
Kynurenine Pathway—An Underestimated Factor Modulating Innate Immunity in Sepsis-Induced Acute Kidney Injury?


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Models of SAKI
2.1. Sepsis–Cecal Ligation, Puncture (CLP)
2.2. Cecal Content Injection (CCI)
2.3. LPS-Induced Sepsis
3. Role of Kidney Cells in SAKI
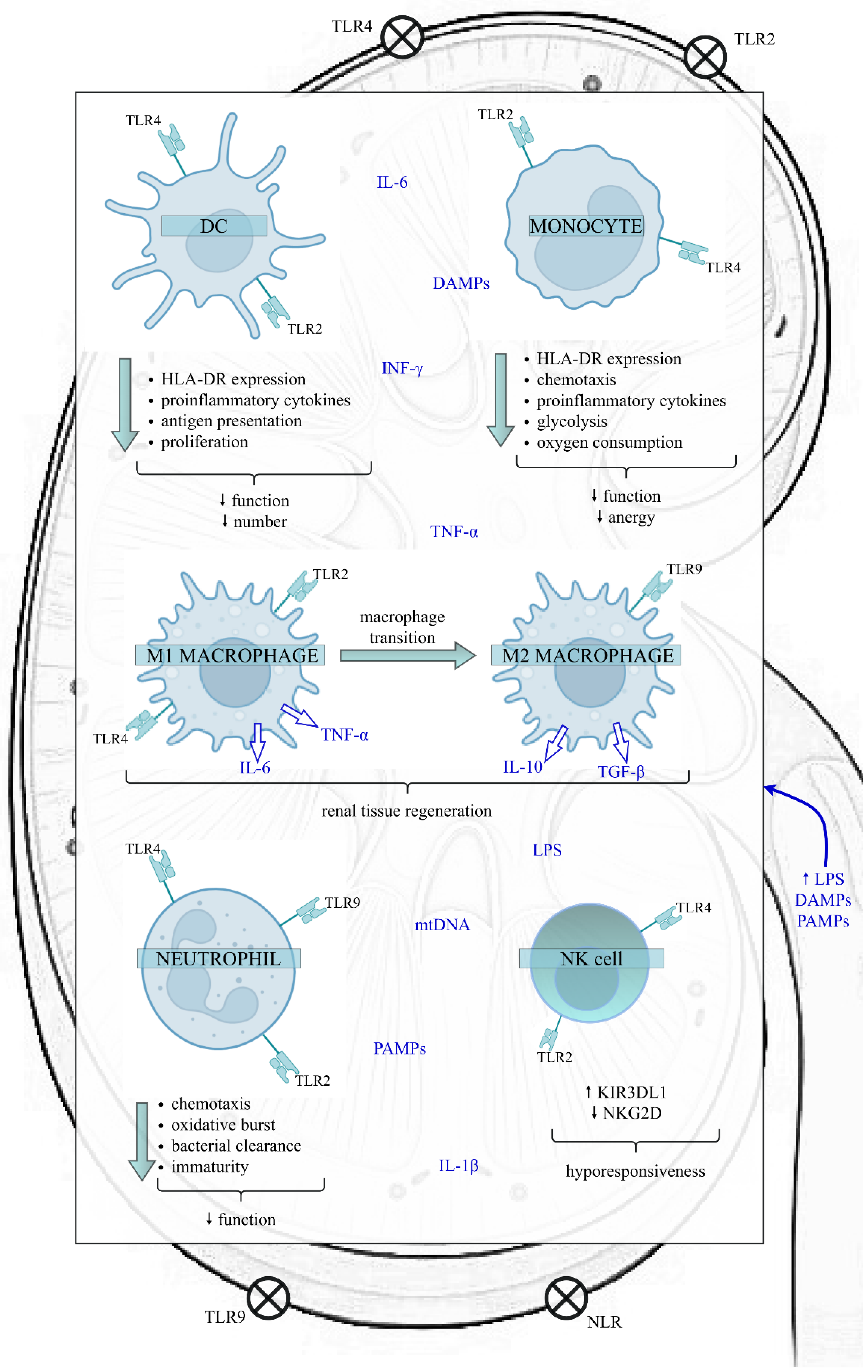
4. Innate Immunity in SAKI
4.1. DCs in SAKI
4.2. Monocytes/Macrophages in SAKI
4.3. Neutrophils in SAKI
4.4. NK Cells in SAKI
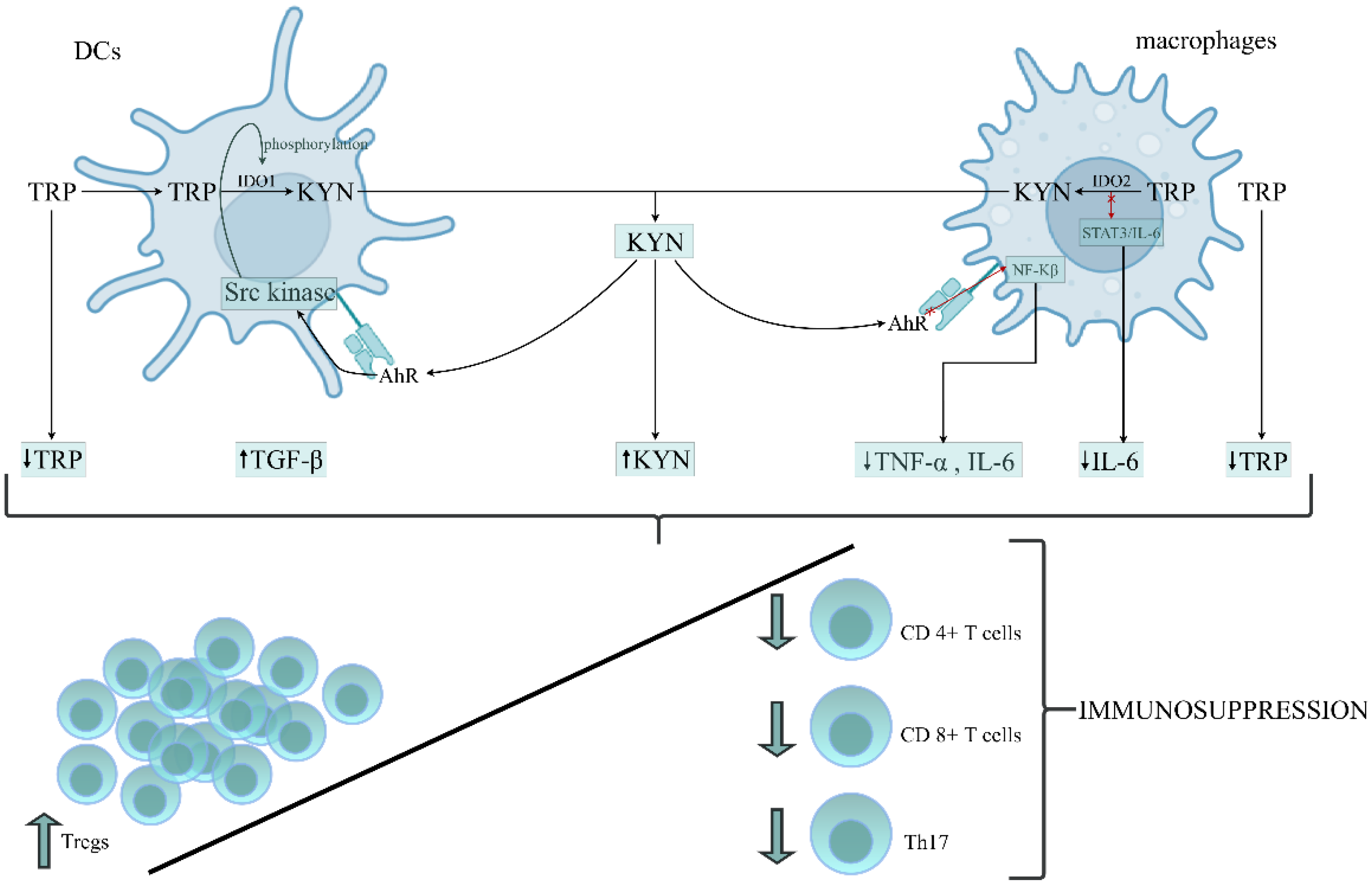
5. Kynurenine Pathway (KP) Activation in SAKI
6. IDO1, KP Activation and Innate Immunity in Sepsis
6.1. Data on Human Studies
6.2. Data on Experimental Sepsis Model
6.3. IDO1, KP Activation and Sepsis-Induced Immunosuppression
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1-MT | 1-methyl-D-tryptophan |
| 3-HKYN | 3-hydroxykynurenine |
| AhR | aryl hydrocarbon receptor |
| AKI | acute kidney injury |
| AP-1 | activator protein 1 |
| APCs | antigen-presenting cells |
| ATP | adenosine triphosphate |
| BTK | Bruton’s tyrosine kinase |
| CCI | cecal content injection |
| CLP | cecal ligation, puncture |
| CpG-DNA | cytosine-phosphate-guanine dinucleotides present in bacterial DNA |
| CVVH | continuous veno-venous haemofiltration |
| CXCL1 | C-X-C Motif Chemokine Ligand 1 |
| CXCR2 | C-X-C Motif Chemokine Receptor 2 |
| DAMPs | damage-associated molecular patterns |
| DCs | dendritic cells |
| FLT3L | FMS-related tyrosine kinase 3 ligand |
| fMLP | N-formyl-methionyl-leucyl-phenylalanine |
| GCN2K | general control nonderepressible 2 kinases |
| GFR | glomerular filtration rate |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| HMGB1 | high-mobility group box 1 |
| i.v. | intravenously |
| ICAM-1 | intercellular adhesion molecule 1 |
| ICU | intensive care unit |
| IDO1 | indoleamine 2,3-dioxygenase 1 |
| IDO2 | indoleamine 2,3-dioxygenase 2 |
| IFN-ɣ | interferon-gamma |
| IGFBP7 | insulin-like growth factor-binding protein 7 |
| IL-12 | interleukin 12 |
| IL-17A | interleukin-17A |
| IL-6 | interleukin 6 |
| IL-8 | interleukin 8 |
| iNOS | inducible nitric oxide synthase |
| IRAKM | IL-1 receptor-associated kinase M |
| ITK | IL-2 inducible T-cell kinase |
| KP | kynurenine pathway |
| KYN | kynurenine |
| KYNA | kynurenic acid |
| LPS | lipopolysaccharide |
| Mac-1 | macrophage-1 antigen |
| MCP-1 | monocyte chemoattractant protein-1 |
| MIF | migration inhibitory factor |
| MPO | myeloperoxidase |
| mtDNA | mitochondrial DNA |
| mTOR | mammalian target of rapamycin |
| MyD88 | Myeloid Differentiation Factor 88 |
| NETs | neutrophil extracellular traps |
| NFAT | nuclear factor of activated T cells |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGAL | neutrophil gelatinase-associated lipocalin |
| NK | natural killer |
| NLRs | nucleotide-binding oligomerization domain-like receptors |
| NOX-2 | neutrophils possess enzymes-NADPH oxidase |
| PAMPs | pathogen-associated molecular patterns |
| PDL1 | programmed death-ligand 1 |
| PPAR-α | peroxisome proliferator-activated receptor α |
| QUIN | quinolinic acid |
| ROS | reactive oxygen species |
| RTECs | renal tubular epithelial cells |
| SAKI | sepsis-induced AKI |
| SIRS | systemic inflammatory response syndrome |
| STAT3 | signal transducer and activator of transcription 3 |
| Syk | spleen tyrosine kinase |
| TDO2 | tryptophan 2,3-dioxygenase 2 |
| TGF-β | transforming growth factor β |
| Th17 | T helper 17 |
| Th2 | T helper 2 |
| TIMP-2 | tissue inhibitor of metalloproteinase-2 |
| TLR4 | Toll-like receptor 4 |
| TLRs | Toll-like receptors |
| TNF-α | tumor necrosis factor α |
| Tregs | regulatory T cells |
| TRP | trypthofan |
| VCAM-1 | vascular cell adhesion molecule 1 |
| WT | Wild-type mice |
References
- Bouchard, J.; Acharya, A.; Cerda, J.; Maccariello, E.R.; Madarasu, R.C.; Tolwani, A.J.; Liang, X.; Fu, P.; Liu, Z.H.; Mehta, R.L. A prospective international multicenter study of AKI in the intensive care unit. Clin. J. Am. Soc. Nephrol. 2015, 10, 1324–1331. [Google Scholar] [CrossRef] [Green Version]
- Hoste, E.A.J.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Bouchard, J.; Soroko, S.B.; Ikizler, T.A.; Paganini, E.P.; Chertow, G.M.; Himmelfarb, J. Sepsis as a cause and consequence of acute kidney injury: Program to Improve Care in Acute Renal Disease. Intensive Care Med. 2011, 37, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- See, E.J.; Jayasinghe, K.; Glassford, N.; Bailey, M.; Johnson, D.W.; Polkinghorne, K.R.; Toussaint, N.D.; Bellomo, R. Long-term risk of adverse outcomes after acute kidney injury: A systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019, 95, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W.; Wang, W. Acute Renal Failure and Sepsis. N. Engl. J. Med. 2004, 351, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Langenberg, C.; Gobe, G.; Hood, S.; May, C.N.; Bellomo, R. Renal histopathology during experimental septic acute kidney injury and recovery. Crit. Care Med. 2014, 42, e58–e67. [Google Scholar] [CrossRef] [PubMed]
- Prowle, J.R.; Ishikawa, K.; May, C.N.; Bellomo, R. Renal plasma flow and glomerular filtration rate duringacute kidney injury in man. Ren. Fail. 2010, 32, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Brenner, M.; Schaer, G.L.; Mallory, D.L.; Suffredini, A.F.; Parillo, J.E. Detection of renal blood flow abnormalities in septic and critically ill patients using a newly designed indwelling thermodilution renal vein catheter. Chest 1990, 98, 170–179. [Google Scholar] [CrossRef]
- Langenberg, C.; Bellomo, R.; May, C.; Wan, L.; Egi, M.; Morgera, S. Renal blood flow in sepsis. Crit. Care 2005, 9, R363. [Google Scholar] [CrossRef] [Green Version]
