
Targeting Heat-Shock Protein 90 in Cancer: An Update on Combination Therapy


Abstract
:1. Introduction
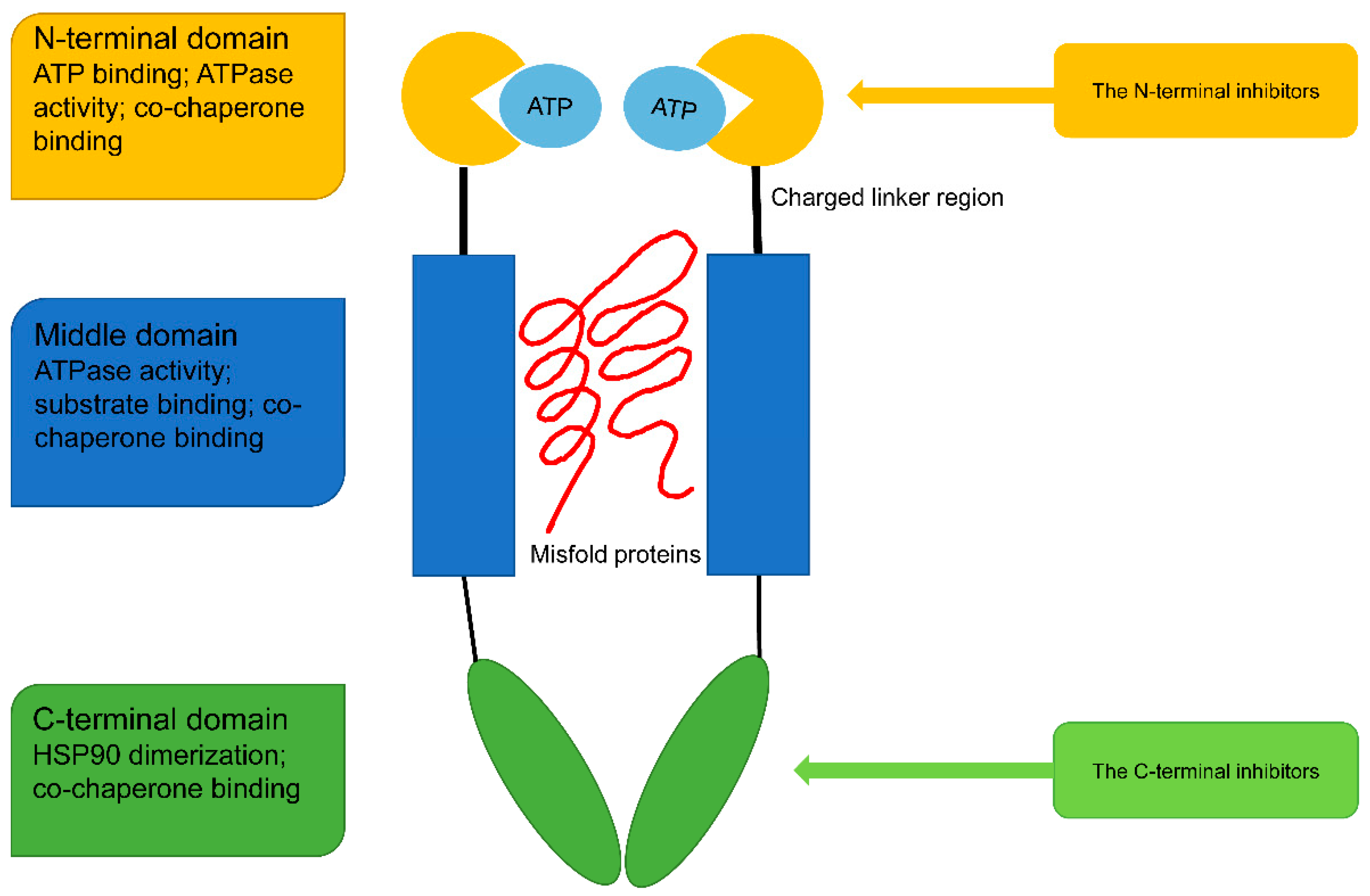
2. Structure and Function of HSP90
3. HSP90 Clients
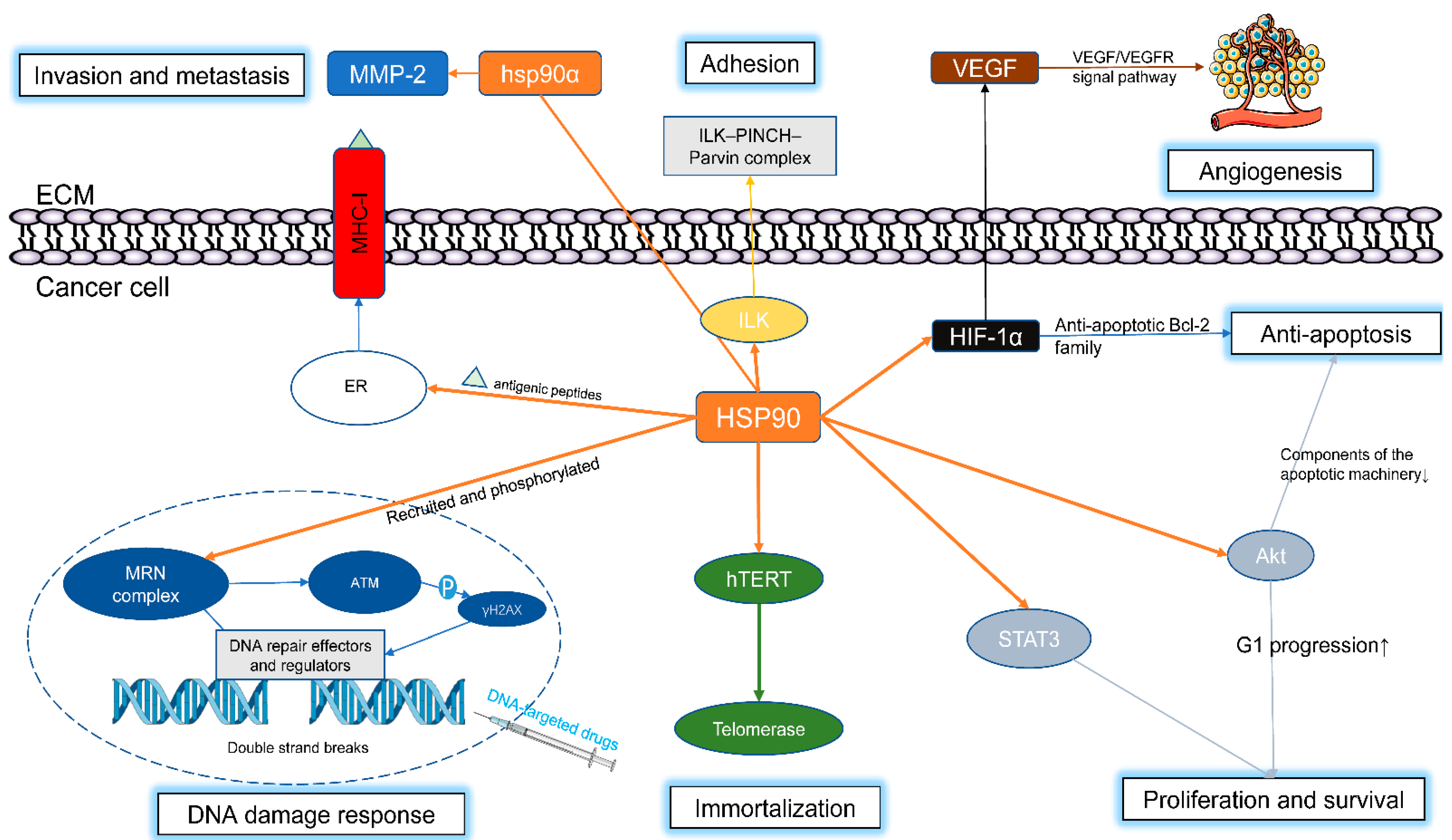
4. Roles of HSP90 in Cancer
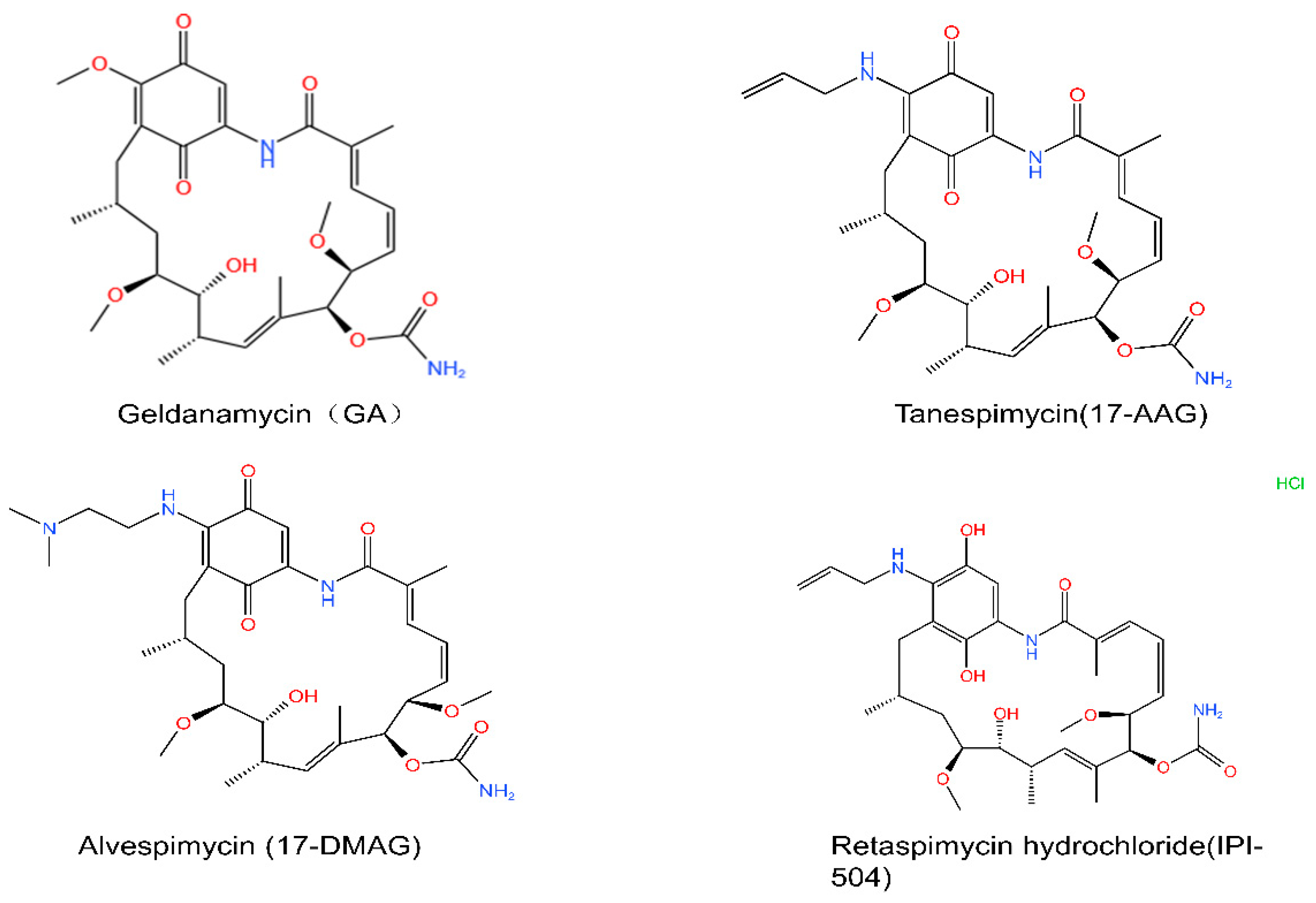
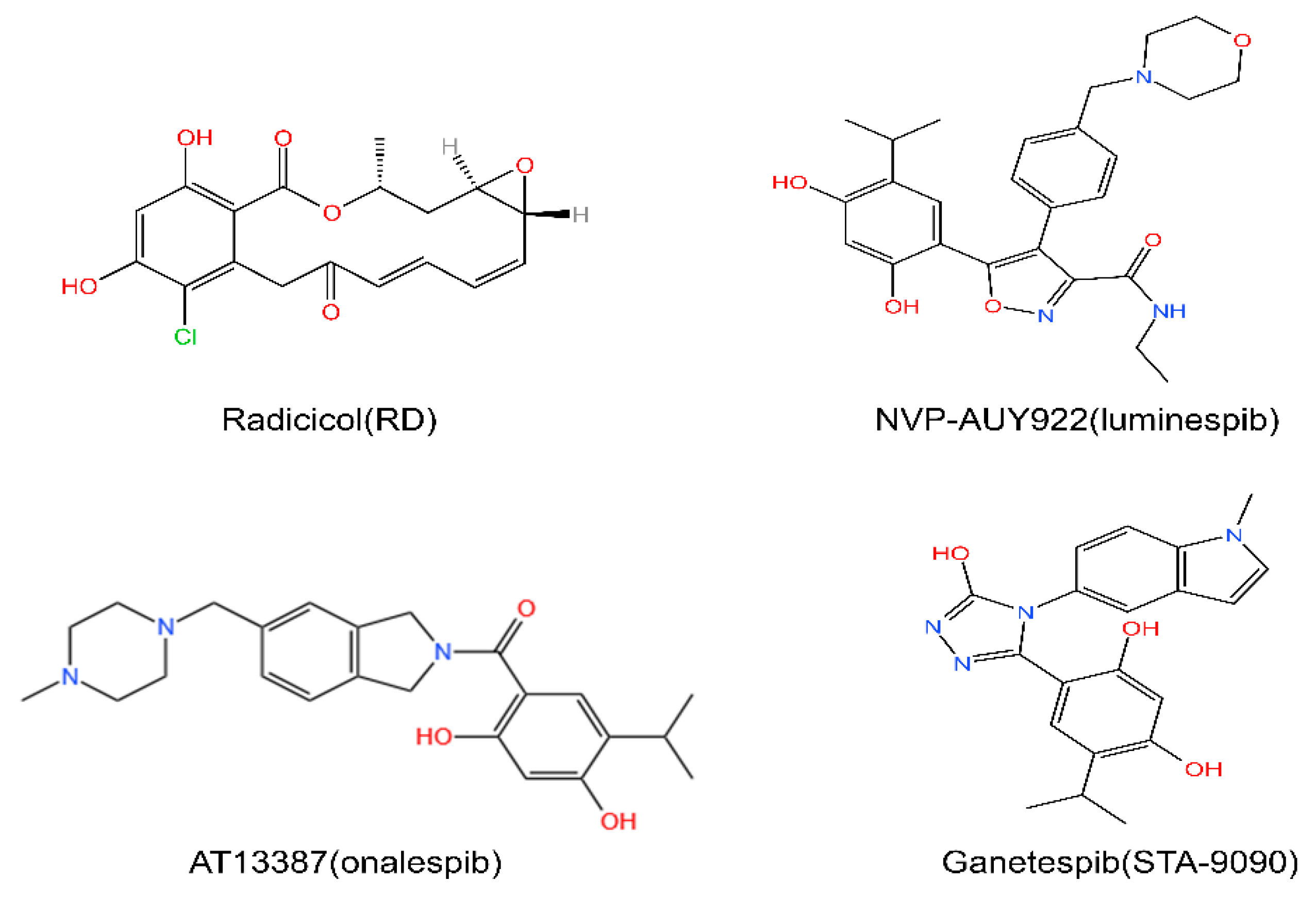
5. HSP90 Inhibitors
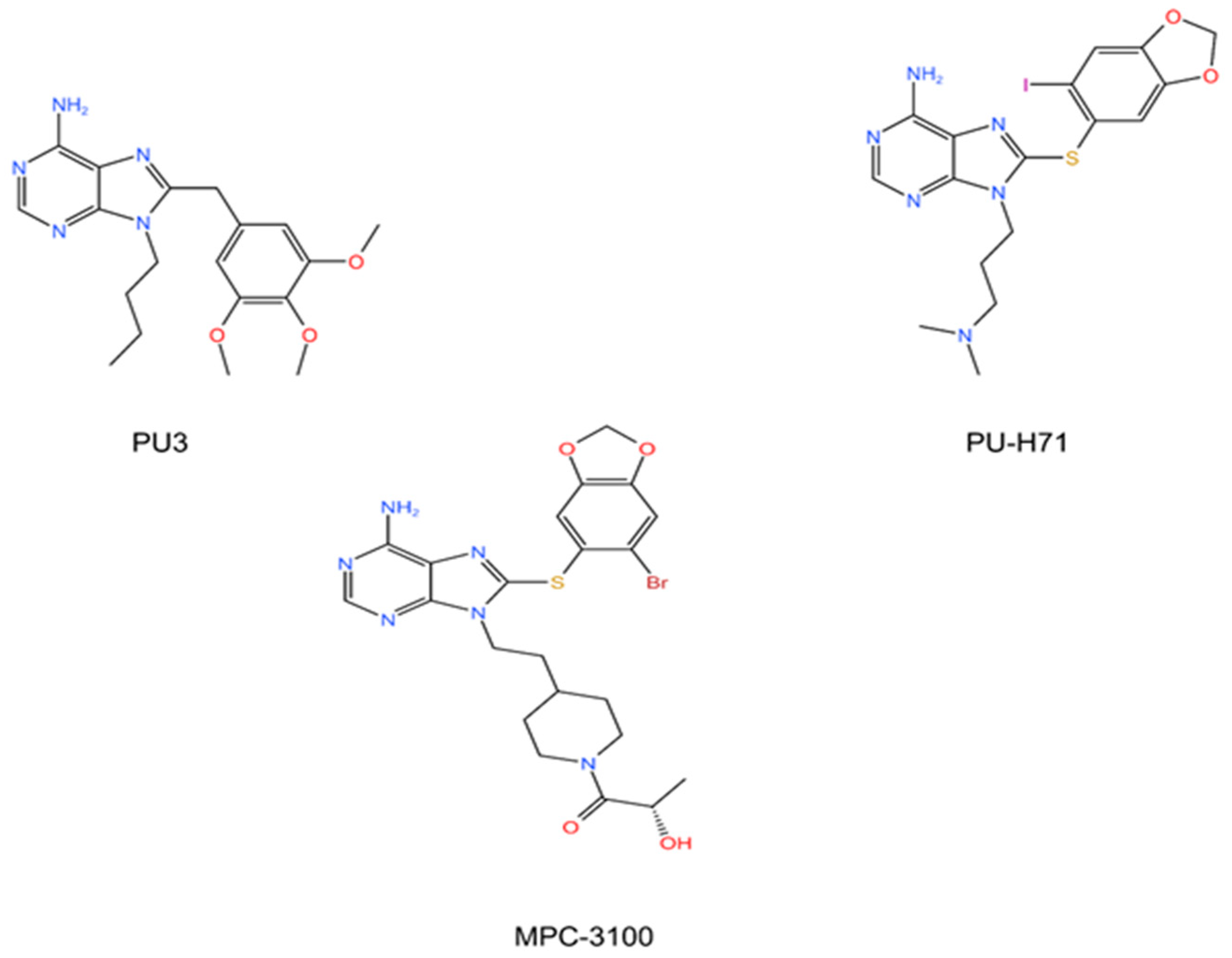
5.1. The N-Terminal Inhibitors
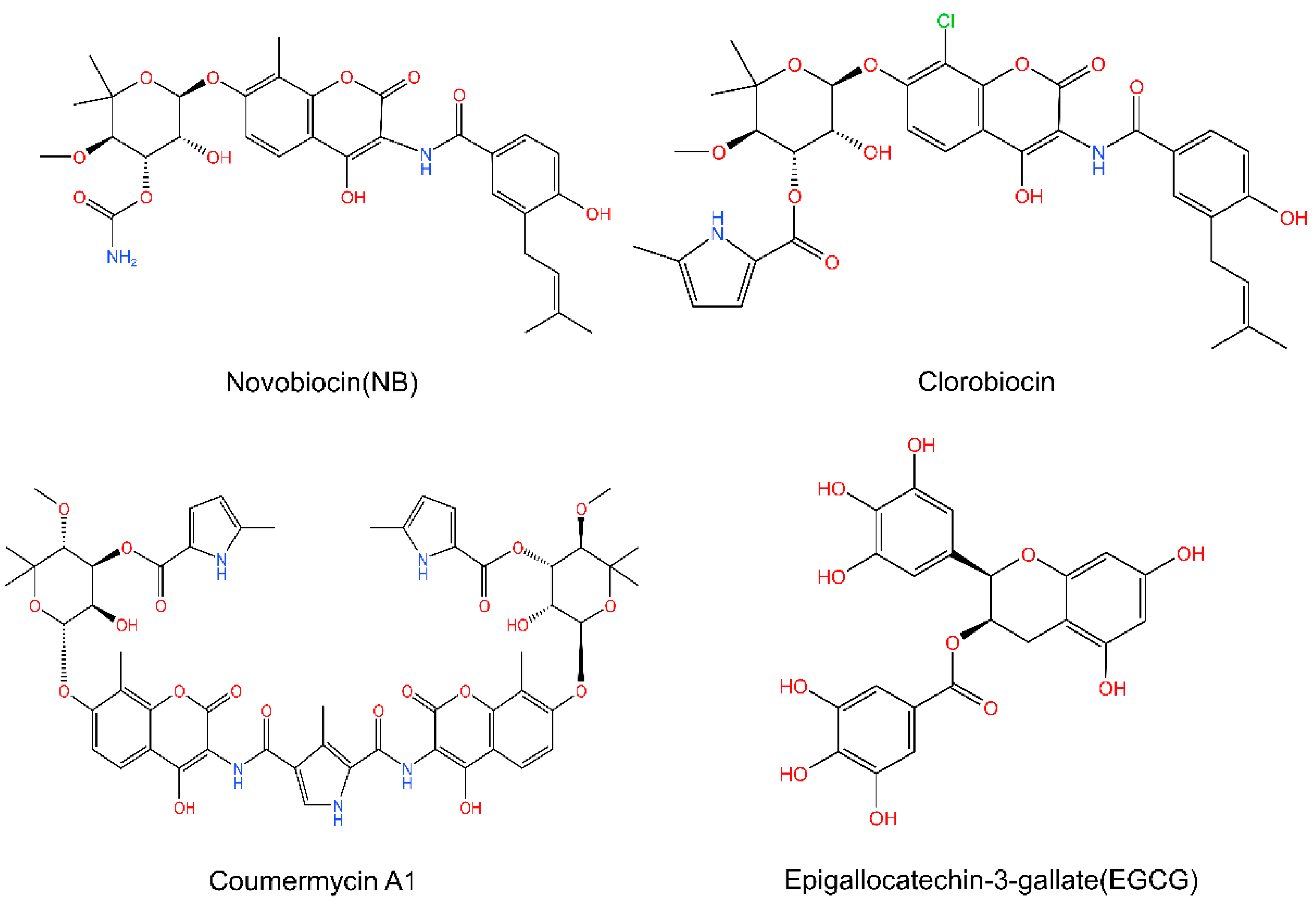
5.2. The C-Terminal Inhibitors
6. Hsp90 Inhibitors as Potential Therapeutic Agents
- As mentioned above, the HSP90 clients contains a wide range of carcinogenic proteins, such as Akt, Bcr-Abl, C-Raf, B-Raf, EGFR, Her-2, HIF-1α, Met, VEGFR, and mutated p53. Most of these clients are key transducers in signaling pathways important for the development and progression of tumors. Hence, inhibiting HSP90 can simultaneously affect a wide range of client proteins, thereby shutting down multiple oncogenic signaling pathways.
- Cancer cells live in a much harsher environment. In addition to common pressures, such as hypoxia, low pH, and malnutrition, tumor cells face external pressures, including mutated client genes and proteins, altered signal pathways and the extra need for nutrients. Therefore, tumor cells depend more on HSP90 to maintain growth/survival than normal cells.
