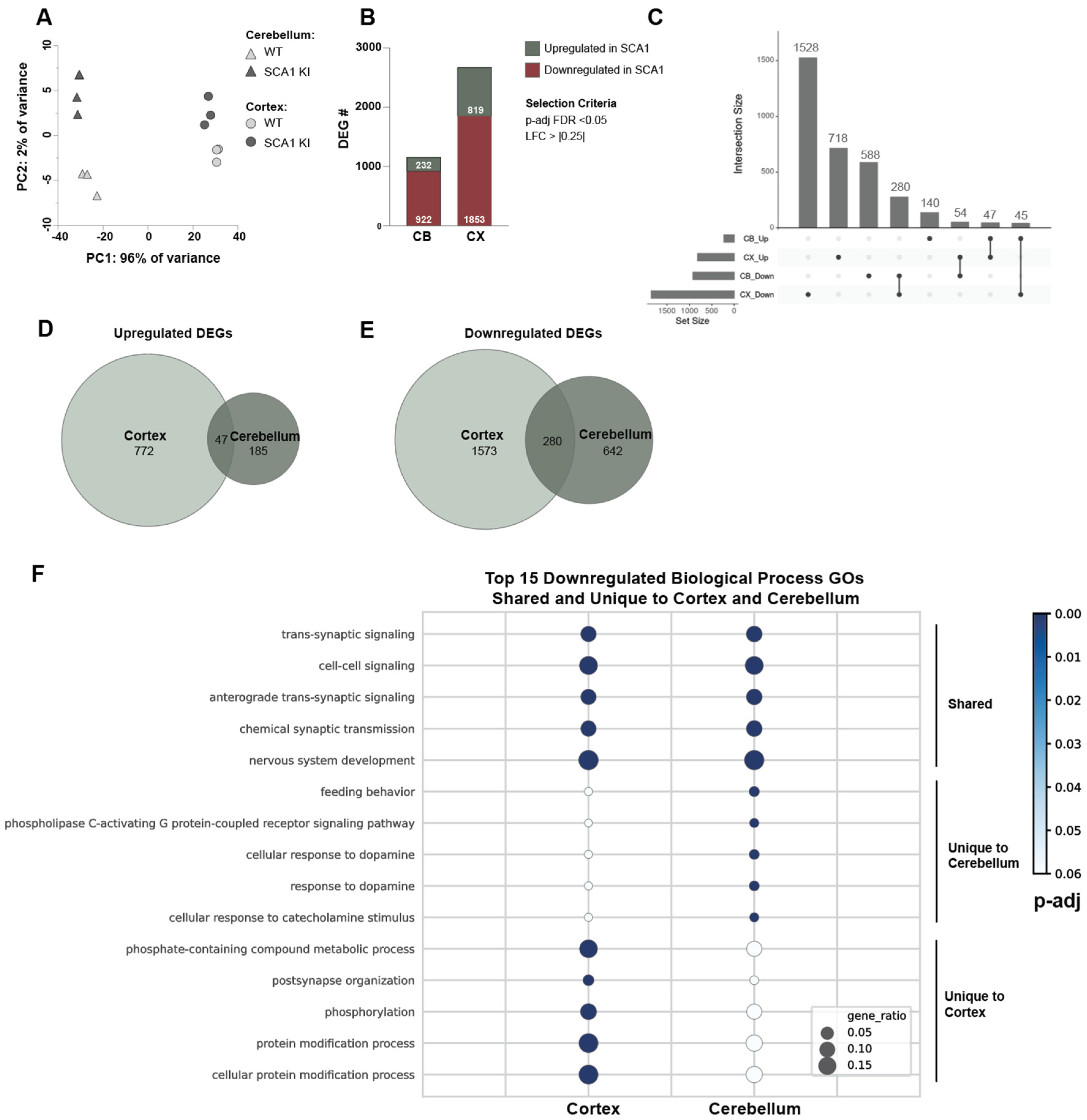
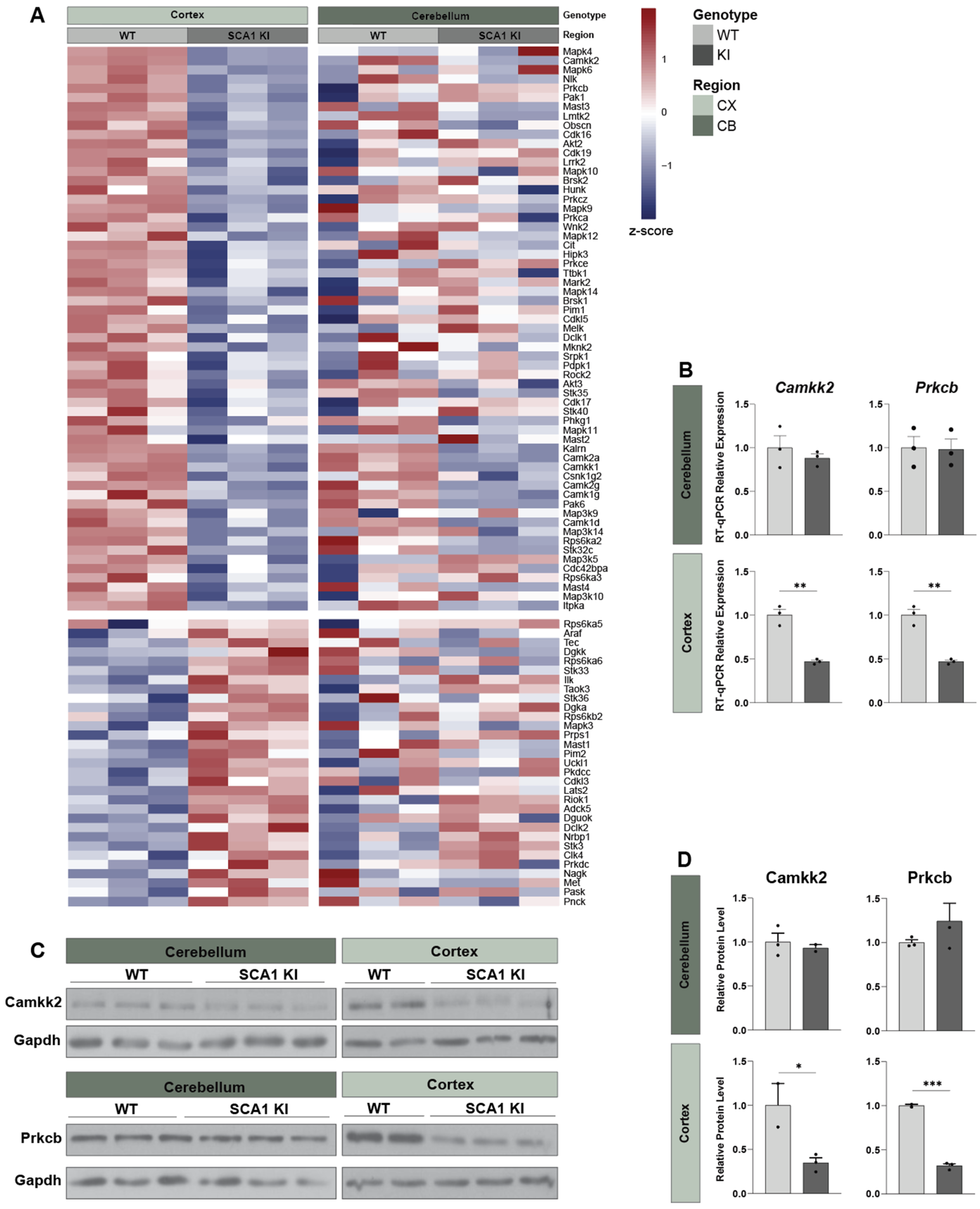
3.3. Transcriptomic Alterations in the SCA1 KI Mouse Cortex
3.4. Cross-Tissue Comparison of the Cortex and Cerebellum Identifies Shared and Unique Transcriptomic Response to SCA1 Disease
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide Repeat Disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef]
- Globas, C.; du Montcel, S.T.; Baliko, L.; Boesch, S.; Depondt, C.; DiDonato, S.; Durr, A.; Filla, A.; Klockgether, T.; Mariotti, C.; et al. Early symptoms in spinocerebellar ataxia type 1, 2, 3, and 6. Mov. Disord. 2008, 23, 2232–2238. [Google Scholar] [CrossRef]
- Luo, L.; Wang, J.; Lo, R.Y.; Figueroa, K.P.; Pulst, S.M.; Kuo, P.H.; Perlman, S.; Wilmot, G.; Gomez, C.M.; Schmahmann, J.; et al. The Initial Symptom and Motor Progression in Spinocerebellar Ataxias. Cerebellum 2017, 16, 615–622. [Google Scholar] [CrossRef]
- Burk, K.; Globas, C.; Bosch, S.; Klockgether, T.; Zuhlke, C.; Daum, I.; Dichgans, J. Cognitive deficits in spinocerebellar ataxia type 1, 2, and 3. J. Neurol. 2003, 250, 207–211. [Google Scholar] [CrossRef]
- Fancellu, R.; Paridi, D.; Tomasello, C.; Panzeri, M.; Castaldo, A.; Genitrini, S.; Soliveri, P.; Girotti, F. Longitudinal study of cognitive and psychiatric functions in spinocerebellar ataxia types 1 and 2. J. Neurol. 2013, 260, 3134–3143. [Google Scholar] [CrossRef]
- Ma, J.; Wu, C.; Lei, J.; Zhang, X. Cognitive impairments in patients with spinocerebellar ataxia types 1, 2 and 3 are positively correlated to the clinical severity of ataxia symptoms. Int. J. Clin. Exp. Med. 2014, 7, 5765–5771. [Google Scholar]
- Burk, K.; Bosch, S.; Globas, C.; Zuhlke, C.; Daum, I.; Klockgether, T.; Dichgans, J. Executive dysfunction in spinocerebellar ataxia type 1. Eur. Neurol 2001, 46, 43–48. [Google Scholar] [CrossRef]
- Orr, H.T.; Chung, M.Y.; Banfi, S.; Kwiatkowski Jr, T.J.; Servadio, A.; Beaudet, A.L.; McCall, A.E.; Duvick, L.A.; Ranum, L.P.; Zoghbi, H.Y. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993, 4, 221–226. [Google Scholar] [CrossRef]
- Servadio, A.; Koshy, B.; Armstrong, D.; Antalffy, B.; Orr, H.T.; Zoghbi, H.Y. Expression analysis of the ataxin-1 protein in tissues from normal and spinocerebellar ataxia type 1 individuals. Nat. Genet. 1995, 10, 94–98. [Google Scholar] [CrossRef]