- Singer, M.; Deutschman, C.S.; Seymour, C.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA J. Am. Med. Assoc. 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Van Der Poll, T.; Van De Veerdonk, F.L.; Scicluna, B.P.; Netea, M.G. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 2017, 17, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Immunosuppression in sepsis: A novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 2013, 13, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Venet, F.; Monneret, G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat. Rev. Nephrol. 2018, 14, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Rosen, S.; Heyman, S.N. Difficulties in understanding human “acute tubular necrosis”: Limited data and flawed animal models. Kidney Int. 2001, 60, 1220–1224. [Google Scholar] [CrossRef] [Green Version]
- Fink, M.P. Animal models of sepsis. Virulence 2014, 5, 143–153. [Google Scholar] [CrossRef]
- Tsuji, N.; Tsuji, T.; Ohashi, N.; Kato, A.; Fujigaki, Y.; Yasuda, H. Role of mitochondrial DNA in septic AKI via toll-like receptor 9. J. Am. Soc. Nephrol. 2016, 27, 2009–2020. [Google Scholar] [CrossRef] [Green Version]
- Dejager, L.; Pinheiro, I.; Dejonckheere, E.; Libert, C. Cecal ligation and puncture: The gold standard model for polymicrobial sepsis? Trends Microbiol. 2011, 19, 198–208. [Google Scholar] [CrossRef]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009, 4, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Yim, H.S.; Choi, K.M.; Kim, B.; Jung, I.D.; Park, Y.M.; Kang, Y.K.; Lee, M.G. Effect of 1-methyl-D-tryptophan and adoptive transfer of dendritic cells on polymicrobial sepsis induced by cecal content injection. Microbiol. Immunol. 2013, 57, 633–639. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Hu, Z.; Su, J.; Huo, Y.; Tan, B.; Wang, X.; Liu, Y. Small interfering RNA targeting toll-like receptor 9 protects mice against polymicrobial septic acute kidney injury. Nephron Exp. Nephrol. 2012, 122, 51–61. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Mizutani, T.; Negishi, H.; Shimada, N.; Suzuki, N.; Ohba, Y.; Takaoka, A.; Yeh, W.C.; Taniguchi, T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 15416–15421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar] [PubMed]
- Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Chernikov, V.P.; Prusov, A.N.; Kireev, I.I.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mitochondrial damage and mitochondria-targeted antioxidant protection in LPS-induced acute kidney injury. Antioxidants 2019, 8, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remick, D.G.; Newcomb, D.E.; Bolgos, G.L.; Call, D.R. Comparison of the mortality and inflammatory response of two models of sepsis: Lipopolysaccharide vs. cecal ligation and puncture. Shock 2000, 13, 110–116. [Google Scholar] [CrossRef]
- Rittirsch, D.; Hoesel, L.M.; Ward, P.A. The disconnect between animal models of sepsis and human sepsis. J. Leukoc. Biol. 2007, 81, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosmann, M.; Ward, P.A. The inflammatory response in sepsis. Trends Immunol. 2013, 34, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Umbro, I.; Gentile, G.; Tinti, F.; Muiesan, P.; Mitterhofer, A.P. Recent advances in pathophysiology and biomarkers of sepsis-induced acute kidney injury. J. Infect. 2016, 72, 131–142. [Google Scholar] [CrossRef]
- Gonçalves, G.M.; Zamboni, D.S.; Cĝmara, N.O.S. The role of innate immunity in septic acute kidney injuries. Shock 2010, 34, 22–26. [Google Scholar] [CrossRef]
- Fry, D.E. Sepsis, systemic inflammatory response, and multiple organ dysfunction: The mystery continues. Am. Surg. 2012, 78, 1–8. [Google Scholar] [CrossRef]
- Kalakeche, R.; Hato, T.; Rhodes, G.; Dunn, K.W.; El-Achkar, T.M.; Plotkin, Z.; Sandoval, R.M.; Dagher, P.C. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J. Am. Soc. Nephrol. 2011, 22, 1505–1516. [Google Scholar] [CrossRef]
- Dellepiane, S.; Marengo, M.; Cantaluppi, V. Detrimental cross-talk between sepsis and acute kidney injury: New pathogenic mechanisms, early biomarkers and targeted therapies. Crit. Care 2016, 20, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, B.; Krick, S.; Dhillon, N.; Lerner, S.M.; Ames, S.; Bromberg, J.S.; Lin, M.; Walsh, L.; Vella, J.; Fischereder, M.; et al. Donor toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc. Natl. Acad. Sci. USA 2009, 106, 3390–3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfs, T.G.A.M.; Buurman, W.A.; van Schadewijk, A.; de Vries, B.; Daemen, M.A.R.C.; Hiemstra, P.S.; van ’t Veer, C. In Vivo Expression of Toll-Like Receptor 2 and 4 by Renal Epithelial Cells: IFN-γ and TNF-α Mediated Up-Regulation During Inflammation. J. Immunol. 2002, 168, 1286–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, H.; Leelahavanichkul, A.; Tsunoda, S.; Dear, J.W.; Takahashi, Y.; Ito, S.; Hu, X.; Zhou, H.; Doi, K.; Childs, R.; et al. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2008, 294, F1050–F1058. [Google Scholar] [CrossRef] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Kwan, B.C.H.; Chow, K.M.; Leung, C.B.; Law, M.C.; Cheng, P.M.S.; Yu, V.; Li, P.K.T.; Szeto, C.C. Circulating bacterial-derived DNA fragments as a marker of systemic inflammation in peritoneal dialysis. Nephrol. Dial. Transplant. 2013, 28, 2139–2145. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Sun, H.; Xu, Y.; Xing, G.; Wang, X. Curcumin plays a protective role against septic acute kidney injury by regulating the TLR9 signaling pathway. Transl. Androl. Urol. 2021, 10, 2103–2112. [Google Scholar] [CrossRef]
- Plitas, G.; Burt, B.M.; Nguyen, H.M.; Bamboat, Z.M.; DeMatteo, R.P. Toll-like receptor 9 inhibition reduces mortality in polymicrobial sepsis. J. Exp. Med. 2008, 205, 1277–1283. [Google Scholar] [CrossRef] [Green Version]
