
Central Roles of STAT3-Mediated Signals in Onset and Development of Cancers: Tumorigenesis and Immunosurveillance


Abstract
:1. Introduction
2. STAT3 Signal in Cancer Cells
2.1. Pancreatic Cancer
2.2. Colorectal Cancer
2.3. Prostate Cancer
2.4. Breast Cancer
2.5. Head and Neck Cancer
2.6. Lung Cancer
3. STAT3 in Cells of the TME
3.1. Immune Cells
3.2. Non-Immune Cells
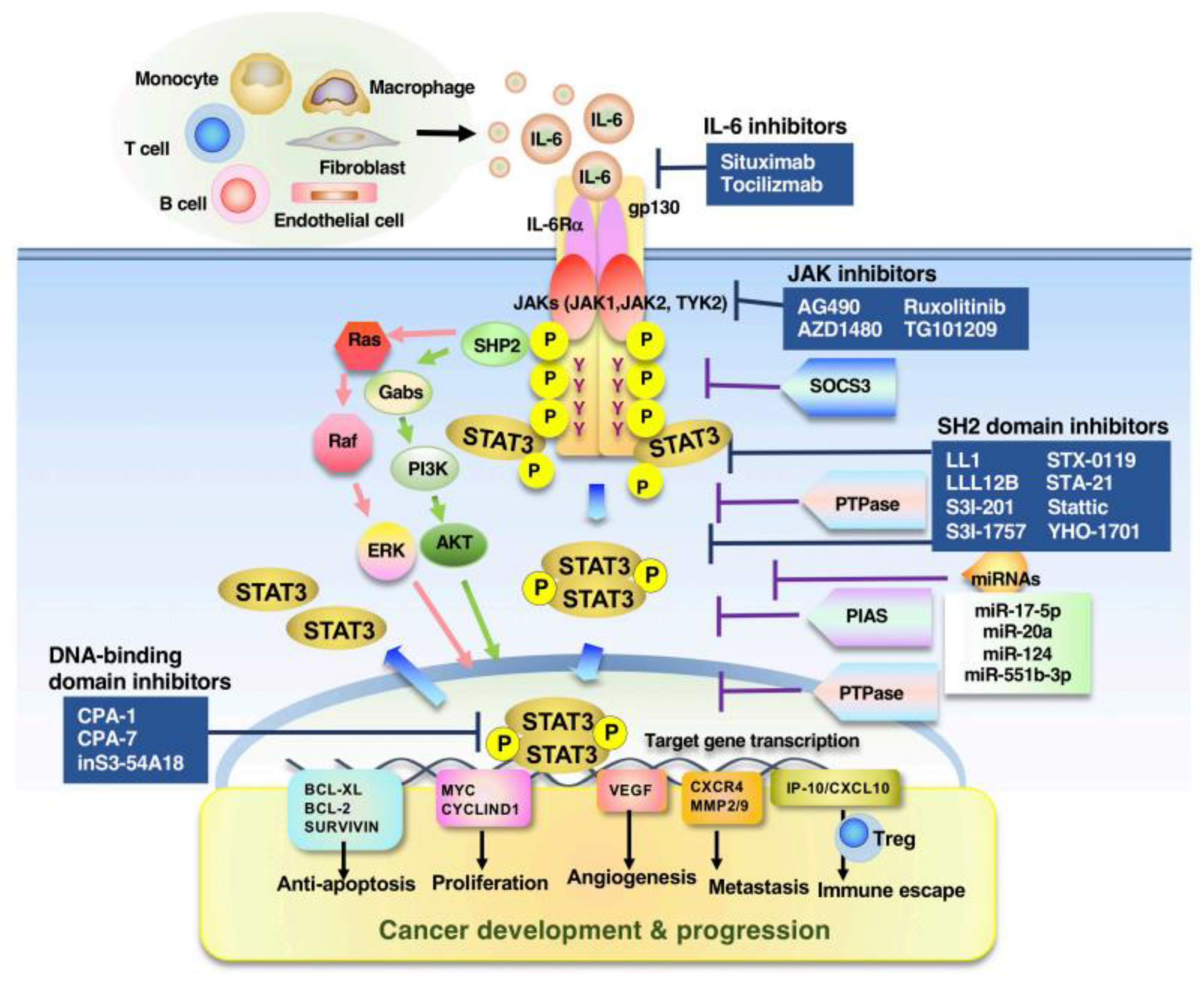
4. Promising Target Molecules in STAT3-Associated Tumors
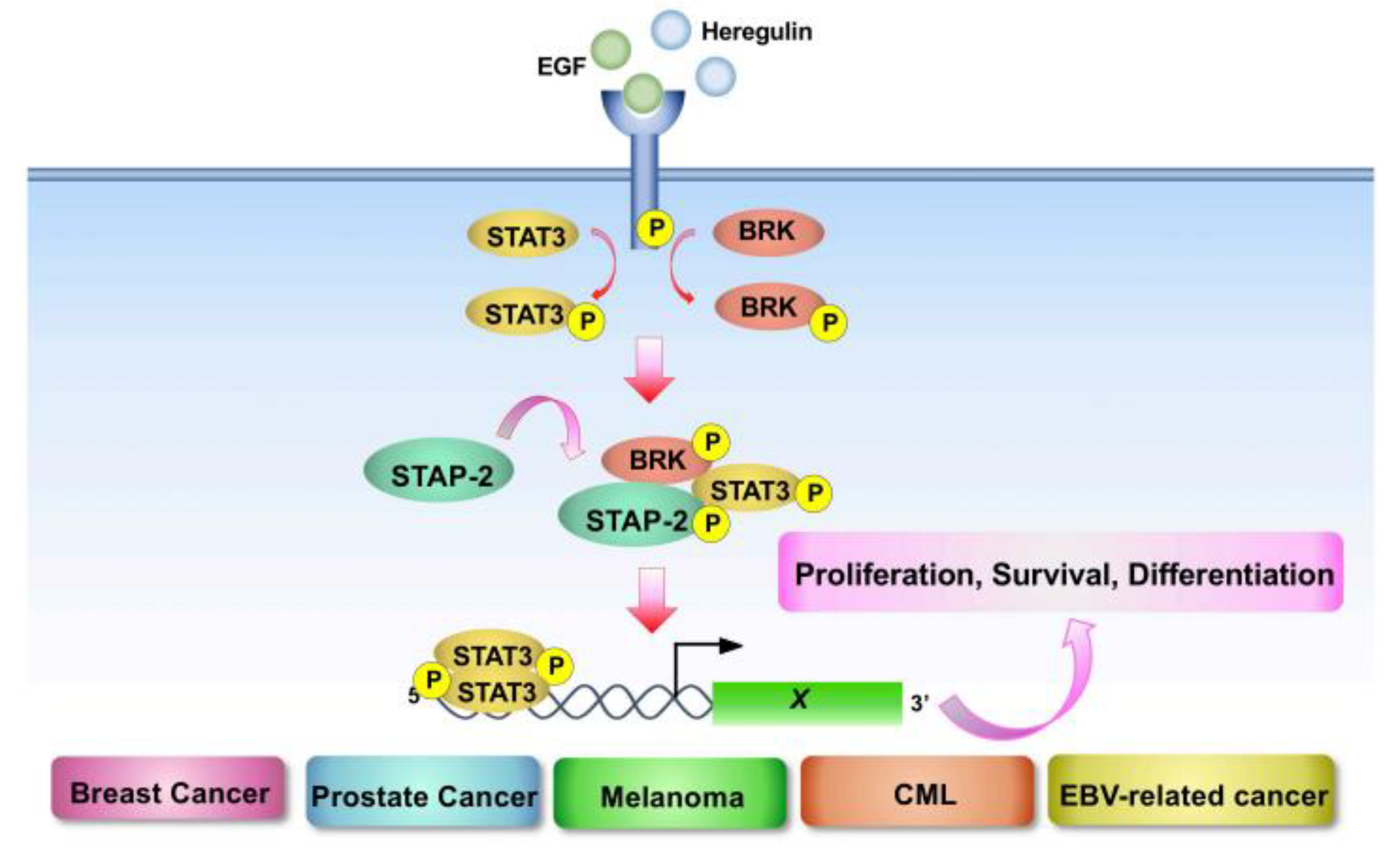
4.1. STAP-2
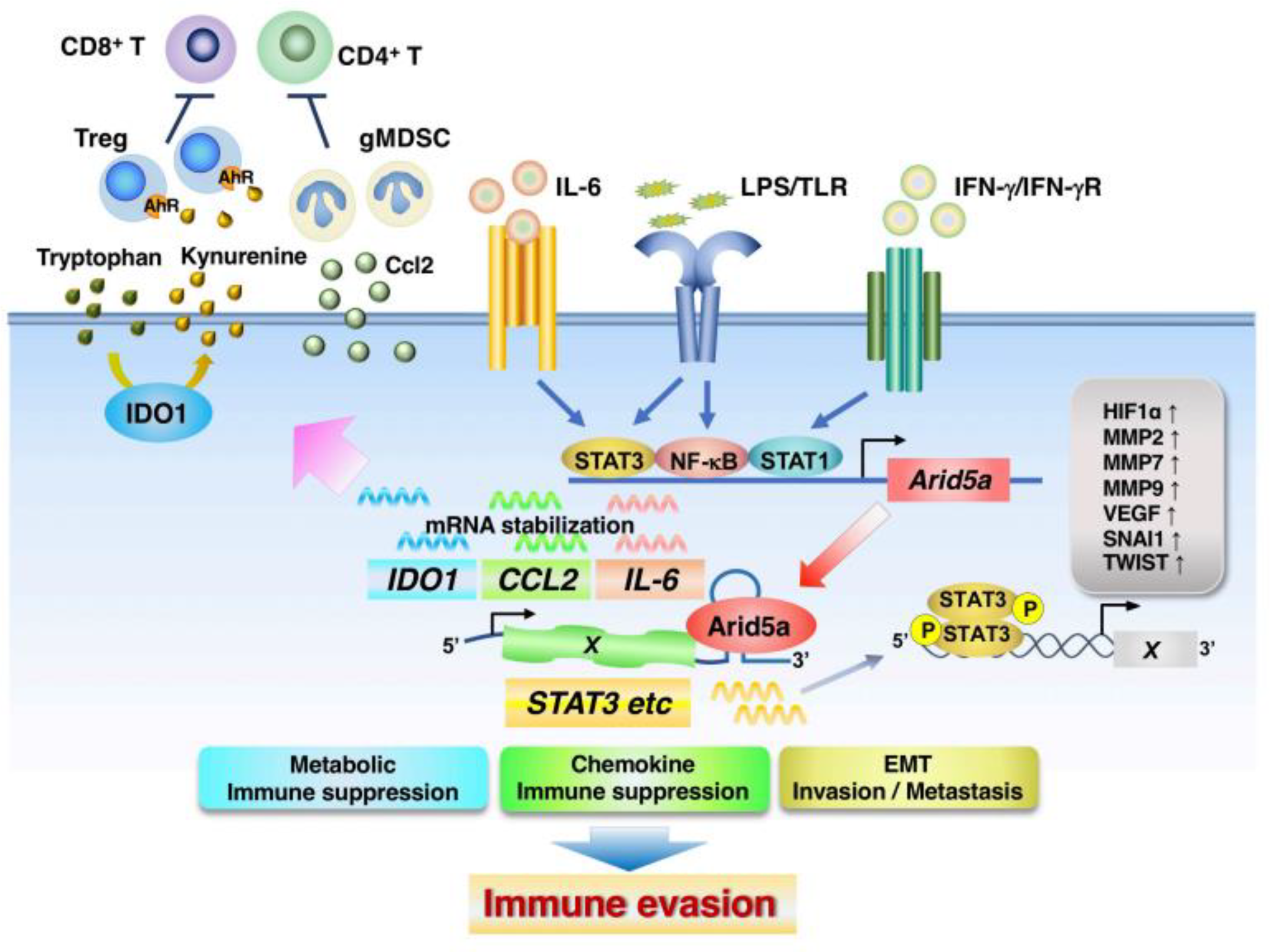
4.2. ARID5A
5. Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Murakami, M.; Kamimura, D.; Hirano, T. Pleiotropy and Specificity: Insights from the Interleukin 6 Family of Cytokines. Immunity 2019, 50, 812–831. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Kishimoto, T.; Kang, S. IL-6 Revisited: From Rheumatoid Arthritis to CAR T Cell Therapy and COVID-19. Annu. Rev. Immunol. 2022, 40, 323–348. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer–using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Hanada, T.; Yoshimura, A. Suppressors of cytokine signaling and immunity. Nat. Immunol. 2003, 4, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, S.E.; De Souza, D.; Fabri, L.J.; Corbin, J.; Willson, T.A.; Zhang, J.G.; Silva, A.; Asimakis, M.; Farley, A.; Nash, A.D.; et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc. Natl. Acad. Sci. USA 2000, 97, 6493–6498. [Google Scholar] [CrossRef] [Green Version]
- Boccaccio, C.; Ando, M.; Tamagnone, L.; Bardelli, A.; Michieli, P.; Battistini, C.; Comoglio, P.M. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 1998, 391, 285–288. [Google Scholar] [CrossRef]
- Quesnelle, K.M.; Boehm, A.L.; Grandis, J.R. STAT-mediated EGFR signaling in cancer. J. Cell. Biochem. 2007, 102, 311–319. [Google Scholar] [CrossRef]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Lu, R.; Lee, H.; Zhang, W.; Zhang, C.; Deng, J.; Liu, Y.; Shen, S.; Wagner, K.U.; Forman, S.; et al. G-protein-coupled receptor agonist BV8/prokineticin-2 and STAT3 protein form a feed-forward loop in both normal and malignant myeloid cells. J. Biol. Chem. 2013, 288, 13842–13849. [Google Scholar] [CrossRef] [Green Version]
- Hossain, D.M.; Dos Santos, C.; Zhang, Q.; Kozlowska, A.; Liu, H.; Gao, C.; Moreira, D.; Swiderski, P.; Jozwiak, A.; Kline, J.; et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood 2014, 123, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Kortylewski, M.; Kujawski, M.; Herrmann, A.; Yang, C.; Wang, L.; Liu, Y.; Salcedo, R.; Yu, H. Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res. 2009, 69, 2497–2505. [Google Scholar] [CrossRef] [Green Version]
- Eyking, A.; Ey, B.; Rünzi, M.; Roig, A.I.; Reis, H.; Schmid, K.W.; Gerken, G.; Podolsky, D.K.; Cario, E. Toll-like receptor 4 variant D299G induces features of neoplastic progression in Caco-2 intestinal cells and is associated with advanced human colon cancer. Gastroenterology 2011, 141, 2154–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, K.; Miyata, H.; Tanaka, K.; Hamano, R.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; Nakajima, K.; Takiguchi, S.; Mori, M.; et al. Let-7 expression is a significant determinant of response to chemotherapy through the regulation of IL-6/STAT3 pathway in esophageal squamous cell carcinoma. Clin. Cancer Res. 2012, 18, 5144–5153. [Google Scholar] [CrossRef] [Green Version]
- Navarro, A.; Diaz, T.; Martinez, A.; Gaya, A.; Pons, A.; Gel, B.; Codony, C.; Ferrer, G.; Martinez, C.; Montserrat, E.; et al. Regulation of JAK2 by miR-135a: Prognostic impact in classic Hodgkin lymphoma. Blood 2009, 114, 2945–2951. [Google Scholar] [CrossRef]
- Du, L.; Subauste, M.C.; DeSevo, C.; Zhao, Z.; Baker, M.; Borkowski, R.; Schageman, J.J.; Greer, R.; Yang, C.R.; Suraokar, M.; et al. miR-337-3p and its targets STAT3 and RAP1A modulate taxane sensitivity in non-small cell lung cancers. PLoS ONE 2012, 7, e39167. [Google Scholar] [CrossRef]
- Priceman, S.J.; Kujawski, M.; Shen, S.; Cherryholmes, G.A.; Lee, H.; Zhang, C.; Kruper, L.; Mortimer, J.; Jove, R.; Riggs, A.D.; et al. Regulation of adipose tissue T cell subsets by Stat3 is crucial for diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 13079–13084. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Liu, Y.; Lee, H.; Herrmann, A.; Zhang, W.; Zhang, C.; Shen, S.; Priceman, S.J.; Kujawski, M.; Pal, S.K.; et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell 2012, 21, 642–654. [Google Scholar] [CrossRef] [Green Version]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24- stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Herrmann, A.; Cherryholmes, G.; Kowolik, C.; Buettner, R.; Pal, S.; Yu, H.; Müller-Newen, G.; Jove, R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014, 74, 1227–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, T.; Oritani, K. STAP-2 adaptor protein regulates multiple steps of immune and inflammatory responses. Biol. Pharm. Bull. 2021, 44, 895–901. [Google Scholar] [CrossRef]
- Hashimoto, S.; Kishimoto, T. Roles of RNA-binding proteins in immune diseases and cancer. Semin. Cancer Biol. 2022; in press. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Grivennikov, S.I.; Karin, M. The unholy trinity: Inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell 2011, 19, 429–431. [Google Scholar] [CrossRef] [Green Version]
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, B.J.; Grail, D.; Nheu, T.; Najdovska, M.; Wang, B.; Waring, P.; Inglese, M.; McLoughlin, R.M.; Jones, S.A.; Topley, N.; et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat. Med. 2005, 11, 845–852. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Lambert, A.W.; Weinberg, R.A. Linking Emt Programmes to Normal and Neoplastic Epithelial Stem Cells. Nat. Rev. Cancer 2021, 21, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Gawlik-Rzemieniewska, N.; Bednarek, I. The role of NANOG transcriptional factor in the development of malignant phenotype of cancer cells. Cancer Biol. Ther. 2016, 17, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal. 2013, 25, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Mitsunaga, S.; Ikeda, M.; Shimizu, S.; Ohno, I.; Furuse, J.; Inagaki, M.; Higashi, S.; Kato, H.; Terao, K.; Ochiai, A. Serum levels of IL-6 and IL-1beta can predict the efficacy of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer 2013, 108, 2063–2069. [Google Scholar] [CrossRef] [Green Version]