- Watase, K.; Weeber, E.J.; Xu, B.; Antalffy, B.; Yuva-Paylor, L.; Hashimoto, K.; Kano, M.; Atkinson, R.; Sun, Y.; Armstrong, D.L.; et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 2002, 34, 905–919. [Google Scholar] [CrossRef]
- Rub, U.; Burk, K.; Timmann, D.; den Dunnen, W.; Seidel, K.; Farrag, K.; Brunt, E.; Heinsen, H.; Egensperger, R.; Bornemann, A.; et al. Spinocerebellar ataxia type 1 (SCA1): New pathoanatomical and clinico-pathological insights. Neuropathol. Appl. Neurobiol. 2012, 38, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Seidel, K.; Siswanto, S.; Brunt, E.R.; den Dunnen, W.; Korf, H.W.; Rub, U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012, 124, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.; Sima, A.A.; Junck, L.; Kluin, K.J.; Koeppe, R.A.; Lohman, M.E.; Little, R. Spinocerebellar ataxia type 1 with multiple system degeneration and glial cytoplasmic inclusions. Ann. Neurol. 1996, 39, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, Y.; Schut, L.; Kish, S.J. Structural and immunocytochemical features of olivopontocerebellar atrophy caused by the spinocerebellar ataxia type 1 (SCA-1) mutation define a unique phenotype. Acta Neuropathol. 1995, 90, 572–581. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Bates, G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet 2003, 361, 1642–1644. [Google Scholar] [CrossRef]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef]
- Berke, S.J.; Paulson, H.L. Protein aggregation and the ubiquitin proteasome pathway: Gaining the UPPer hand on neurodegeneration. Curr. Opin. Genet. Dev. 2003, 13, 253–261. [Google Scholar] [CrossRef]
- Nussbaum, R.L.; Ellis, C.E. Alzheimer’s disease and Parkinson’s disease. N. Engl. J. Med. 2003, 348, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Olmos, V.; Gogia, N.; Luttik, K.; Haidery, F.; Lim, J. The extra-cerebellar effects of spinocerebellar ataxia type 1 (SCA1): Looking beyond the cerebellum. Cell Mol. Life Sci. 2022, 79, 404. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Barreto, J.; Fryer, J.D.; Shaw, C.A.; Orr, H.T.; Zoghbi, H.Y. Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet. 2010, 6, e1001021. [Google Scholar] [CrossRef]