- Cantaluppi, V.; Weber, V.; Lauritano, C.; Figliolini, F.; Beltramo, S.; Biancone, L.; De Cal, M.; Cruz, D.; Ronco, C.; Segoloni, G.P.; et al. Protective effect of resin adsorption on septic plasma-induced tubular injury. Crit. Care 2010, 14, R4. [Google Scholar] [CrossRef] [Green Version]
- Mariano, F.; Cantaluppi, V.; Stella, M.; Romanazzi, G.M.; Assenzio, B.; Cairo, M.; Biancone, L.; Triolo, G.; Ranieri, V.M.; Camussi, G. Circulating plasma factors induce tubular and glomerular alterations in septic burns patients. Crit. Care 2008, 12, R42. [Google Scholar] [CrossRef] [Green Version]
- Lerolle, N.; Nochy, D.; Guérot, E.; Bruneval, P.; Fagon, J.Y.; Diehl, J.L.; Hill, G. Histopathology of septic shock induced acute kidney injury: Apoptosis and leukocytic infiltration. Intensive Care Med. 2010, 36, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantaluppi, V.; Medica, D.; Quercia, A.D.; Dellepiane, S.; Figliolini, F.; Virzì, G.M.; Brocca, A.; Quaglia, M.; Marengo, M.; Olivieri, C.; et al. Perfluorocarbon solutions limit tubular epithelial cell injury and promote CD133+ kidney progenitor differentiation: Potential use in renal assist devices for sepsis-associated acute kidney injury and multiple organ failure. Nephrol. Dial. Transplant. 2018, 33, 1110–1121. [Google Scholar] [CrossRef]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA is released by shock and activates neutrophils via P38 map kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Cantaluppi, V.; Quercia, A.D.; Dellepiane, S.; Ferrario, S.; Camussi, G.; Biancone, L. Interaction between systemic inflammation and renal tubular epithelial cells. Nephrol. Dial. Transplant. 2014, 29, 2004–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Zhang, J.; Tian, J.; Virzì, G.M.; Digvijay, K.; Cueto, L.; Yin, Y.; Rosner, M.H.; Ronco, C. Mitochondria in sepsis-induced AKI. J. Am. Soc. Nephrol. 2019, 30, 1151–1161. [Google Scholar] [CrossRef]
- Pathak, E.; MacMillan-Crow, L.A.; Mayeux, P.R. Role of mitochondrial oxidants in an in vitro model of sepsis-induced renal injury. J. Pharmacol. Exp. Ther. 2012, 340, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Quoilin, C.; Mouithys-Mickalad, A.; Lécart, S.; Fontaine-Aupart, M.P.; Hoebeke, M. Evidence of oxidative stress and mitochondrial respiratory chain dysfunction in an in vitro model of sepsis-induced kidney injury. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 1790–1800. [Google Scholar] [CrossRef] [Green Version]
- Kashani, K.; Al-Khafaji, A.; Ardiles, T.; Artigas, A.; Bagshaw, S.M.; Bell, M.; Bihorac, A.; Birkhahn, R.; Cely, C.M.; Chawla, L.S.; et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit. Care 2013, 17, R25. [Google Scholar] [CrossRef] [Green Version]
- Gomez, H.; Ince, C.; De Backer, D.; Pickkers, P.; Payen, D.; Hotchkiss, J.; Kellum, J.A. A unified theory of sepsis-induced acute kidney injury: Inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 2014, 41, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Post, E.H.; Kellum, J.A.; Bellomo, R.; Vincent, J.L. Renal perfusion in sepsis: From macro- to microcirculation. Kidney Int. 2017, 91, 45–60. [Google Scholar] [CrossRef]
- Ergin, B.; Kapucu, A.; Demirci-Tansel, C.; Ince, C. The renal microcirculation in sepsis. Nephrol. Dial. Transplant. 2015, 30, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calzavacca, P.; Evans, R.G.; Bailey, M.; Bellomo, R.; May, C.N. Cortical and medullary tissue perfusion and oxygenation in experimental septic acute kidney injury. Crit. Care Med. 2015, 43, e431–e439. [Google Scholar] [CrossRef] [PubMed]
- Yatim, K.M.; Gosto, M.; Humar, R.; Williams, A.L.; Oberbarnscheidt, M.H. Renal dendritic cells sample blood-borne antigen and guide T-cell migration to the kidney by means of intravascular processes. Kidney Int. 2016, 90, 818–827. [Google Scholar] [CrossRef]
- Strother, R.K.; Danahy, D.B.; Kotov, D.I.; Kucaba, T.A.; Zacharias, Z.R.; Griffith, T.S.; Legge, K.L.; Badovinac, V.P. Polymicrobial Sepsis Diminishes Dendritic Cell Numbers and Function Directly Contributing to Impaired Primary CD8 T Cell Responses In Vivo. J. Immunol. 2016, 197, 4301–4311. [Google Scholar] [CrossRef]
- Grimaldi, D.; Louis, S.; Pène, F.; Sirgo, G.; Rousseau, C.; Claessens, Y.E.; Vimeux, L.; Cariou, A.; Mira, J.P.; Hosmalin, A.; et al. Profound and persistent decrease of circulating dendritic cells is associated with ICU-acquired infection in patients with septic shock. Intensive Care Med. 2011, 37, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.Y.; Qi, D.; Yu, C.; Zhao, F.; Liu, T.; Zhang, Z.K.; Yang, M.Y.; Zhang, L.M.; Chen, D.Q.; Du, Y. Paeonol protects endotoxin-induced acute kidney injury: Potential mechanism of inhibiting TLR4-NF-κB signal pathway. Oncotarget 2016, 7, 39497–39510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Schaller, M.A.; Dou, Y.; Hogaboam, C.M.; Kunkel, S.L. Dendritic cells at the interface of innate and acquired immunity: The role for epigenetic changes. J. Leukoc. Biol. 2008, 83, 439–446. [Google Scholar] [CrossRef]
- Wen, H.; Dou, Y.; Hogaboam, C.M.; Kunkel, S.L. Epigenetic regulation of dendritic cell-derived interleukin-12 facilitates immunosuppression after a severe innate immune response. Blood 2008, 111, 1797–1804. [Google Scholar] [CrossRef]
- Poehlmann, H.; Schefold, J.C.; Zuckermann-Becker, H.; Volk, H.D.; Meisel, C. Phenotype changes and impaired function of dendritic cell subsets in patients with sepsis: A prospective observational analysis. Crit. Care 2009, 13, R119. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Zhao, Z.; Zhu, W.; Yang, T.; Deng, X.; Bao, R. CD155 blockade improves survival in experimental sepsis by reversing dendritic cell dysfunction. Biochem. Biophys. Res. Commun. 2017, 490, 283–289. [Google Scholar] [CrossRef]