- Denley, S.M.; Jamieson, N.B.; McCall, P.; Oien, K.A.; Morton, J.P.; Carter, C.R.; Edwards, J.; McKay, C.J. Activation of the IL-6R/Jak/stat pathway is associated with a poor outcome in resected pancreatic ductal adenocarcinoma. J. Gastrointestin. Surg. 2013, 17, 887–898. [Google Scholar] [CrossRef]
- Scholz, A.; Heinze, S.; Detjen, K.M.; Peters, M.; Welzel, M.; Hauff, P.; Schirner, M.; Wiedenmann, B.; Rosewicz, S. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 2003, 125, 891–905. [Google Scholar] [CrossRef]
- Huang, C.; Huang, R.; Chang, W.; Jiang, T.; Huang, K.; Cao, J.; Sun, X.; Qiu, Z. The expression and clinical significance of pSTAT3, VEGF and VEGF-C in pancreatic adenocarcinoma. Neoplasma 2012, 59, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Yang, G.; Jiang, T.; Zhu, G.; Li, H.; Qiu, Z. The effects and mechanisms of blockage of STAT3 signaling pathway on IL-6 inducing EMT in human pancreatic cancer cells in vitro. Neoplasma 2011, 58, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Miyatsuka, T.; Kaneto, H.; Shiraiwa, T.; Matsuoka, T.A.; Yamamoto, K.; Kato, K.; Nakamura, Y.; Akira, S.; Takeda, K.; Kajimoto, Y.; et al. Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev. 2006, 20, 1435–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, A.; Wang, S.C.; Morris, J.P., 4th; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J., 3rd. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef] [Green Version]
- Rose-John, S.; Mitsuyama, K.; Matsumoto, S.; Thaiss, W.M.; Scheller, J. Interleukin-6 trans-signaling and colonic cancer associated with inflammatory bowel disease. Curr. Pharm. Des. 2009, 15, 2095–2103. [Google Scholar] [CrossRef]
- Belluco, C.; Nitti, D.; Frantz, M.; Toppan, P.; Basso, D.; Plebani, M.; Lise, M.; Jessup, J.M. Interleukin-6 blood level is associated with circulating carcinoembryonic antigen and prognosis in patients with colorectal cancer. Ann. Surg. Oncol. 2000, 7, 133–138. [Google Scholar] [CrossRef]
- Chung, Y.C.; Chang, Y.F. Serum interleukin-6 levels reflect the disease status of colorectal cancer. J. Surg. Oncol. 2003, 83, 222–226. [Google Scholar] [CrossRef]
- Kusaba, T.; Nakayama, T.; Yamazumi, K.; Yakata, Y.; Yoshizaki, A.; Inoue, K.; Nagayasu, T.; Sekine, I. Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncol. Rep. 2006, 15, 1445–1451. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, T.; Baba, Y.; Yamauchi, M.; Kuchiba, A.; Nosho, K.; Shima, K.; Tanaka, N.; Huttenhower, C.; Frank, D.A.; Fuchs, C.S.; et al. STAT3 expression, molecular features, inflammation patterns, and prognosis in a database of 724 colorectal cancers. Clin. Cancer Res. 2011, 17, 1452–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Wang, A.; Chen, J.; Liu, X.; Wang, G. Relationship between expression and prognostic ability of PTEN, STAT3 and VEGF-C in colorectal cancer. Exp. Ther Med. 2012, 4, 633–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, C.; Fantini, M.C.; Schramm, C.; Lehr, H.A.; Wirtz, S.; Nikolaev, A.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004, 21, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The microbiome and human cancer. Science 2021, 371, eabc4552. [Google Scholar] [CrossRef]
- Hu, B.; Elinav, E.; Huber, S.; Strowig, T.; Hao, L.; Hafemann, A.; Jin, C.; Wunderlich, C.; Wunderlich, T.; Eisenbarth, S.C.; et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 9862–9867. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Fantini, M.C.; Wirtz, S.; Nikolaev, A.; Lehr, H.A.; Galle, P.R.; Rose-John, S.; Neurath, M.F. IL-6 signaling promotes tumor growth in colorectal cancer. Cell Cycle 2005, 4, 217–220. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef] [Green Version]
- Fenton, J.I.; Hursting, S.D.; Perkins, S.N.; Hord, N.G. Interleukin-6 production induced by leptin treatment promotes cell proliferation in an Apc (Min/+) colon epithelial cell line. Carcinogenesis 2006, 27, 1507–1515. [Google Scholar] [CrossRef] [Green Version]
- Musteanu, M.; Blaas, L.; Mair, M.; Schlederer, M.; Bilban, M.; Tauber, S.; Esterbauer, H.; Mueller, M.; Casanova, E.; Kenner, L.; et al. Stat3 is a negative regulator of intestinal tumor progression in Apc(Min) mice. Gastroenterology 2010, 138, 1003–1011.e5. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.C.; Lee, S.E.; Quinley, C.; Kim, H.; Herdman, S.; Corr, M.; Raz, E. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to- carcinoma transition in Apcmin/+ mice via regulation of Snail-1 (SNAI) protein stability. J. Biol. Chem. 2012, 287, 18182–18189. [Google Scholar] [CrossRef] [Green Version]
- Ernst, M.; Putoczki, T.L. Targeting IL-11 signaling in colon cancer. Oncotarget 2013, 4, 1860–1861. [Google Scholar] [CrossRef] [Green Version]
- Dong, C. TH17 cells in development: An updated view of their molecular identity and genetic programming. Nat. Rev. Immunol. 2008, 8, 337–348. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pagès, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Nguyen, D.P.; Li, J.; Tewari, A.K. Inflammation and prostate cancer: The role of interleukin 6 (IL-6). BJU Int. 2014, 113, 986–992. [Google Scholar] [CrossRef]
- Furuya, Y.; Nishio, R.; Junicho, A.; Nagakawa, O.; Fuse, H. Serum interleukin-11 in patients with benign prostatic hyperplasia and prostate cancer. Int. Urol. Nephrol. 2005, 37, 69–71. [Google Scholar] [CrossRef]
- Campbell, C.L.; Jiang, Z.; Savarese, D.M.; Savarese, T.M. Increased expression of the interleukin-11 receptor and evidence of STAT3 activation in prostate carcinoma. Am. J. Pathol. 2001, 158, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Zurita, A.J.; Troncoso, P.; Cardo-Vila, M.; Logothetis, C.J.; Pasqualini, R.; Arap, W. Combinatorial screenings in patients: The interleukin-11 receptor alpha as a candidate target in the progression of human prostate cancer. Cancer Res. 2004, 64, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Torres-Roca, J.F.; DeSilvio, M.; Mora, L.B.; Khor, L.Y.; Hammond, E.; Ahmad, N.; Jove, R.; Forman, J.; Lee, R.J.; Sandler, H.; et al. Activated STAT3 as a correlate of distant metastasis in prostate cancer: A secondary analysis of Radiation Therapy Oncology Group 86-10. Urology 2007, 69, 505–509. [Google Scholar] [CrossRef]
- Liu, X.; He, Z.; Li, C.H.; Huang, G.; Ding, C.; Liu, H. Correlation analysis of JAK-STAT pathway components on prognosis of patients with prostate cancer. Pathol. Oncol. Res. 2012, 18, 17–23. [Google Scholar] [CrossRef]