- As mentioned earlier, HSP90 is involved in many malignant behaviors, such as proliferation, metastasis/invasion, antiapoptosis, angiogenesis, and therapeutic resistance. Inhibition of HSP90 may simultaneously reverse multiple malignant behaviors of tumors.
- (Hsp90 often presents in a latent, uncomplex state in normal tissues. However, in tumor cells, Hsp90 entirely exists in multichaperone complexes with high ATPase activity and hence has high affinity for ATP and substrates. Thus, tumor cells are likely more sensitive to HSP90 inhibitors [59].
7. HSP90 Combination Therapy
7.1. Chemotherapy
7.2. Radiotherapy
7.3. Immunotherapy
7.4. Protein Kinase Inhibitors
7.5. Proteasome Inhibitors
7.6. Histone Deacetylase Inhibitors
7.7. Other HSP Inhibitors
7.8. Others
8. Conclusions and Future Decisions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef] [PubMed]
- Gvozdenov, Z.; Kolhe, J.; Freeman, B.C. The Nuclear and DNA-Associated Molecular Chaperone Network. Cold Spring Harb. Perspect. Biol. 2019, 11, a034009. [Google Scholar] [CrossRef] [PubMed]
- Zuehlke, A.; Johnson, J.L.; Gestwicki, J.E. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 2010, 93, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Xu, W.; Neckers, L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 2003, 3, 213–217. [Google Scholar] [CrossRef]