- Roquilly, A.; McWilliam, H.E.G.; Jacqueline, C.; Tian, Z.; Cinotti, R.; Rimbert, M.; Wakim, L.; Caminschi, I.; Lahoud, M.H.; Belz, G.T.; et al. Local Modulation of Antigen-Presenting Cell Development after Resolution of Pneumonia Induces Long-Term Susceptibility to Secondary Infections. Immunity 2017, 47, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roquilly, A.; Villadangos, J.A. The role of dendritic cell alterations in susceptibility to hospital-acquired infections during critical-illness related immunosuppression. Mol. Immunol. 2015, 68, 120–123. [Google Scholar] [CrossRef]
- Sester, D.P.; Stacey, K.J.; Sweet, M.J.; Beasley, S.J.; Cronau, S.L.; Hume, D.A. The actions of bacterial DNA on murine macrophages. J. Leukoc. Biol. 1999, 66, 542–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, Y.; Tsuji, T.; Nagata, S.; Tsuji, N.; Fujikura, T.; Ohashi, N.; Kato, A.; Miyajima, H.; Yasuda, H. IL-17A activated by Toll-like receptor 9 contributes to the development of septic acute kidney injury. Am. J. Physiol. Ren. Physiol. 2020, 318, F238–F247. [Google Scholar] [CrossRef]
- Mócsai, A.; Ruland, J.; Tybulewicz, V.L.J. The SYK tyrosine kinase: A crucial player in diverse biological functions. Nat. Rev. Immunol. 2010, 10, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Al-Harbi, N.O.; Nadeem, A.; Ahmad, S.F.; Alanazi, M.M.; Aldossari, A.A.; Alasmari, F. Amelioration of sepsis-induced acute kidney injury through inhibition of inflammatory cytokines and oxidative stress in dendritic cells and neutrophils respectively in mice: Role of spleen tyrosine kinase signaling. Biochimie 2019, 158, 102–110. [Google Scholar] [CrossRef]
- Kaplan, J.; Nowell, M.; Chima, R.; Zingarelli, B. Pioglitazone reduces inflammation through inhibition of NF-κB in polymicrobial sepsis. Innate Immun. 2014, 20, 519–528. [Google Scholar] [CrossRef]
- Gasparini, C.; Feldmann, M. NF-κB as a Target for Modulating Inflammatory Responses. Curr. Pharm. Des. 2012, 18, 5735–5745. [Google Scholar] [CrossRef]
- Ní Gabhann, J.; Spence, S.; Wynne, C.; Smith, S.; Byrne, J.C.; Coffey, B.; Stacey, K.; Kissenpfennig, A.; Johnston, J.; Jefferies, C.A. Defects in acute responses to TLR4 in Btk-deficient mice result in impaired dendritic cell-induced IFN-γ production by natural killer cells. Clin. Immunol. 2012, 142, 373–382. [Google Scholar] [CrossRef]
- Crofford, L.J.; Nyhoff, L.E.; Sheehan, J.H.; Kendall, P.L. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev. Clin. Immunol. 2016, 12, 763–773. [Google Scholar]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Ibrahim, K.E.; Alqahtani, F.; Alanazi, W.A.; Mahmood, H.M.; Alsanea, S.; Attia, S.M. Bruton’s tyrosine kinase inhibition attenuates oxidative stress in systemic immune cells and renal compartment during sepsis-induced acute kidney injury in mice. Int. Immunopharmacol. 2021, 90, 107123. [Google Scholar] [CrossRef] [PubMed]
- Pázmándi, K.; Sütő, M.; Fekete, T.; Varga, A.; Boldizsár, E.; Boldogh, I.; Bácsi, A. Oxidized base 8-oxoguanine, a product of DNA repair processes, contributes to dendritic cell activation. Free Radic. Biol. Med. 2019, 143, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.W.; Gresnigt, M.S.; Joosten, L.A.B.; Netea, M.G. Cellular metabolism of myeloid cells in sepsis. J. Leukoc. Biol. 2017, 101, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.C.; Scicluna, B.P.; Arts, R.J.W.; Gresnigt, M.S.; Lachmandas, E.; Giamarellos-Bourboulis, E.J.; Kox, M.; Manjeri, G.R.; Wagenaars, J.A.L.; Cremer, O.L.; et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol. 2016, 17, 406–413. [Google Scholar] [CrossRef]
- Weisheit, C.K.; Klüners, A.; Wild, L.; Casalter, A.; Heilmann-Heimbach, S.; Sivalingam, S.; Kleiner, J.L.; Ehrentraut, S.F.; Hoeft, A.; Frede, S.; et al. Sustained Immunoparalysis in Endotoxin-Tolerized Monocytic Cells. Mediat. Inflamm. 2020, 2020, 8294342. [Google Scholar] [CrossRef]
- Shalova, I.N.; Lim, J.Y.; Chittezhath, M.; Zinkernagel, A.S.; Beasley, F.; Hernández-Jiménez, E.; Toledano, V.; Cubillos-Zapata, C.; Rapisarda, A.; Chen, J.; et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity 2015, 42, 484–498. [Google Scholar] [CrossRef] [Green Version]
- Landelle, C.; Lepape, A.; Voirin, N.; Tognet, E.; Venet, F.; Bohé, J.; Vanhems, P.; Monneret, G. Low monocyte human leukocyte antigen-DR is independently associated with nosocomial infections after septic shock. Intensive Care Med. 2010, 36, 1859–1866. [Google Scholar] [CrossRef]
- Shao, R.; Fang, Y.; Yu, H.; Zhao, L.; Jiang, Z.; Li, C.S. Monocyte programmed death ligand-1 expression after 3-4 days of sepsis is associated with risk stratification and mortality in septic patients: A prospective cohort study. Crit. Care 2016, 20, 124. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Ma, J.; Sheng, L.; Zhang, D.; Chen, X.; Yang, J.; Wang, D. Total coumarins from Hydrangea paniculata show renal protective effects in lipopolysaccharide-induced acute kidney injury via anti-inflammatory and antioxidant activities. Front. Pharmacol. 2017, 8, 872. [Google Scholar] [CrossRef] [Green Version]
- Aslan, A.; Van Den Heuvel, M.C.; Stegeman, C.A.; Popa, E.R.; Leliveld, A.M.; Molema, G.; Zijlstra, J.G.; Moser, J.; Van Meurs, M. Kidney histopathology in lethal human sepsis. Crit. Care 2018, 22, 359. [Google Scholar] [CrossRef] [Green Version]
- Han, H.I.; Skvarca, L.B.; Espiritu, E.B.; Davidson, A.J.; Hukriede, N.A. The role of macrophages during acute kidney injury: Destruction and repair. Pediatr. Nephrol. 2019, 34, 561–569. [Google Scholar] [CrossRef]