- Tam, L.; McGlynn, L.M.; Traynor, P.; Mukherjee, R.; Bartlett, J.M.; Edwards, J. Expression levels of the JAK/STAT pathway in the transition from hormone-sensitive to hormone-refractory prostate cancer. Br. J. Cancer 2007, 97, 378–383. [Google Scholar] [CrossRef]
- Wu, C.T.; Hsieh, C.C.; Lin, C.C.; Chen, W.C.; Hong, J.H.; Chen, M.F. Significance of IL-6 in the transition of hormone-resistant prostate cancer and the induction of myeloid-derived suppressor cells. J. Mol. Med. 2012, 90, 1343–1355. [Google Scholar] [CrossRef]
- Ammirante, M.; Luo, J.L.; Grivennikov, S.; Nedospasov, S.; Karin, M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010, 464, 302–305. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Domenech, J.; Oliva, C.; Rovira, A.; Codony-Servat, J.; Bosch, M.; Filella, X.; Montagut, C.; Tapia, M.; Campás, C.; Dang, L.; et al. Interleukin 6, a nuclear factor-kappaB target, predicts resistance to docetaxel in hormone-independent prostate cancer and nuclear factor-kappaB inhibition by PS-1145 enhances docetaxel antitumor activity. Clin. Cancer Res. 2006, 12, 5578–5586. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, T.; Junicho, A.; Yamamoto, T.; Kishi, H.; Korkmaz, K.; Saatcioglu, F.; Fuse, H.; Muraguchi, A. Cross-talk between signal transducer and activator of transcription 3 and androgen receptor signaling in prostate carcinoma cells. Biochem. Biophys. Res. Commun. 2001, 283, 179–187. [Google Scholar] [CrossRef]
- Kroon, P.; Berry, P.A.; Stower, M.J.; Rodrigues, G.; Mann, V.M.; Simms, M.; Bhasin, D.; Chettiar, S.; Li, C.; Li, P.K.; et al. JAK- STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013, 73, 5288–5298. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.J.; Adachi, I. Serum interleukin-6 levels correlate to tumor progression and prognosis in metastatic breast carcinoma. Anticancer Res. 1999, 19, 1427–1432. [Google Scholar] [PubMed]
- Salgado, R.; Junius, S.; Benoy, I.; Van Dam, P.; Vermeulen, P.; Van Marck, E.; Huget, P.; Dirix, L.Y. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int. J. Cancer 2003, 103, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef]
- Dethlefsen, C.; Hojfeldt, G.; Hojman, P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res. Treat. 2013, 138, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rincon, M.; Broadwater, G.; Harris, L.; Crocker, A.; Weaver, D.; Dressler, L.; Berry, D.; Sutton, L.; Michaelson, R.; Messino, M.; et al. Interleukin-6, multidrug resistance protein-1 expression and response to paclitaxel in women with metastatic breast cancer: Results of cancer and leukemia group B trial 159806. Breast Cancer Res. Treat. 2006, 100, 301–308. [Google Scholar] [CrossRef]
- Widschwendter, A.; Tonko-Geymayer, S.; Welte, T.; Daxenbichler, G.; Marth, C.; Doppler, W. Prognostic significance of signal transducer and activator of transcription 1 activation in breast cancer. Clin. Cancer Res. 2002, 8, 3065–3074. [Google Scholar]
- Sato, T.; Neilson, L.M.; Peck, A.R.; Liu, C.; Tran, T.H.; Witkiewicz, A.; Hyslop, T.; Nevalainen, M.T.; Sauter, G.; Rui, H. Signal transducer and activator of transcription-3 and breast cancer prognosis. Am. J. Cancer Res. 2011, 1, 347–355. [Google Scholar]
- Ren, L.; Wang, X.; Dong, Z.; Liu, J.; Zhang, S. Bone metastasis from breast cancer involves elevated IL-11 expression and the gp130/STAT3 pathway. Med. Oncol. 2012, 30, 634. [Google Scholar] [CrossRef]
- Zhou, B.; Damrauer, J.S.; Bailey, S.T.; Hadzic, T.; Jeong, Y.; Clark, K.; Fan, C.; Murphy, L.; Lee, C.Y.; Troester, M.A.; et al. Erythropoietin promotes breast tumorigenesis through tumor-initiating cell self- renewal. J. Clin. Investig. 2014, 124, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Karakasheva, T.A.; Lin, E.W.; Tang, Q.; Qiao, W.; Waldron, T.; Soni, M.; Klein-Szanto, A.J.; Sahu, V.; Basu, D.; Ohashi, S.; et al. IL-6 mediates cross-talk between tumor cells and activated fibroblasts in the tumor microenvironment. Cancer Res. 2018, 78, 4957–4970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.S.; Chen, W.C.; Lu, C.H.; Chen, M.F. The prognosis of head and neck squamous cell carcinoma related to immunosuppressive tumor microenvironment regulated by IL-6 signaling. Oral. Oncol. 2019, 91, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.L.; Grandis, J.R.; Bauman, J.E. The STAT3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations. Oral. Oncol. 2016, 56, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Lui, V.W.; Peyser, N.D.; Ng, P.K.; Hritz, J.; Zeng, Y.; Lu, Y.; Li, H.; Wang, L.; Gilbert, B.R.; General, I.J.; et al. Frequent mutation of receptor protein tyrosine phosphatases provides a mechanism for STAT3 hyperactivation in head and neck cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1114–1119. [Google Scholar] [CrossRef] [Green Version]
- Peyser, N.D.; Du, Y.; Li, H.; Lui, V.; Xiao, X.; Chan, T.A.; Grandis, J.R. Loss-of-function PTPRD mutations lead to increased STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. PLoS ONE 2015, 10, e0135750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin Grandis, J.; Melhem, M.F.; Gooding, W.E.; Day, R.; Holst, V.A.; Wagener, M.M.; Drenning, S.D.; Tweardy, D.J. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J. Natl. Cancer Inst. 1998, 90, 824–832. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zhang, F.; Zhang, W.; He, J.; Zhao, Y.; Chen, X. Prognostic role of epidermal growth factor receptor in head and neck cancer: A meta-analysis. J. Surg. Oncol. 2013, 108, 387–397. [Google Scholar] [CrossRef]
- Alsahafi, E.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019, 10, 540. [Google Scholar] [CrossRef] [Green Version]
- Madoz-Gúrpide, J.; Zazo, S.; Chamizo, C.; Cassado, V.; Caramés, C.; Gavín, E.; Cristóbal, I.; García-Foncillas, J.; Rojo, F. Activation of MET pathway predicts poor outcome to cetuximab in patients with recurrent or metastatic head and neck cancer. J. Transl. Med. 2015, 13, 282. [Google Scholar] [CrossRef]
- Arnold, L.; Enders, J.; Thomas, S.M. Activated HGF-c-Met axis in head and neck cancer. Cancers 2017, 9, 169. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Parker, J.S.; Ely, K.; Carter, J.; Yi, Y.; Murphy, B.A.; Ang, K.K.; EI-Naggar, A.K.; Zanation, A.M.; Cmelak, A.J.; et al. Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor-kappaB signaling as characteristics of a high-risk head and neck squamous cell carcinoma. Cancer Res. 2006, 66, 8210–8218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipper, J.H.; Frixen, U.H.; Behrens, J.; Unger, A.; Jahnke, K.; Birchmeier, W. E-cadherin expression in squamous cell carcinomas of head and neck: Inverse correlation with tumor dedifferentiation and lymph node metastasis. Cancer Res. 1991, 51, 6328–6337. [Google Scholar] [PubMed]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef] [Green Version]