- Žáčková, M.; Moučková, D.; Lopotová, T.; Ondráčková, Z.; Klamová, H.; Moravcová, J. Hsp90—A Potential Prognostic Marker in Cml. Blood Cells Mol. Dis. 2013, 50, 184–189. [Google Scholar] [CrossRef]
- Patel, K.; Wen, J.; Magliocca, K.; Muller, S.; Liu, Y.; Chen, Z.G.; Saba, N.; Diaz, R. Heat Shock Protein 90 (Hsp90) is Overexpressed in P16-Negative Oropharyngeal Squamous Cell Carcinoma, and Its Inhibition in Vitro Potentiates the Effects of Chemoradiation. Cancer Chemother. Pharmacol. 2014, 74, 1015–1022. [Google Scholar] [CrossRef]
- Jhaveri, K.; Ochiana, S.O.; Dunphy, M.P.; Gerecitano, J.F.; Corben, A.D.; Peter, R.I.; Janjigian, Y.Y.; Gomes-DaGama, E.M.; Koren, J.; Modi, S.; et al. Heat shock protein 90 inhibitors in the treatment of cancer: Current status and future directions. Expert Opin. Investig. Drugs 2014, 23, 611–628. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Mollapour, M.; Prodromou, C.; Lee, C.-T.; Panaretou, B.; Yoshida, S.; Mayer, M.P.; Neckers, L.M. Charged linker sequence modulates eukaryotic heat shock protein 90 (Hsp90) chaperone activity. Proc. Natl. Acad. Sci. USA 2012, 109, 2937–2942. [Google Scholar] [CrossRef]
- Chène, P. ATPases as drug targets: Learning from their structure. Nat. Rev. Drug Discov. 2002, 1, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.; Prodromou, C.; Hu, B.; Vaughan, C.; Roe, S.M.