- Gottlieb, R.A. Cell death pathways in acute ischemia/reperfusion injury. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 233–238. [Google Scholar] [CrossRef]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.S.; Ruhrberg, C.; Cantley, L.G. Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, R.; Wang, C.; Zhang, F.; Zhao, M.; Liu, S.; Liao, G.; Li, L.; Chen, Y.; Cheng, J.; Liu, J.; et al. Peritoneal M2 macrophage transplantation as a potential cell therapy for enhancing renal repair in acute kidney injury. J. Cell. Mol. Med. 2020, 24, 3314–3327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Z.; Wang, X.; Wang, Y.; Niu, A.; Wang, S.; Zou, C.; Harris, R.C. IL-4/IL-13–mediated polarization of renal macrophages/dendritic cells to an M2a phenotype is essential for recovery from acute kidney injury. Kidney Int. 2017, 91, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.Z.; Yao, B.; Yang, S.; Jiang, L.; Wang, S.; Fan, X.; Yin, H.; Wong, K.; Miyazawa, T.; Chen, J.; et al. CSF-1 signaling mediates recovery from acute kidney injury. J. Clin. Investig. 2012, 122, 4519–4532. [Google Scholar] [CrossRef] [Green Version]
- Belliere, J.; Casemayou, A.; Ducasse, L.; Zakaroff-Girard, A.; Martins, F.; Iacovoni, J.S.; Guilbeau-Frugier, C.; Buffin-Meyer, B.; Pipy, B.; Chauveau, D.; et al. Specific macrophage subtypes influence the progression of rhabdomyolysis-induced kidney injury. J. Am. Soc. Nephrol. 2015, 26, 1363–1377. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.; Guo, A.; Han, X.; Wu, S.; Chen, C.; Luo, C.; Li, H.; Li, S.; Hei, Z. Aerosol inhalation of a hydrogen-rich solution restored septic renal function. Aging 2019, 11, 12097–12113. [Google Scholar] [CrossRef]
- Pohl, J.; Papathanasiou, M.; Heisler, M.; Stock, P.; Kelm, M.; Hendgen-Cotta, U.B.; Rassaf, T.; Luedike, P. Renal replacement therapy neutralizes elevated MIF levels in septic shock. J. Intensive Care 2016, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- Cairo, G.; Recalcati, S.; Mantovani, A.; Locati, M. Iron trafficking and metabolism in macrophages: Contribution to the polarized phenotype. Trends Immunol. 2011, 32, 241–247. [Google Scholar] [CrossRef]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Mertens, C.; Kuchler, L.; Sola, A.; Guiteras, R.; Grein, S.; Brüne, B.; Von Knethen, A.; Jung, M. Macrophage-derived iron-bound lipocalin-2 correlates with renal recovery markers following sepsis-induced kidney damage. Int. J. Mol. Sci. 2020, 21, 7527. [Google Scholar] [CrossRef] [PubMed]
- Mertens, C.; Mora, J.; Ören, B.; Grein, S.; Winslow, S.; Scholich, K.; Weigert, A.; Malmström, P.; Forsare, C.; Fernö, M.; et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology 2018, 7, e1408751. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kaplan, M.J. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat. Rev. Nephrol. 2016, 12, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Margraf, A.; Ley, K.; Zarbock, A. Neutrophil Recruitment: From Model Systems to Tissue-Specific Patterns. Trends Immunol. 2019, 40, 613–634. [Google Scholar] [CrossRef]
- Herter, J.M.; Rossaint, J.; Spieker, T.; Zarbock, A. Adhesion molecules involved in neutrophil recruitment during sepsis-induced acute kidney injury. J. Innate Immun. 2014, 6, 597–606. [Google Scholar] [CrossRef]
- Rossaint, J.; Spelten, O.; Kässens, N.; Mueller, H.; Van Aken, H.K.; Singbartl, K.; Zarbock, A. Acute loss of renal function attenuates slow leukocyte rolling and transmigration by interfering with intracellular signaling. Kidney Int. 2011, 80, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Sônego, F.; Castanheira, F.V.e.S.; Ferreira, R.G.; Kanashiro, A.; Leite, C.A.V.G.; Nascimento, D.C.; Colón, D.F.; Borges, V. de F.; Alves-Filho, J.C.; Cunha, F.Q. Paradoxical roles of the neutrophil in sepsis: Protective and deleterious. Front. Immunol. 2016, 7, 155. [Google Scholar] [CrossRef] [Green Version]
- Castoldi, A.; Braga, T.T.; Correa-Costa, M.; Aguiar, C.F.; Bassi, Ê.J.; Correa-Silva, R.; Elias, R.M.; Salvador, F.; Moraes-Vieira, P.M.; Cenedeze, M.A.; et al. TLR2, TLR4 and the Myd88 signaling pathway are crucial for neutrophil migration in acute kidney injury induced by sepsis. PLoS ONE 2012, 7, e37584. [Google Scholar] [CrossRef]
- Bonavia, A.; Singbartl, K. A review of the role of immune cells in acute kidney injury. Pediatr. Nephrol. 2018, 33, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, S.; Pickkers, P.; Bouw, M.P.W.J.M.; Draisma, A.; van der Hoeven, J.G.; Peters, W.H.M.; Smits, P.; Russel, F.G.M.; Masereeuw, R. Upregulation of renal inducible nitric oxide synthase during human endotoxemia and sepsis is associated with proximal tubule injury. Clin. J. Am. Soc. Nephrol. 2006, 1, 853–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.A.; Noel, S.; Sadasivam, M.; Hamad, A.R.A.; Rabb, H. Role of Immune Cells in Acute Kidney Injury and Repair. Nephron 2017, 137, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Demaret, J.; Venet, F.; Friggeri, A.; Cazalis, M.-A.; Plassais, J.; Jallades, L.; Malcus, C.; Poitevin-Later, F.; Textoris, J.; Lepape, A.; et al. Marked alterations of neutrophil functions during sepsis-induced immunosuppression. J. Leukoc. Biol. 2015, 98, 1081–1090. [Google Scholar] [CrossRef]
- Hampson, P.; Dinsdale, R.J.; Wearn, C.M.; Bamford, A.L.; Bishop, J.R.B.; Hazeldine, J.; Moiemen, N.S.; Harrison, P.; Lord, J.M. Neutrophil dysfunction, immature granulocytes, and cell-free DNA are early biomarkers of sepsis in burn-injured patients: A prospective observational cohort study. Ann. Surg. 2017, 265, 1241–1249. [Google Scholar] [CrossRef]
- Groeneveld, K.M.; Koenderman, L.; Warren, B.L.; Jol, S.; Leenen, L.P.H.; Hietbrink, F. Early decreased neutrophil responsiveness is related to late onset sepsis in multitrauma patients: An international cohort study. PLoS ONE 2017, 12, e0180145. [Google Scholar] [CrossRef] [Green Version]
- Grégoire, M.; Tadié, J.-M.; Uhel, F.; Gacouin, A.; Piau, C.; Bone, N.; Le Tulzo, Y.; Abraham, E.; Tarte, K.; Zmijewski, J.W. Frontline Science: HMGB1 induces neutrophil dysfunction in experimental sepsis and in patients who survive septic shock. J. Leukoc. Biol. 2017, 101, 1281–1287. [Google Scholar] [CrossRef]