- Peltanova, B.; Raudenska, M.; Masarik, M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: A systematic review. Mol. Cancer 2019, 18, 63. [Google Scholar] [CrossRef]
- Zeng, Q.; Chen, S.; You, Z.; Yang, F.; Carey, T.E.; Saims, D.; Wang, C.Y. Hepatocyte growth factor inhibits anoikis in head and neck squamous cell carcinoma cells by activation of ERK and Akt signaling independent of NFκB. J. Biol. Chem. 2002, 277, 25203–25208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neiva, K.G.; Zhang, Z.; Miyazawa, M.; Warner, K.A.; Karl, E.; Nör, J.E. Cross talk initiated by endothelial cells enhances migration and inhibits anoikis of squamous cell carcinoma cells through STAT3/Akt/ERK signaling. Neoplasia 2009, 11, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Ernst, M.; Poh, A.R. Multicellular Effects of STAT3 in Non-small Cell Lung Cancer: Mechanistic Insights and Therapeutic Opportunities. Cancers 2021, 13, 6228. [Google Scholar] [CrossRef]
- Barrera, L.; Montes-Servín, E.; Barrera, A.; Ramírez-Tirado, L.A.; Salinas-Parra, F.; Bañales-Méndez, J.L.; Sandoval-Ríos, M.; Arrieta, Ó. Cytokine profile determined by data-mining analysis set into clusters of non-small-cell lung cancer patients according to prognosis. Ann. Oncol. 2015, 26, 428–435. [Google Scholar] [CrossRef]
- Pine, S.R.; Mechanic, L.E.; Enewold, L.; Chaturvedi, A.K.; Katki, H.A.; Zheng, Y.-L.; Bowman, E.D.; Engels, E.A.; Caporaso, N.E.; Harris, C.C. Increased levels of circulating interleukin 6, interleukin 8, C-reactive protein, and risk of lung cancer. J. Natl. Cancer Inst. 2011, 103, 1112–1122. [Google Scholar] [CrossRef]
- Ujiie, H.; Tomida, M.; Akiyama, H.; Nakajima, Y.; Okada, D.; Yoshino, N.; Takiguchi, Y.; Tanzawa, H. Serum hepatocyte growth factor and interleukin-6 are effective prognostic markers for non-small cell lung cancer. Anticancer Res. 2012, 32, 3251–3258. [Google Scholar]
- Song, L.; Smith, M.A.; Doshi, P.; Sasser, K.; Fulp, W.; Altiok, S.; Haura, E.B. Antitumorefficacyoftheanti-interleukin-6 (IL-6) antibody siltuximab in mouse xenograft models of lung cancer. J. Thorac. Oncol. 2014, 9, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpagnano, G.E.; Resta, O.; Foschino-Barbaro, M.P.; Gramiccioni, E.; Carpagnano, F. Interleukin-6 is increased in breath condensate of patients with non-small cell lung cancer. Int. J. Biol. Markers 2002, 17, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Kimura, H.; Yokota, S.; Yamamoto, Y.; Hashimoto, T.; Nakagawa, M.; Ito, M.; Ogura, T. Effect of IL-6 elevation in malignant pleural effusion on hyperfibrinogenemia in lung cancer patients. Jpn. J. Clin. Oncol. 2000, 30, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.H.; Hsiao, C.F.; Yeh, Y.M.; Chang, G.C.; Tsai, Y.H.; Chen, Y.M.; Huang, M.S.; Chen, H.L.; Li, Y.J.; Yang, P.C.; et al. Circulating interleukin-6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int. J. Cancer 2013, 132, 1977–1985. [Google Scholar] [CrossRef]
- Silva, E.M.; Mariano, V.S.; Pastrez, P.R.A.; Pinto, M.C.; Castro, A.G.; Syrjanen, K.J.; Longatto-Filho, A. High systemic IL-6 is associated with worse prognosis in patients with non-small cell lung cancer. PLoS ONE 2017, 12, e0181125. [Google Scholar] [CrossRef]
- Zhao, M.; Liu, Y.; Liu, R.; Qi, J.; Hou, Y.; Chang, J.; Ren, L. Upregulation of IL-11, an IL-6 Family Cytokine, Promotes Tumor Progression and Correlates with Poor Prognosis in Non-Small Cell Lung Cancer. Cell Physiol. Biochem. 2018, 45, 2213–2224. [Google Scholar] [CrossRef]
- Wu, J.; Chen, J.; Lv, X.; Yang, Q.; Yao, S.; Zhang, D.; Chen, J. Clinical value of serum and exhaled breath condensate inflammatory factor IL-11 levels in non-small cell lung cancer: Clinical value of IL-11 in non-small cell lung cancer. Int. J. Biol. Markers 2021, 36, 64–76. [Google Scholar] [CrossRef]
- Hosoda, H.; Izumi, H.; Tukada, Y.; Takagiwa, J.; Chiaki, T.; Yano, M.; Arai, H. Plasma hepatocyte growth factor elevation may be associated with early metastatic disease in primary lung cancer patients. Ann. Thorac. Cardiovasc. Surg. 2012, 18, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Haura, E.B.; Zheng, Z.; Song, L.; Cantor, A.; Bepler, G. Activated epidermal growth factor receptor-Stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 8288–8294. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Ceja, S.G.; Reyes-Maldonado, E.; Vázquez-Manríquez, M.E.; López-Luna, J.J.; Belmont, A.; Gutiérrez-Castellanos, S. Differential expression of STAT5 and Bcl-xL, and high expression of Neu and STAT3 in non-small-cell lung carcinoma. Lung Cancer 2006, 54, 163–168. [Google Scholar] [CrossRef]
- Yin, Z.; Zhang, Y.; Li, Y.; Lv, T.; Liu, J.; Wang, X. Prognostic significance of STAT3 expression and its correlation with chemoresis-tance of non-small cell lung cancer cells. Acta Histochem. 2012, 114, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.J.; Jin, F.G.; Liu, T.G.; Fu, E.Q.; Xie, Y.H.; Sun, R.L. Overexpression of STAT3 potentiates growth, survival, and radioresistance of non-small-cell lung cancer (NSCLC) cells. J. Surg. Res. 2011, 171, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.M.; Kwon, O.J.; Hong, Y.K.; Kim, J.H.; Solca, F.; Ha, S.J.; Soo, R.A.; Christensen, J.G.; Lee, J.H.; Cho, B.C. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol. Cancer 2012, 11, 2254–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Jin, Z.; Liu, Z.; Sun, L.; Wang, L.; Li, K. Correlation of activated STAT3 expression with clinicopathologic features in lung adenocarcinoma and squamous cell carcinoma. Mol. Diagn. Ther. 2011, 15, 347–352. [Google Scholar] [CrossRef]
- Ai, T.; Wang, Z.; Zhang, M.; Zhang, L.; Wang, N.; Li, W.; Song, L. Expression and prognostic relevance of STAT3 and cyclin D1 in non-small cell lung cancer. Int. J. Biol. Markers 2012, 27, 132–138. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, C.; Zhang, J. Dominant negative STAT3 suppresses the growth and invasion capability of human lung cancer cells. Mol. Med. Rep. 2009, 2, 819–824. [Google Scholar]