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Structural and Functional Analysis of the Middle Segment of Hsp90: Implications for ATP Hydrolysis and Client Protein and Cochaperone Interactions. Mol. Cell 2003, 11, 647–658. [Google Scholar] [CrossRef]
- Soti, C.; Vermes, A.; Haystead, T.A.; Csermely, P. Comparative analysis of the ATP-binding sites of Hsp90 by nucleotide affinity cleavage: A distinct nucleotide specificity of the C-terminal ATP-binding site. Eur. J. Biochem. 2003, 270, 2421–2428. [Google Scholar] [CrossRef]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta 2012, 1823, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, S.; Tavecchio, M.; Chae, Y.C.; Liu, Q.; Brice, A.K.; Thakur, M.L.; Languino, L.R.; Altieri, D.C. Deletion of the mitochondrial chaperone TRAP-1 uncovers global reprogramming of metabolic networks. Cell Rep. 2014, 8, 671–677. [Google Scholar] [CrossRef]
- Calderwood, S.K. Molecular Cochaperones: Tumor Growth and Cancer Treatment. Scientifica 2013, 2013, 217513. [Google Scholar] [CrossRef]
- Roe, S.M.; Ali, M.M.U.; Meyer, P.; Vaughan, C.K.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. The Mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 2004, 116, 87–98. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Pearl, L.H.; Prodromou, C.; Workman, P. The Hsp90 molecular chaperone: An open and shut case for treatment. Biochem. J. 2008, 410, 439–453. [Google Scholar] [CrossRef]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef]