- Wang, J.F.; Wang, Y.P.; Xie, J.; Zhao, Z.Z.; Gupta, S.; Guo, Y.; Jia, S.H.; Parodo, J.; Marshall, J.C.; Deng, X.M. Upregulated PD-L1 delays human neutrophil apoptosis and promotes lung injury in an experimental mouse model of sepsis. Blood 2021, 138, 806–810. [Google Scholar] [CrossRef]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Ibrahim, K.E.; Sarawi, W.; Attia, S.M.; Alasmari, A.F.; Alqarni, S.A.; Alfradan, A.S.; Bakheet, S.A.; et al. Role of ITK signaling in acute kidney injury in mice: Amelioration of acute kidney injury associated clinical parameters and attenuation of inflammatory transcription factor signaling in CD4+ T cells by ITK inhibition. Int. Immunopharmacol. 2021, 99, 108028. [Google Scholar] [CrossRef]
- Takasu, O.; Gaut, J.P.; Watanabe, E.; To, K.; Fagley, R.E.; Sato, B.; Jarman, S.; Efimov, I.R.; Janks, D.L.; Srivastava, A.; et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am. J. Respir. Crit. Care Med. 2013, 187, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Törnblom, S.; Nisula, S.; Vaara, S.T.; Poukkanen, M.; Andersson, S.; Pettilä, V.; Pesonen, E. Neutrophil activation in septic acute kidney injury: A post hoc analysis of the FINNAKI study. Acta Anaesthesiol. Scand. 2019, 63, 1390–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Törnblom, S.; Nisula, S.; Vaara, S.T.; Poukkanen, M.; Andersson, S.; Pettilä, V.; Pesonen, E. Early prolonged neutrophil activation in critically ill patients with sepsis. Innate Immun. 2021, 27, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.E.; Rickassel, C.; Healy, H.; Kassianos, A.J. Natural killer cells in kidney health and disease. Front. Immunol. 2019, 10, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, S.; Nakashima, H.; Nakashima, M.; Kinoshita, M. Antitumor immunity produced by the liver kupffer cells, NK Cells, NKT cells, and CD8 + CD122 + T cells. Clin. Dev. Immunol. 2011, 2011, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emoto, M.; Miyamoto, M.; Yoshizawa, I.; Emoto, Y.; Schaible, U.E.; Kita, E.; Kaufmann, S.H.E. Critical Role of NK Cells Rather Than Vα14 + NKT Cells in Lipopolysaccharide-Induced Lethal Shock in Mice. J. Immunol. 2002, 169, 1426–1432. [Google Scholar] [CrossRef] [Green Version]
- Kerr, A.R.; Kirkham, L.A.S.; Kadioglu, A.; Andrew, P.W.; Garside, P.; Thompson, H.; Mitchell, T.J. Identification of a detrimental role for NK cells in pneumococcal pneumonia and sepsis in immunocompromised hosts. Microbes Infect. 2005, 7, 845–852. [Google Scholar] [CrossRef]
- Badgwell, B.; Parihar, R.; Magro, C.; Dierksheide, J.; Russo, T.; Carson, W.E. Natural killer cells contribute to the lethality of a murine model of Escherichi coli infection. Surgery 2002, 132, 205–212. [Google Scholar] [CrossRef]
- Etogo, A.O.; Nunez, J.; Lin, C.Y.; Toliver-Kinsky, T.E.; Sherwood, E.R. NK but Not CD1-Restricted NKT Cells Facilitate Systemic Inflammation during Polymicrobial Intra-Abdominal Sepsis. J. Immunol. 2008, 180, 6334–6345. [Google Scholar] [CrossRef] [Green Version]
- Uchida, T.; Ito, S.; Kumagai, H.; Oda, T.; Nakashima, H.; Seki, S. Roles of natural killer T cells and natural killer cells in kidney injury. Int. J. Mol. Sci. 2019, 20, 2487. [Google Scholar] [CrossRef] [Green Version]
- Uchida, T.; Seki, S.; Oda, T. Infections, reactions of natural killer T cells and natural killer cells, and kidney injury. Int. J. Mol. Sci. 2022, 23, 479. [Google Scholar] [CrossRef] [PubMed]
- Forel, J.M.; Chiche, L.; Thomas, G.; Mancini, J.; Farnarier, C.; Cognet, C.; Guervilly, C.; Daumas, A.; Vély, F.; Xéridat, F.; et al. Phenotype and Functions of Natural Killer Cells in Critically-Ill Septic Patients. PLoS ONE 2012, 7, e50446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peraldi, M.-N.; Berrou, J.; Dulphy, N.; Seidowsky, A.; Haas, P.; Boissel, N.; Metivier, F.; Randoux, C.; Kossari, N.; Guérin, A.; et al. Oxidative Stress Mediates a Reduced Expression of the Activating Receptor NKG2D in NK Cells from End-Stage Renal Disease Patients. J. Immunol. 2009, 182, 1696–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwatani, H.; Nagasawa, Y.; Yamamoto, R.; Iio, K.; Mizui, M.; Horii, A.; Kitahara, T.; Inohara, H.; Kumanogoh, A.; Imai, E.; et al. CD16+CD56+ cells are a potential culprit for hematuria in IgA nephropathy. Clin. Exp. Nephrol. 2015, 19, 216–224. [Google Scholar] [CrossRef]
- Law, B.M.P.; Wilkinson, R.; Wang, X.; Kildey, K.; Lindner, M.; Rist, M.J.; Beagley, K.; Healy, H.; Kassianos, A.J. Interferon-γ production by tubulointerstitial human CD56bright natural killer cells contributes to renal fibrosis and chronic kidney disease progression. Kidney Int. 2017, 92, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Badawy, A.A.B. Kynurenine pathway of tryptophan metabolism: Regulatory and functional aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [Green Version]
- Harden, J.L.; Egilmez, N.K. Indoleamine 2,3-dioxygenase and dendritic cell tolerogenicity. Immunol. Investig. 2012, 41, 738–764. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.F.; Wang, H.S.; Wang, H.; Zhang, F.; Wang, K.F.; Guo, Q.; Zhang, G.; Cai, S.H.; Du, J. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: Focus on macrophage polarization of THP-1 cells. Cell. Immunol. 2014, 289, 42–48. [Google Scholar] [CrossRef]
- Odemuyiwa, S.O.; Ghahary, A.; Li, Y.; Puttagunta, L.; Lee, J.E.; Musat-Marcu, S.; Ghahary, A.; Moqbel, R. Cutting Edge: Human Eosinophils Regulate T Cell Subset Selection through Indoleamine 2,3-Dioxygenase. J. Immunol. 2004, 173, 5909–5913. [Google Scholar] [CrossRef]
- De Ravin, S.S.; Zarember, K.A.; Long-Priel, D.; Chan, K.C.; Fox, S.D.; Gallin, J.I.; Kuhns, D.B.; Malech, H.L. Tryptophan/kynurenine metabolism in human leukocytes is independent of superoxide and is fully maintained in chronic granulomatous disease. Blood 2010, 116, 1755–1760. [Google Scholar] [CrossRef] [Green Version]