- Blakely, C.M.; Pazarentzos, E.; Olivas, V.; Asthana, S.; Yan, J.J.; Tan, I.; Hrustanovic, G.; Chan, E.; Lin, L.; Neel, D.S.; et al. NF-κB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 2015, 11, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.P.; Chang, Q.; Mao, N.; Daly, L.A.; Vogel, R.; Chan, T.; Liu, S.H.; Bournazou, E.; Schori, E.; Zhang, H.; et al. JAK2 inhibition sensitizes resistant EGFR-mutant lung adenocarcinoma to tyrosine kinase inhibitors. Sci. Signal. 2016, 9, ra33. [Google Scholar] [CrossRef] [Green Version]
- Chaib, I.; Karachaliou, N.; Pilotto, S.; Codony Servat, J.; Cai, X.; Li, X.; Drozdowskyj, A.; Servat, C.C.; Yang, J.; Hu, C.; et al. Co-activation of STAT3 and YES- associated protein 1 (YAP1) pathway in EGFR-mutant NSCLC. J. Natl. Cancer Inst. 2017, 109, djx014. [Google Scholar] [CrossRef]
- Shou, J.; You, L.; Yao, J.; Xie, J.; Jing, J.; Jing, Z.; Jiang, L.; Sui, X.; Pan, H.; Han, W. Cyclosporine A sensitizes human non- small cell lung cancer cells to gefitinib through inhibition of STAT3. Cancer Lett. 2016, 379, 124–133. [Google Scholar] [CrossRef]
- Song, L.; Rawal, B.; Nemeth, J.A.; Haura, E.B. JAK1 activates STAT3 activity in non-small-cell lung cancer cells and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling. Mol. Cancer Ther. 2011, 10, 481–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Perez, L.; Chang, Q.; Gao, S.P.; Kris, M.G.; Riely, G.J.; Bromberg, J. A phase 1/2 trial of ruxolitinib and erlotinib in patients with EGFR-mutant lung adenocarcinomas with acquired resistance to erlotinib. J. Thorac. Oncol. 2017, 12, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [Green Version]
- Xiang, M.; Kim, H.; Ho, V.T.; Walker, S.R.; Bar-Natan, M.; Anahtar, M.; Liu, S.; Toniolo, P.A.; Kroll, Y.; Jones, N.; et al. Gene expression-based discovery of atovaquone as a STAT3 inhibitor and anticancer agent. Blood 2016, 128, 1845–1853. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.G.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef]
- Herrmann, A.; Priceman, S.J.; Swiderski, P.; Kujawski, M.; Xin, H.; Cherryholmes, G.A.; Zhang, W.; Zhang, C.; Lahtz, C.; Kowolik, C.; et al. CTLA4 aptamer delivers STAT3 siRNA to tumor-associated and malignant T cells. J. Clin. Investig. 2014, 124, 2977–2987. [Google Scholar] [CrossRef] [Green Version]
- Yue, C.; Shen, S.; Deng, J.; Priceman, S.J.; Li, W.; Huang, A.; Yu, H. STAT3 in CD8+T cells inhibits their tumor accumulation by downregulating CXCR3/CXCL10 Axis. Cancer Immunol. Res. 2015, 3, 864–870. [Google Scholar] [CrossRef] [Green Version]
- Schmetterer, K.G.; Neunkirchner, A.; Wojta-Stremayr, D.; Leitner, J.; Steinberger, P.; Pickl, W.F. STAT3 governs hyporesponsiveness and granzyme B-dependent suppressive capacity in human CD4+ T cells. FASEB J. 2015, 29, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihara, M.; Ogura, H.; Ueda, N.; Tsuruoka, M.; Kitabayashi, C.; Tsuji, F.; Aono, H.; Ishihara, K.; Huseby, E.; Betz, U.A.; et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int. Immunol. 2007, 19, 695–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.J.; Grosso, J.F.; Yen, H.R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting edge: An in vivo requirement for STAT3 signaling in Th17 development and Th17-dependent autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnerlich, J.L.; Mitchem, J.B.; Weir, J.S.; Sankpal, N.V.; Kashiwagi, H.; Belt, B.A.; Porembka, M.R.; Herndon, J.M.; Eberlein, T.J.; Goedegebuure, P.; et al. Induction of Th17 cells in the tumor microenvironment improves survival in a murine model of pancreatic cancer. J. Immunol. 2010, 185, 4063–4071. [Google Scholar] [CrossRef] [Green Version]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef] [Green Version]
- Austin, J.W.; Lu, P.; Majumder, P.; Ahmed, R.; Boss, J.M. STAT3, STAT4, NFATc1, and CTCF regulate PD-1 through multiple novel regulatory regions in murine T cells. J. Immunol. 2014, 192, 4876–4886. [Google Scholar] [CrossRef]
- Celada, L.J.; Kropski, J.A.; Herazo-Maya, J.D.; Luo, W.; Creecy, A.; Abad, A.T.; Chioma, O.S.; Lee, G.; Hassell, N.E.; Shaginurova, G.I.; et al. PD-1 up-regulation on CD4+ T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci. Transl. Med. 2018, 10, eaar8356. [Google Scholar] [CrossRef] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Korn, T.; Mitsdoerffer, M.; Croxford, A.L.; Awasthi, A.; Dardalhon, V.A.; Galileos, G.; Vollmar, P.; Stritesky, G.L.; Kaplan, M.H.; Waisman, A.; et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 18460–18465. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, M.; Nakano, M.; Terabe, F.; Kawahata, H.; Ohkawara, T.; Han, Y.; Ripley, B.; Serada, S.; Nishikawa, T.; Kimura, A.; et al. The influence of excessive IL-6 production in vivo on the development and function of Foxp3+ regulatory T cells. J. Immunol. 2011, 186, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.; Santner-Nanan, B.; Hu, M.; Skarratt, K.; Lee, C.H.; Stormon, M.; Wong, M.; Fuller, S.J.; Nanan, R. IL-10 potentiates differentiation of human induced regulatory T cells via STAT3 and Foxo1. J. Immunol. 2015, 195, 3665–3674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Giurisato, E.; Xu, Q.; Lonardi, S.; Telfer, B.; Russo, I.; Pearson, A.; Finegan, K.G.; Wang, W.; Wang, J.; Gray, N.S.; et al. Myeloid ERK5 deficiency suppresses tumor growth by blocking protumor macrophage polarization via STAT3 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E2801–E2810. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liao, D.; Chen, C.; Liu, Y.; Chuang, T.H.; Xiang, R.; Markowitz, D.; Reisfeld, R.A.; Luo, Y. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells 2013, 31, 248–258. [Google Scholar] [CrossRef]
- Takaishi, K.; Komohara, Y.; Tashiro, H.; Ohtake, H.; Nakagawa, T.; Katabuchi, H.; Takeya, M. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Sci. 2010, 101, 2128–2136. [Google Scholar] [CrossRef]
- Yan, D.; Wang, H.W.; Bowman, R.L.; Joyce, J.A. STAT3 and STAT6 signaling pathways synergize to promote cathepsin secretion from macrophages via IRE1α activation. Cell Rep. 2016, 16, 2914–2927. [Google Scholar] [CrossRef] [Green Version]
- Wölfle, S.J.; Strebovsky, J.; Bartz, H.; Sähr, A.; Arnold, C.; Kaiser, C.; Dalpke, A.H.; Heeg, K. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur. J. Immunol. 2011, 41, 413–424. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, D.M.; Pal, S.K.; Moreira, D.; Duttagupta, P.; Zhang, Q.; Won, H.; Jones, J.; D’Apuzzo, M.; Forman, S.; Kortylewski, M. TLR9-targeted STAT3 silencing abrogates immunosuppressive activity of myeloid- derived suppressor cells from prostate cancer patients. Clin. Cancer Res. 2015, 21, 3771–3782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabritz, J.; Judd, L.M.; Chalinor, H.V.; Menheniott, T.R.; Giraud, A.S. Altered gp130 signalling ameliorates experimental colitis via myeloid cell-specific STAT3 activation and myeloid-derived suppressor cells. Sci. Rep. 2016, 6, 20584. [Google Scholar] [CrossRef]