- Villodre, E.S.; Kipper, F.C.; Pereira, M.B.; Lenz, G. Roles of OCT4 in tumorigenesis, cancer therapy resistance and prognosis. Cancer Treat. Rev. 2016, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Lipton, A. SRC kinase inhibition: Targeting bone metastases and tumor growth in prostate and breast cancer. Cancer Treat. Rev. 2010, 36, 177–184. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.-P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Grbovic, O.M.; Basso, A.D.; Sawai, A.; Ye, Q.; Friedlander, P.; Solit, D.; Rosen, N. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 57–62. [Google Scholar] [CrossRef]
- Peiris, M.N.; Meyer, A.N.; Nelson, K.N.; Bisom-Rapp, E.W.; Donoghue, D.J. Oncogenic fusion protein BCR-FGFR1 requires the breakpoint cluster region-mediated oligomerization and chaperonin Hsp90 for activation. Haematologica 2020, 105, 1262–1273. [Google Scholar] [CrossRef]
- Li, T.; Mehraein-Ghomi, F.; Forbes, M.E.; Namjoshi, S.V.; Ballard, E.A.; Song, Q.; Chou, P.C.; Wang, X.; Parker Kerrigan, B.C.; Lang, F.F.; et al. HSP90-CDC37 functions as a chaperone for the oncogenic FGFR3-TACC3 fusion. Mol. Ther. 2022, 30, 1610–1627. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef]