- Kai, S.; Goto, S.; Tahara, K.; Sasaki, A.; Kawano, K.; Kitano, S. Inhibition of indoleamine 2,3-dioxygenase suppresses NK cell activity and accelerates tumor growth. J. Exp. Ther. Oncol. 2003, 3, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Krupa, A.; Kowalska, I. The kynurenine pathway—new linkage between innate and adaptive immunity in autoimmune endocrinopathies. Int. J. Mol. Sci. 2021, 22, 9879. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, W.; Kocki, T.; Pilat, J.; Parada-Turska, J.; Malbrain, M.L.N.G. Changes in plasma kynurenic acid concentration in septic shock patients undergoing continuous veno-venous haemofiltration. Inflammation 2014, 37, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwaki, T.; Bennion, B.G.; Stenson, E.K.; Lynn, J.C.; Otinga, C.; Djukovic, D.; Raftery, D.; Fei, L.; Wong, H.R.; Liles, W.C.; et al. PPARα contributes to protection against metabolic and inflammatory derangements associated with acute kidney injury in experimental sepsis. Physiol. Rep. 2019, 7, e14078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794. [Google Scholar] [CrossRef] [Green Version]
- Agudelo, L.Z.; Femenía, T.; Orhan, F.; Porsmyr-Palmertz, M.; Goiny, M.; Martinez-Redondo, V.; Correia, J.C.; Izadi, M.; Bhat, M.; Schuppe-Koistinen, I.; et al. Skeletal muscle PGC-1α1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 2014, 159, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Däubener, W.; Mackenzie, C.R. IFN-γ activated indoleamine 2,3-dioxygenase activity in human cells is an antiparasitic and an antibacterial effector mechanism. Adv. Exp. Med. Biol. 2000, 467, 517–524. [Google Scholar]
- Niño-Castro, A.; Abdullah, Z.; Popov, A.; Thabet, Y.; Beyer, M.; Knolle, P.; Domann, E.; Chakraborty, T.; Schmidt, S.V.; Schultze, J.L. The IDO1-induced kynurenines play a major role in the antimicrobial effect of human myeloid cells against Listeria monocytogenes. Innate Immun. 2014, 20, 401–411. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Tattevin, P.; Monnier, D.; Tribut, O.; Dulong, J.; Bescher, N.; Mourcin, F.; Uhel, F.; Le Tulzo, Y.; Tarte, K. Enhanced indoleamine 2,3-dioxygenase activity in patients with severe sepsis and septic shock. J. Infect. Dis. 2010, 201, 956–966. [Google Scholar] [CrossRef]
- Huttunen, R.; Syrjänen, J.; Aittoniemi, J.; Oja, S.S.; Raitala, A.; Laine, J.; Pertovaara, M.; Vuento, R.; Huhtala, H.; Hurme, M. High activity of indoleamine 2,3 dioxygenase enzyme predicts disease severity and case fatality in bacteremic patients. Shock 2010, 33, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Schefold, J.C.; Zeden, J.P.; Pschowski, R.; Hammoud, B.; Fotopoulou, C.; Hasper, D.; Fusch, G.; Von Haehling, S.; Volk, H.D.; Meisel, C.; et al. Treatment with granulocytemacrophage colony-stimulating factor is associated with reduced indoleamine 2,3-dioxygenase activity and kynurenine pathway catabolites in patients with severe sepsis and septic shock. Scand. J. Infect. Dis. 2010, 42, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Meisel, C.; Schefold, J.C.; Pschowski, R.; Baumann, T.; Hetzger, K.; Gregor, J.; Weber-Carstens, S.; Hasper, D.; Keh, D.; Zuckermann, H.; et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: A double-blind, randomized, placebo-controlled multicenter trial. Am. J. Respir. Crit. Care Med. 2009, 180, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.D.; Jeong, Y.I.; Lee, C.M.; Noh, K.T.; Jeong, S.K.; Chun, S.H.; Choi, O.H.; Park, W.S.; Han, J.; Shin, Y.K.; et al. COX-2 and PGE2 signaling is essential for the regulation of IDO expression by curcumin in murine bone marrow-derived dendritic cells. Int. Immunopharmacol. 2010, 10, 760–768. [Google Scholar] [CrossRef]
- Jung, I.D.; Lee, M.-G.; Chang, J.H.; Lee, J.S.; Jeong, Y.-I.; Lee, C.-M.; Park, W.S.; Han, J.; Seo, S.-K.; Lee, S.Y.; et al. Blockade of Indoleamine 2,3-Dioxygenase Protects Mice against Lipopolysaccharide-Induced Endotoxin Shock. J. Immunol. 2009, 182, 3146–3154. [Google Scholar] [CrossRef] [Green Version]
- Hoshi, M.; Osawa, Y.; Ito, H.; Ohtaki, H.; Ando, T.; Takamatsu, M.; Hara, A.; Saito, K.; Seishima, M. Blockade of indoleamine 2,3-dioxygenase reduces mortality from peritonitis and sepsis in mice by regulating functions of CD11b+ peritoneal cells. Infect. Immun. 2014, 82, 4487–4495. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Huang, L.; Bradley, J.; Liu, K.; Bardhan, K.; Ron, D.; Mellor, A.L.; Munn, D.H.; McGaha, T.L. GCN2-Dependent Metabolic Stress Is Essential for Endotoxemic Cytokine Induction and Pathology. Mol. Cell. Biol. 2014, 34, 428–438. [Google Scholar] [CrossRef] [Green Version]
- Osuchowski, M.F.; Welch, K.; Siddiqui, J.; Remick, D.G. Circulating Cytokine/Inhibitor Profiles Reshape the Understanding of the SIRS/CARS Continuum in Sepsis and Predict Mortality. J. Immunol. 2006, 177, 1967–1974. [Google Scholar] [CrossRef]
- Ploder, M.; Spittler, A.; Kurz, K.; Neurauter, G.; Pelinka, L.E.; Roth, E.; Fuchs, D. Accelerated tryptophan degradation predicts poor survival in trauma and sepsis patients. Int. J. Tryptophan Res. 2010, 3, 61–67. [Google Scholar] [CrossRef]
- Schäfer, S.T.; Gessner, S.; Scherag, A.; Rump, K.; Frey, U.H.; Siffert, W.; Westendorf, A.M.; Steinmann, J.; Peters, J.; Adamzik, M. Hydrocortisone fails to abolish NF-κB1 protein nuclear translocation in deletion allele carriers of the NFKB1 promoter polymorphism (-94ins/delATTG) and is associated with increased 30-day mortality in septic shock. PLoS ONE 2014, 9, e104953. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.P.; Levy, M.; Rhodes, A.; Annane, D.; Gerlach, H.; Opal, S.M.; Sevransky, J.E.; Sprung, C.L.; Douglas, I.S.; Jaeschke, R.; et al. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013, 39, 165–228. [Google Scholar] [CrossRef] [PubMed]