- Sumida, K.; Ohno, Y.; Ohtake, J.; Kaneumi, S.; Kishikawa, T.; Takahashi, N.; Taketomi, A.; Kitamura, H. IL-11 induces differentiation of myeloid-derived suppressor cells through activation of STAT3 signalling pathway. Sci. Rep. 2015, 5, 13650. [Google Scholar] [CrossRef]
- Takeda, K.; Clausen, B.E.; Kaisho, T.; Tsujimura, T.; Terada, N.; Förster, I.; Akira, S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 1999, 10, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Panopoulos, A.D.; Zhang, L.; Snow, J.W.; Jones, D.M.; Smith, A.M.; El Kasmi, K.C.; Liu, F.; Goldsmith, M.A.; Link, D.C.; Murray, P.J.; et al. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood 2006, 108, 3682–3690. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Nguyen-Jackson, H.; Panopoulos, A.D.; Li, H.S.; Murray, P.J.; Watowich, S.S. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood 2010, 116, 2462–2471. [Google Scholar] [CrossRef] [Green Version]
- Gotthardt, D.; Putz, E.M.; Straka, E.; Kudweis, P.; Biaggio, M.; Poli, V.; Strobl, B.; Müller, M.; Sexl, V. Loss of STAT3 in murine NK cells enhances NK cell-dependent tumor surveillance. Blood 2014, 124, 2370–2379. [Google Scholar] [CrossRef]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Koskela, H.; Leblanc, F.; Peng Ng, K.; Olson, T.; Przychodzen, B.; Afable, M.; Gomez-Segui, I.; et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T cell large granular lymphocyte leukemia. Blood 2012, 120, 3048–3057. [Google Scholar] [CrossRef]
- Küçük, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat. Commun. 2015, 6, 6025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.D.; Watt, F.M. Fibroblast Heterogeneity: Implications for Human Disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, J.L.; Bondre, C.; Gladstone, H.B.; Brown, P.O.; Chang, H.Y. Anatomic Demarcation by Positional Variation in Fibroblast Gene Expression Programs. PLoS Genet. 2006, 2, e119. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Viros, A.; Hooper, S.; Mitter, R.; Féral, C.C.; Cook, M.; et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef] [Green Version]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [Green Version]
- Nagathihalli, N.S.; Castellanos, J.A.; Shi, C.; Beesetty, Y.; Reyzer, M.L.; Caprioli, R.; Chen, X.; Walsh, A.J.; Skala, M.C.; Moses, H.L.; et al. Signal transducer and activator of transcription 3, mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic cancer. Gastroenterology 2015, 149, 1932–1943. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, R.J.; Knight, D.A.; Richards, C.D.; Prêle, C.M.; Lau, H.L.; Jarnicki, A.G.; Jones, J.; Bozinovski, S.; Vlahos, R.; Thiem, S.; et al. Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol. Med. 2012, 4, 939–951. [Google Scholar] [CrossRef]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 2010, 15, 166–179. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer- associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 422. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Huang, G.; Wang, R.; Pan, Y.; He, Z.; Chu, X.; Song, H.; Chen, L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 38408. [Google Scholar] [CrossRef] [PubMed]
- Minoguchi, M.; Minoguchi, S.; Aki, D.; Joo, A.; Yamamoto, T.; Yumioka, T.; Matsuda, T.; Yoshimura, A. STAP-2/BKS, an adaptor/docking protein, modulates STAT3 activation in acute-phase response through its YXXQ motif. J. Biol. Chem. 2003, 278, 11182–11189. [Google Scholar] [CrossRef] [Green Version]
- Brauer, P.M.; Tyner, A.L. Building a better understanding of the intra- cellular tyrosine kinase PTK6—BRK by BRK. Biochim. Biophys Acta 2010, 1806, 66–73. [Google Scholar]
- Ikeda, O.; Sekine, Y.; Mizushima, A.; Nakasuji, M.; Miyasaka, Y.; Yamamoto, C.; Muromoto, R.; Nanbo, A.; Oritani, K.; Yoshimura, A.; et al. Interactions of STAP-2 with Brk and STAT3 participate in cell growth of human breast cancer cells. J. Biol. Chem. 2010, 285, 38093–38103. [Google Scholar] [CrossRef] [Green Version]
- Kitai, Y.; Iwakami, M.; Saitoh, K.; Togi, S.; Isayama, S.; Sekine, Y.; Muromoto, R.; Kashiwakura, J.I.; Yoshimura, A.; Oritani, K.; et al. STAP-2 protein promotes prostate cancer growth by enhancing epidermal growth factor receptor stabilization. J. Biol. Chem. 2017, 292, 19392–19399. [Google Scholar] [CrossRef] [Green Version]
- Heo, T.-H.; Wahler, J.; Suh, N. Potential therapeutic implications of IL-6/IL-6R/gp130-targeting agents in breast cancer. Oncotarget 2016, 7, 15460–15473. [Google Scholar] [CrossRef] [Green Version]
- Grimley, P.M.; Dong, F.; Rui, H. Stat5a and Stat5b: Fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev. 1999, 10, 131–157. [Google Scholar] [CrossRef]
- Li, X.; Lu, Y.; Liang, K.; Hsu, J.M.; Albarracin, C.; Mills, G.B.; Hung, M.C.; Fan, Z. Brk/PTK6 sustains activated EGFR signaling through inhibiting EGFR degradation and transactivating EGFR. Oncogene 2012, 31, 4372–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyati, K.K.; Zaman, M.M.-U.; Sharma, P.; Kishimoto, T. Arid5a, an RNA-binding protein in immune regulation: RNA stability, inflammation, and autoimmunity. Trends Immunol. 2020, 41, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Ripley, B.; Nishimura, R.; Mino, T.; Takeuchi, O.; Shioi, G.; Kiyonari, H.; Kishimoto, T. Arid5a controls IL-6 mRNA stability, which contributes to elevation of IL-6 level in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 9409–9414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Kagami, S.; Sanayama, Y.; Ikeda, K.; Suto, A.; Kashiwakuma, D.; Furuta, S.; Iwamoto, I.; Nonaka, K.; Ohara, O.; et al. AT-rich-interactive domain-containing protein 5A functions as a negative regulator of retinoic acid receptor-related orphan nuclear receptor γt-induced Th17 cell differentiation. Arthritis Rheumatol. 2014, 66, 1185–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, K.; Ripley, B.; Nyati, K.K.; Dubey, P.K.; Zaman, M.M.; Hanieh, H.; Higa, M.; Yamashita, K.; Standley, D.M.; Mashima, T.; et al. Arid5a regulates naive CD4+ T cell fate through selective stabilization of Stat3 mRNA. J. Exp. Med. 2016, 213, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, G.; Tekguc, M.; Wing, J.B.; Hashimoto, A.; Okuzaki, D.; Hirata, T.; Sasaki, A.; Itokazu, T.; Handa, H.; Sugino, H.; et al. Arid5a Promotes Immune Evasion by Augmenting Tryptophan Metabolism and Chemokine Expression. Cancer Immunol. Res. 2021, 9, 862–876. [Google Scholar] [CrossRef]