- Kim, R.H.; Kim, R.; Chen, W.; Hu, S.; Shin, K.H.; Park, N.H.; Kang, M.K. Association of hsp90 to the hTERT promoter is necessary for hTERT expression in human oral cancer cells. Carcinogenesis 2008, 29, 2425–2431. [Google Scholar] [CrossRef]
- Niu, M.; Zhang, B.; Li, L.; Su, Z.; Pu, W.; Zhao, C.; Wei, L.; Lian, P.; Lu, R.; Wang, R.; et al. Targeting HSP90 Inhibits Proliferation and Induces Apoptosis Through AKT1/ERK Pathway in Lung Cancer. Front. Pharmacol. 2022, 12, 4113. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Basso, A.D.; Olshen, A.B.; Scher, H.I.; Rosen, N. Inhibition of heat shock protein 90 function down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer Res. 2003, 63, 2139–2144. [Google Scholar]
- Aoyagi, Y.; Fujita, N.; Tsuruo, T. Stabilization of integrin-linked kinase by binding to Hsp90. Biochem. Biophys. Res. Commun. 2005, 331, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Ochel, H.J.; Schulte, T.W.; Nguyen, P.; Trepel, J.; Neckers, L. The benzoquinone ansamycin geldanamycin stimulates proteolytic degradation of focal adhesion kinase. Mol. Genet. Metab. 1999, 66, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, G.P.; Zakka, K.M.; Landry, J.C.; Shaib, W.L.; Lesinski, G.B.; El-Rayes, B.F. Inhibition of HSP90 overcomes resistance to chemotherapy and radiotherapy in pancreatic cancer. Int. J. Cancer 2019, 145, 1529–1537. [Google Scholar] [CrossRef]