- Fatokun, A.A.; Hunt, N.H.; Ball, H.J. Indoleamine 2,3-dioxygenase 2 (IDO2) and the kynurenine pathway: Characteristics and potential roles in health and disease. Amino Acids 2013, 45, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.C.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.; Abe, H.; Tsuruta, S.; Chiba, S.; Fujii-Kuriyama, Y.; Sekiya, T.; Morita, R.; Yoshimura, A. Aryl hydrocarbon receptor protects against bacterial infection by promoting macrophage survival and reactive oxygen species production. Int. Immunol. 2014, 26, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Yamasuge, W.; Imai, S.; Kunisawa, K.; Hoshi, M.; Fujigaki, H.; Mouri, A.; Nabeshima, T.; Saito, K. Lipopolysaccharide shock reveals the immune function of indoleamine 2,3-dioxygenase 2 through the regulation of IL-6/stat3 signalling. Sci. Rep. 2018, 8, 15917. [Google Scholar] [CrossRef] [Green Version]
- Croitoru-Lamoury, J.; Lamoury, F.M.J.; Caristo, M.; Suzuki, K.; Walker, D.; Takikawa, O.; Taylor, R.; Brew, B.J. Interferon-γ regulates the proliferation and differentiation of mesenchymal stem cells via activation of indoleamine 2,3 dioxygenase (IDO). PLoS ONE 2011, 6, e14698. [Google Scholar] [CrossRef] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Salazar, F.; Awuah, D.; Negm, O.H.; Shakib, F.; Ghaemmaghami, A.M. The role of indoleamine 2,3-dioxygenase-aryl hydrocarbon receptor pathway in the TLR4-induced tolerogenic phenotype in human DCs. Sci. Rep. 2017, 7, 43337. [Google Scholar] [CrossRef]
- Venet, F.; Chung, C.S.; Kherouf, H.; Geeraert, A.; Malcus, C.; Poitevin, F.; Bohé, J.; Lepape, A.; Ayala, A.; Monneret, G. Increased circulating regulatory T cells (CD4+CD25 +CD127-) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med. 2009, 35, 678–686. [Google Scholar] [CrossRef] [Green Version]
- Darcy, C.J.; Davis, J.S.; Woodberry, T.; McNeil, Y.R.; Stephens, D.P.; Yeo, T.W.; Anstey, N.M. An observational cohort study of the kynurenine to tryptophan ratio in sepsis: Association with impaired immune and microvascular function. PLoS ONE 2011, 6, e21185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, H.; Mimura, J.; Oshima, M.; Okawa, H.; Kanno, J.; Igarashi, K.; Gonzalez, F.J.; Ikuta, T.; Kawajiri, K.; Fujii-Kuriyama, Y. Hypersensitivity of Aryl Hydrocarbon Receptor-Deficient Mice to Lipopolysaccharide-Induced Septic Shock. Mol. Cell. Biol. 2009, 29, 6391–6400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirthgen, E.; Otten, W.; Tuchscherer, M.; Tuchscherer, A.; Domanska, G.; Brenmoehl, J.; Günther, J.; Ohde, D.; Weitschies, W.; Seidlitz, A.; et al. Effects of 1-methyltryptophan on immune responses and the kynurenine pathway after lipopolysaccharide challenge in pigs. Int. J. Mol. Sci. 2018, 19, 3009. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Sorrentino, C.; Denison, M.S.; Kolaja, K.; Fielden, M.R. Induction of Cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: Results of large scale screening of pharmaceuticals and toxicants in vivo and In Vitro. Mol. Pharmacol. 2007, 71, 1475–1486. [Google Scholar] [CrossRef]
- Moyer, B.J.; Rojas, I.Y.; Murray, I.A.; Lee, S.; Hazlett, H.F.; Perdew, G.H.; Tomlinson, C.R. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors activate the aryl hydrocarbon receptor. Toxicol. Appl. Pharmacol. 2017, 323, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Kiank, C.; Zeden, J.P.; Drude, S.; Domanska, G.; Fusch, G.; Otten, W.; Schuett, C. Psychological stress-induced, IDO1-dependent tryptophan catabolism: Implications on immunosuppression in mice and humans. PLoS ONE 2010, 5, e11825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [Green Version]
- Barth, M.C.; Ahluwalia, N.; Anderson, T.J.T.; Hardy, G.J.; Sinha, S.; Alvarez-Cardona, J.A.; Pruitt, I.E.; Rhee, E.P.; Colvin, R.A.; Gerszten, R.E. Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. J. Biol. Chem. 2009, 284, 19189–19195. [Google Scholar] [CrossRef] [Green Version]
- Agaugué, S.; Perrin-Cocon, L.; Coutant, F.; André, P.; Lotteau, V. 1-Methyl-Tryptophan Can Interfere with TLR Signaling in Dendritic Cells Independently of IDO Activity. J. Immunol. 2006, 177, 2061–2071. [Google Scholar] [CrossRef] [Green Version]
- Lehner, M.D.; Ittner, J.; Bundschuh, D.S.; Van Rooijen, N.; Wendel, A.; Hartung, T. Improved innate immunity of endotoxin-tolerant mice increases resistance to Salmonella enterica serovar typhimurium infection despite attenuated cytokine response. Infect. Immun. 2001, 69, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Dudek, K.; Bednarek, D. Cellular immune response of pigeons in the conditions of endotoxin fever and pyrogenic tolerance. Pol. J. Vet. Sci. 2011, 14, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.P.; Shih, C.C.; Lin, C.Y.; Hua, C.C.; Chuang, D.Y. Serial increase of IL-12 response and human leukocyte antigen-DR expression in severe sepsis survivors. Crit. Care 2011, 15, R224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazier, W.J.; Hall, M.W. Immunoparalysis and Adverse Outcomes from Critical Illness. Pediatr. Clin. N. Am. 2008, 55, 647–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krupa, A.; Krupa, M.M.; Pawlak, K. Kynurenine Pathway—An Underestimated Factor Modulating Innate Immunity in Sepsis-Induced Acute Kidney Injury? Cells 2022, 11, 2604. https://doi.org/10.3390/cells11162604
Krupa A, Krupa MM, Pawlak K. Kynurenine Pathway—An Underestimated Factor Modulating Innate Immunity in Sepsis-Induced Acute Kidney Injury? Cells. 2022; 11(16):2604. https://doi.org/10.3390/cells11162604
Chicago/Turabian StyleKrupa, Anna, Mikolaj M. Krupa, and Krystyna Pawlak. 2022. "Kynurenine Pathway—An Underestimated Factor Modulating Innate Immunity in Sepsis-Induced Acute Kidney Injury?" Cells 11, no. 16: 2604. https://doi.org/10.3390/cells11162604