- Ingram, M.; Wozniak, E.A.L.; Duvick, L.; Yang, R.; Bergmann, P.; Carson, R.; O’Callaghan, B.; Zoghbi, H.Y.; Henzler, C.; Orr, H.T. Cerebellar Transcriptome Profiles of ATXN1 Transgenic Mice Reveal SCA1 Disease Progression and Protection Pathways. Neuron 2016, 89, 1194–1207. [Google Scholar] [CrossRef]
- Lin, X.; Antalffy, B.; Kang, D.; Orr, H.T.; Zoghbi, H.Y. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat. Neurosci 2000, 3, 157–163. [Google Scholar] [CrossRef]
- Driessen, T.M.; Lee, P.J.; Lim, J. Molecular pathway analysis towards understanding tissue vulnerability in spinocerebellar ataxia type 1. Elife 2018, 7, e39981. [Google Scholar] [CrossRef]
- Friedrich, J.; Kordasiewicz, H.B.; O’Callaghan, B.; Handler, H.P.; Wagener, C.; Duvick, L.; Swayze, E.E.; Rainwater, O.; Hofstra, B.; Benneyworth, M.; et al. Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight 2018, 3, 1–18. [Google Scholar] [CrossRef]
- Luttik, K.; Tejwani, L.; Ju, H.; Driessen, T.; Smeets, C.; Edamakanti, C.R.; Khan, A.; Yun, J.; Opal, P.; Lim, J. Differential effects of Wnt-beta-catenin signaling in Purkinje cells and Bergmann glia in spinocerebellar ataxia type 1. Proc. Natl. Acad. Sci. USA 2022, 119, e2208513119. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Hulsen, T. BioVenn- an R and Python package for the comparison and visualization of biological lists using area-proportional Venn diagrams. Data Sci. 2021, 4, 51–61. [Google Scholar] [CrossRef]
- Hulsen, T. BioVenn: Create Area-Proportional Venn Diagrams from Biological Lists; 2021. Available online: Cran.r-project.org/package=BioVenn (accessed on 1 May 2022).
- Khan, A.; Mathelier, A. Intervene: A tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinform. 2017, 18, 287. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids. Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Kull, M.; Peterson, H.; Hansen, J.; Vilo, J. g:Profiler--a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids. Res. 2007, 35, W193–W200. [Google Scholar] [CrossRef]
- Allen Mouse Brain Atlas [Mouse, Sagittal]; Allen Institute for Brain Science: 2004; 56p. Available online: Mouse.brain-map.org (accessed on 10 August 2022).
- Bolte, S.; Cordelieres, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Cvetanovic, M.; Ingram, M.; Orr, H.; Opal, P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience 2015, 289, 289–299. [Google Scholar] [CrossRef]