- Nyati, K.K.; Hashimoto, S.; Singh, S.K.; Tekguc, M.; Metwally, H.; Liu, Y.C.; Okuzaki, D.; Gemechu, Y.; Kang, S.; Kishimoto, T. The novel long noncoding RNA AU021063, induced by IL-6/Arid5a signaling, exacerbates breast cancer invasion and metastasis by stabilizing Trib3 and activating the Mek/Erk pathway. Cancer Lett. 2021, 520, 295–306. [Google Scholar] [CrossRef]
- Edinger, A.L.; Thompson, C.B. Antigen-presenting cells control T cell proliferation by regulating amino acid availability. Proc. Natl. Acad. Sci. USA 2002, 99, 1107–1109. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [Green Version]
- Neamah, W.H.; Singh, N.P.; Alghetaa, H.; Abdulla, O.A.; Chatterjee, S.; Busbee, P.B.; Nagarkatti, M.; Nagarkatti, P. Ahr activation leads to massive mobilization of myeloid-derived suppressor cells with immunosuppressive activity through regulation of Cxcr2 and microrna mir-150-5p and mir-543-3p that target anti-inflammatory genes. J. Immunol. 2019, 203, 1830–1844. [Google Scholar] [CrossRef] [PubMed]
- Fein, M.R.; He, X.Y.; Almeida, A.S.; Bružas, E.; Pommier, A.; Yan, R.; Eberhardt, A.; Fearon, D.T.; Van Aelst, L.; Wilkinson, J.E.; et al. Cancer cell Ccr2 orchestrates suppression of the adaptive immune response. J. Exp. Med. 2020, 217, e20181551. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+Cd115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Lei, Z.; Zhao, J.; Gong, W.; Liu, J.; Chen, Z.; Liu, Y.; Li, D.; Yuan, Y.; Zhang, G.M.; et al. Ccl2/Ccr2 Pathway Mediates Recruitment of Myeloid Suppressor Cells to Cancers. Cancer Lett. 2007, 252, 86–92. [Google Scholar] [CrossRef]
- Li, B.H.; Garstka, M.A.; Li, Z.F. Chemokines and Their Receptors Promoting the Recruitment of Myeloid-Derived Suppressor Cells into the Tumor. Mol. Immunol 2020, 117, 201–215. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, H.; Guan, L.; Lai, C.; Yu, W.; Lai, M. LL1, a novel and highly selective STAT3 inhibitor, displays anti-colorectal cancer activities in vitro and in vivo. Br. J. Pharmacol. 2020, 177, 298–313. [Google Scholar] [CrossRef]
- Chen, X.; Pan, L.; Wei, J.; Zhang, R.; Yang, X.; Song, J.; Bai, R.Y.; Fu, S.; Pierson, C.R.; Finlay, J.L.; et al. LLL12B, a small molecule STAT3 inhibitor, induces growth arrest, apoptosis, and enhances cisplatin- mediated cytotoxicity in medulloblastoma cells. Sci. Rep. 2021, 11, 6517. [Google Scholar] [CrossRef]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef] [Green Version]
- Siddiquee, K.A.; Gunning, P.T.; Glenn, M.; Katt, W.P.; Zhang, S.; Schrock, C.; Sebti, S.M.; Jove, R.; Hamilton, A.D.; Turkson, J. An Oxazole-Based Small-Molecule Stat3 Inhibitor Modulates Stat3 Stability and Processing and Induces Antitumor Cell Effects. ACS Chem. Biol. 2007, 2, 787–798. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Pireddu, R.; Yang, H.; Urlam, M.K.; Lawrence, H.R.; Guida, W.C.; Lawrence, N.J.; Sebti, S.M. A Novel Inhibitor of STAT3 Homodimerization Selectively Suppresses STAT3 Activity and Malignant Transformation. Cancer Res. 2013, 73, 1922–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, Y.; Nonomura, C.; Ashizawa, T.; Iizuka, A.; Kondou, R.; Miyata, H.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; et al. The anti-tumor activity of the STAT3 inhibitor STX-0119 occurs via promotion of tumor-infiltrating lymphocyte accumulation in temozolomide-resistant glioblastoma cell line. Immunol. Lett. 2017, 190, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, R.; Wang, S.; Lin, J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4700–4705. [Google Scholar] [CrossRef] [Green Version]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishisaka, F.; Taniguchi, K.; Tsugane, M.; Hirata, G.; Takagi, A.; Asakawa, N.; Kurita, A.; Takahashi, H.; Ogo, N.; Shishido, Y.; et al. Antitumor activity of a novel oral signal transducer and activator of transcription 3 inhibitor YHO-1701. Cancer Sci. 2020, 111, 1774–1784. [Google Scholar] [CrossRef] [Green Version]
- Turkson, J.; Ryan, D.; Kim, J.S.; Zhang, Y.; Chen, Z.; Haura, E.; Laudano, A.; Sebti, S.; Hamilton, A.D.; Jove, R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. [Google Scholar] [CrossRef] [Green Version]
- Turkson, J.; Zhang, S.; Palmer, J.; Kay, H.; Stanko, J.; Mora, L.B.; Sebti, S.; Yu, H.; Jove, R. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol. Cancer Ther. 2004, 3, 1533–1542. [Google Scholar] [CrossRef]
- Liang, M.; Zhan, F.; Zhao, J.; Li, Q.; Wuyang, J.; Mu, G.; Li, D.; Zhang, Y.; Huang, X. CPA-7 influences immune profile and elicits anti-prostate cancer effects by inhibiting activated STAT3. BMC Cancer 2016, 16, 504. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Dong, Z.; Chen, Y.; Wang, F.; Wang, C.J.; Peng, H.; He, Y.; Hangoc, G.; Pollok, K.; Sandusky, G.; et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene 2016, 35, 783–792. [Google Scholar] [CrossRef]
- Nagel-Wolfrum, K.; Buerger, C.; Wittig, I.; Butz, K.; Hoppe-Seyler, F.; Groner, B. The interaction of specific peptide aptamers with the DNA binding domain and the dimerization domain of the transcription factor Stat3 inhibits transactivation and induces apoptosis in tumor cells. Mol. Cancer Res. 2004, 2, 170–182. [Google Scholar] [CrossRef]
- Kobayashi, A.; Tanizaki, Y.; Kimura, A.; Ishida, Y.; Nosaka, M.; Toujima, S.; Kuninaka, Y.; Minami, S.; Ino, K.; Kondo, T. AG490, a Jak2 inhibitor, suppressed the progression of murine ovarian cancer. Eur. J. Pharmacol. 2015, 766, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, E.; Lemoine, M.; Buglio, D.; Katayama, H.; Ji, Y.; Davis, R.E.; Sen, S.; Younes, A. The JAK inhibitor AZD1480 regulates proliferation and immunity in Hodgkin lymphoma. Blood Cancer J. 2011, 1, e46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Takigawa, N.; Ninomiya, T.; Ochi, N.; Yasugi, M.; Honda, Y.; Kubo, T.; Ichihara, E.; Hotta, K.; Tanimoto, M.; et al. Effect of AZD1480 in an epidermal growth factor receptor-driven lung cancer model. Lung Cancer 2014, 83, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, G.S.; Tian, A.; Hebbard, L.; Duan, W.; George, J.; Li, X.; Qiao, L. Tumoricidal Effects of the JAK Inhibitor Ruxolitinib (INC424) on Hepatocellular Carcinoma In Vitro. Cancer Lett. 2013, 341, 224–230. [Google Scholar] [CrossRef]