- Zhang, P.-C.; Liu, X.; Li, M.-M.; Ma, Y.-Y.; Sun, H.-T.; Tian, X.-Y.; Wang, Y.; Liu, M.; Fu, L.-S.; Wang, Y.-F.; et al. AT-533, a novel Hsp90 inhibitor, inhibits breast cancer growth and HIF-1α/VEGF/VEGFR-2-mediated angiogenesis in vitro and in vivo. Biochem. Pharmacol. 2020, 172, 113771. [Google Scholar] [CrossRef]
- Dubrez, L.; Causse, S.; Bonan, N.B.; Dumétier, B.; Garrido, C. Heat-shock proteins: Chaperoning DNA repair. Oncogene 2019, 39, 516–529. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Quanz, M.; Herbette, A.; Sayarath, M.; de Koning, L.; Dubois, T.; Sun, J.S.; Dutreix, M. Heat shock protein 90alpha (Hsp90alpha) is phosphorylated in response to DNA damage and accumulates in repair foci. J. Biol. Chem. 2012, 287, 8803–8815. [Google Scholar] [CrossRef]
- Solier, S.; Kohn, K.W.; Scroggins, B.; Xu, W.; Trepel, J.; Neckers, L.; Pommier, Y. Heat shock protein 90alpha (HSP90alpha), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proc. Natl. Acad. Sci. USA 2012, 109, 12866–12872. [Google Scholar] [CrossRef]
- Ha, K.; Fiskus, W.; Rao, R.; Balusu, R.; Venkannagari, S.; Nalabothula, N.R.; Bhalla, K.N. Hsp90 inhibitor-mediated disruption of chaperone association of ATR with hsp90 sensitizes cancer cells to DNA damage. Mol. Cancer Ther. 2011, 10, 1194–1206. [Google Scholar] [CrossRef] [PubMed]