- Tichanek, F.; Salomova, M.; Jedlicka, J.; Kuncova, J.; Pitule, P.; Macanova, T.; Petrankova, Z.; Tuma, Z.; Cendelin, J. Hippocampal mitochondrial dysfunction and psychiatric-relevant behavioral deficits in spinocerebellar ataxia 1 mouse model. Sci. Rep. 2020, 10, 5418. [Google Scholar] [CrossRef]
- Rüb, U.; Schöls, L.; Paulson, H.; Auburger, G.; Kermer, P.; Jen, J.C.; Seidel, K.; Korf, H.W.; Deller, T. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013, 104, 38–66. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Lamark, T.; Bruun, J.A.; Overvatn, A.; Bjorkoy, G.; Johansen, T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J. Biol. Chem. 2010, 285, 5941–5953. [Google Scholar] [CrossRef] [PubMed]
- Cvetanovic, M.; Patel, J.M.; Marti, H.H.; Kini, A.R.; Opal, P. Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1. Nat. Med. 2011, 17, 1445–1447. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.; Kokubu, H.; Todd, T.W.; Kahle, J.J.; Kim, S.; Richman, R.; Chirala, K.; Orr, H.T.; Zoghbi, H.Y.; Lim, J. Polyglutamine Disease Toxicity Is Regulated by Nemo-like Kinase in Spinocerebellar Ataxia Type 1. J. Neurosci. 2013, 33, 9328–9336. [Google Scholar] [CrossRef]
- Park, J.; Al-Ramahi, I.; Tan, Q.; Mollema, N.; Diaz-Garcia, J.R.; Gallego-Flores, T.; Lu, H.C.; Lagalwar, S.; Duvick, L.; Kang, H.; et al. RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature 2013, 498, 325–331. [Google Scholar] [CrossRef]
- Lee, W.S.; Lavery, L.; Rousseaux, M.W.C.; Rutledge, E.B.; Jang, Y.; Wan, Y.W.; Wu, S.R.; Kim, W.; Al-Ramahi, I.; Rath, S.; et al. Dual targeting of brain region-specific kinases potentiates neurological rescue in Spinocerebellar ataxia type 1. EMBO J. 2021, 40, e106106. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar]
- Song, P.; Li, S.; Wu, H.; Gao, R.; Rao, G.; Wang, D.; Chen, Z.; Ma, B.; Wang, H.; Sui, N.; et al. Parkin promotes proteasomal degradation of p62: Implication of selective vulnerability of neuronal cells in the pathogenesis of Parkinson’s disease. Protein Cell 2016, 7, 114–129. [Google Scholar] [CrossRef]
- Sato, S.; Uchihara, T.; Fukuda, T.; Noda, S.; Kondo, H.; Saiki, S.; Komatsu, M.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci. Rep. 2018, 8, 2813. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, H.; Chen, D.; Gao, F.; Ying, Z.; Wang, G. p62/sequestosome 1 regulates aggresome formation of pathogenic ataxin-3 with expanded polyglutamine. Int. J. Mol. Sci. 2014, 15, 14997–15010. [Google Scholar] [CrossRef]