- Lo, M.C.; Peterson, L.F.; Yan, M.; Cong, X.; Hickman, J.H.; Dekelver, R.C.; Niewerth, D.; Zhang, D.E. JAK inhibitors suppress t(8;21) fusion protein-induced leukemia. Leukemia 2013, 27, 2272–2279. [Google Scholar] [CrossRef] [Green Version]
- Horiguchi, A.; Asano, T.; Kuroda, K.; Sato, A.; Asakuma, J.; Ito, K.; Hayakawa, M.; Sumitomo, M.; Asano, T. STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br. J. Cancer 2010, 102, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Buerger, C.; Nagel-Wolfrum, K.; Kunz, C.; Wittig, I.; Butz, K.; Hoppe-Seyler, F.; Groner, B. Sequence-specific Peptide Aptamers, Interacting with the Intracellular Domain of the Epidermal Growth Factor Receptor, Interfere with Stat3 Activation and Inhibit the Growth of Tumor Cells. J. Biol. Chem. 2003, 278, 37610–37621. [Google Scholar] [CrossRef] [Green Version]
- Ge, H.; Liu, H.; Fu, Z.; Sun, Z. Therapeutic and Preventive Effects of an Epidermal Growth Factor Receptor Inhibitor on Oral Squamous Cell Carcinoma. J. Int. Med. Res. 2012, 40, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.Y.; Wang, M. Molecular mechanisms of action and potential biomarkers of growth inhibition of dasatinib (BMS-354825) on hepatocellular carcinoma cells. BMC Cancer 2013, 13, 267. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Action | Inhibitor/ Compound | Mechanism of Action | Cancer Type | Ref. |
|---|---|---|---|---|
| Direct inhibitors | LL1 | SH2 domain inhibitor | CRC, NSCLC | [217] |
| LLL12B | SH2 domain inhibitor | Medulloblastoma | [218] | |
| S3I-201 | SH2 domain inhibitor | Breast cancer, liver cancer | [219] | |
| S3I-M2001 | SH2 domain inhibitor | Breast cancer | [220] | |
| S31-1757 | SH2 domain inhibitor | Breast cancer, lung cancer | [221] | |
| STX-0119 | SH2 domain inhibitor | Glioblastoma | [222] | |
| STA-21 | SH2 domain inhibitor | Breast cancer | [223] | |
| Stattic | SH2 domain inhibitor | Breast cancer, HNSCC | [224] | |
| YHO-1701 | SH2 domain inhibitor | HNSCC, NSCLC | [225] | |
| PY*LKTK | SH2 domain inhibitor | NIH3T3/v-Src or v-Ras | [226] | |
| CPA-1 | DNA-binding domain inhibitor | Breast cancer, colon cancer, melanoma | [227] | |
| CPA-7 | DNA-binding domain inhibitor | Prostate cancer, breast cancer, colon cancer, melanoma | [227,228] | |
| inS3-54A18 | DNA-binding domain inhibitor | NSCLC | [229] | |
| DBD-1 | DNA-binding domain inhibitor | Melanoma, myeloma | [230] | |
| Indirect inhibitors | AG490 | JAK inhibitor | Ovarian cancer, pancreatic cancer | [231] |
| AZD1480 | JAK inhibitor | Lymphoma, lung cancer | [232,233] | |
| Ruxolitinib | JAK inhibitor | Hepatocellular carcinom | [234] | |
| TG101209 | JAK2 inhibitor | Leukemia | [235] | |
| WP1066 | JAK inhibitor | Renal cell carcinoma | [236] | |
| KDI1 | RTK inhibitor | Vulval and breast cancer | [237] | |
| PD153035 | RTK inhibitor | Oral squamous carcinoma | [238] | |
| Dasatinib | Src inhibitor | Synovial sarcoma, hepatocellular carcinoma, glioma, prostate cancer | [239] |
| Action | Inhibitor/Compound | Type | Cancer Type | Phase | NCT Number |
|---|---|---|---|---|---|
| Direct inhibitors | BBI608 (FDA approved) | Small molecules | Advanced malignancies | I/II | NCT01775423 |
| CRC | III | NCT01830621 | |||
| C188-9 | Small molecules | BC, CRC, HNSCC, HCC, NSCLC, GAC, melanoma, advanced cancer | I | NCT03195699 | |
| OPB-31121 | Small molecules | advanced cancer, solid tumorS | I | NCT00955812 | |
| HCC | I/II | NCT01406574 | |||
| OPB-51602 | Small molecules | Malignant solid tumors | I | NCT01184807 | |
| Hematological malignancies | I | NCT01344876 | |||
| Nasopharyngeal carcinoma | I | NCT02058017 | |||
| OPB-111077 | Small molecules | Acute myeloid leukemia | I | NCT03197714 | |
| Advanced HCC | I | NCT01942083 | |||
| AZD-9150 | Oligonucleotides | Lymphoma | I/II | NCT01563302 | |
| Indirect inhibitors | AZD-1480 | JAK1/2 | Solid tumors | I | NCT01112397 |
| CYT387 | JAK1/2 | Myelofibrosis | I/II | NCT02101268 | |
| PMF, post-PV, post-ET MF | III | NCT03427866 | |||
| Ruxolitinib (FDA approved) | JAK1/2 | Myelofibrosis | III | NCT03427866 | |
| LY2784544 | JAK2 | Myeloproliferative neoplasms | II | NCT01594723 | |
| SB1518 | JAK2 | Myelofibrosis | III | NCT02055781 | |
| Siltuximab (FDA approved) | IL-6R | Multiple myeloma | II | NCT03315026 | |
| Tocilitizumab (FDA approved) | IL-6R | HCC | I/II | NCT02997956 | |
| Combinations | AZD9150, durvalumab (anti-PD-L1) | Direct inhibitors and ICB | NSCLC | II | NCT03334617 |
| PC, CRC, NSCLC | II | NCT02983578 | |||
| Advanced solid tumors, metastatic HNSCC | I/II | NCT02499328 | |||
| Diffuse large B-cell lymphoma | I | NCT02549651 | |||
| BBI608, nivolumab (anti-PD-1) | Direct inhibitors and ICB | Metastatic CRC | II | NCT03647839 | |
| BBI608, pembrolizumab (anti-PD-1) | Direct inhibitors and ICB | Metastatic CRC | I/II | NCT02851004 | |
| Apatinib, SHR-1210 (anti-PD-1) | Indirect inhibitors and ICB | Melanoma | II | NCT03955354 | |
| Bevacizumab, atezolizumab (anti-PD-L1) | Indirect inhibitors and ICB | Unresectable HCC | III | NCT03434379 | |
| Dasatinib, Ipilimumab (anti-CTLA-4) | Indirect inhibitors and ICB | GIST, stage III/IV soft tissue sarcoma | I | NCT01643278 | |
| Dasatinib, nivolumab (anti-PD-1) | Indirect inhibitors and ICB | Philadelphia chromosome positive ALL | I | NCT02819804 | |
| Ruxolitinib, pembrolizumab (anti-PD-1) | Indirect inhibitors and ICB | Hematological malignancies | II | NCT04016116 | |
| Metastatic stage IV TNBC | I | NCT03012230 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashimoto, S.; Hashimoto, A.; Muromoto, R.; Kitai, Y.; Oritani, K.; Matsuda, T. Central Roles of STAT3-Mediated Signals in Onset and Development of Cancers: Tumorigenesis and Immunosurveillance. Cells 2022, 11, 2618. https://doi.org/10.3390/cells11162618
Hashimoto S, Hashimoto A, Muromoto R, Kitai Y, Oritani K, Matsuda T. Central Roles of STAT3-Mediated Signals in Onset and Development of Cancers: Tumorigenesis and Immunosurveillance. Cells. 2022; 11(16):2618. https://doi.org/10.3390/cells11162618
Chicago/Turabian StyleHashimoto, Shigeru, Ari Hashimoto, Ryuta Muromoto, Yuichi Kitai, Kenji Oritani, and Tadashi Matsuda. 2022. "Central Roles of STAT3-Mediated Signals in Onset and Development of Cancers: Tumorigenesis and Immunosurveillance" Cells 11, no. 16: 2618. https://doi.org/10.3390/cells11162618