- Ma, S.; Attarwala, I.Y.; Xie, X.Q. SQSTM1/p62: A Potential Target for Neurodegenerative Disease. ACS Chem. Neurosci. 2019, 10, 2094–2114. [Google Scholar] [CrossRef] [PubMed]
- Nezis, I.P.; Stenmark, H. p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxid. Redox Signal. 2012, 17, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Fenouille, N.; Auberger, P. When autophagy meets cancer through p62/SQSTM1. Am. J. Cancer Res. 2012, 2, 397–413. [Google Scholar] [PubMed]
- Kickstein, E.; Krauss, S.; Thornhill, P.; Rutschow, D.; Zeller, R.; Sharkey, J.; Williamson, R.; Fuchs, M.; Kohler, A.; Glossmann, H.; et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21830–21835. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; Imarisio, S.; Menzies, F.M.; Jimenez-Sanchez, M.; Siddiqi, F.H.; Wu, X.; Renna, M.; O’Kane, C.J.; Crowther, D.C.; Rubinsztein, D.C. CCT complex restricts neuropathogenic protein aggregation via autophagy. Nat. Commun. 2016, 7, 13821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, L.; Chen, S.; Yang, D.; Wang, Y.; Zhang, X.; Wang, Z.; Le, W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 2011, 7, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Babu, J.R.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J. Neurochem. 2005, 94, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T.; Wooten, M.W. Signal integration and diversification through the p62 scaffold protein. Trends Biochem. Sci. 2007, 32, 95–100. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luttik, K.; Olmos, V.; Owens, A.; Khan, A.; Yun, J.; Driessen, T.; Lim, J. Identifying Disease Signatures in the Spinocerebellar Ataxia Type 1 Mouse Cortex. Cells 2022, 11, 2632. https://doi.org/10.3390/cells11172632
Luttik K, Olmos V, Owens A, Khan A, Yun J, Driessen T, Lim J. Identifying Disease Signatures in the Spinocerebellar Ataxia Type 1 Mouse Cortex. Cells. 2022; 11(17):2632. https://doi.org/10.3390/cells11172632
Chicago/Turabian StyleLuttik, Kimberly, Victor Olmos, Ashley Owens, Aryaan Khan, Joy Yun, Terri Driessen, and Janghoo Lim. 2022. "Identifying Disease Signatures in the Spinocerebellar Ataxia Type 1 Mouse Cortex" Cells 11, no. 17: 2632. https://doi.org/10.3390/cells11172632