- Kinzel, L.; Ernst, A.; Orth, M.; Albrecht, V.; Hennel, R.; Brix, N.; Frey, B.; Gaipl, U.S.; Zuchtriegel, G.; Reichel, C.A.; et al. A novel HSP90 inhibitor with reduced hepatotoxicity synergizes with radiotherapy to induce apoptosis, abrogate clonogenic survival, and improve tumor control in models of colorectal cancer. Oncotarget 2016, 7, 43199–43219. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Albrecht, V.; Seidl, K.; Kinzel, L.; Unger, K.; Hess, J.; Kreutzer, L.; Sun, N.; Stegen, B.; Nieto, A.; et al. Inhibition of HSP90 as a Strategy to Radiosensitize Glioblastoma: Targeting the DNA Damage Response and Beyond. Front. Oncol. 2021, 11, 612354. [Google Scholar] [CrossRef] [PubMed]
- Binder, R.J. Heat-shock protein-based vaccines for cancer and infectious disease. Expert Rev. Vaccines 2008, 7, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Blagg, B.S.; Kerr, T.D. Hsp90 inhibitors: Small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Med. Res. Rev. 2006, 26, 310–338. [Google Scholar] [CrossRef]
- Lancet, J.E.; Gojo, I.; Burton, M.; Quinn, M.; Tighe, S.M.; Kersey, K.; Zhong, Z.; Albitar, M.X.; Bhalla, K.; Hannah, A.L.; et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 2010, 24, 699–705. [Google Scholar] [CrossRef]
- Wagner, A.J.; Chugh, R.; Rosen, L.S.; Morgan, J.A.; George, S.; Gordon, M.; Dunbar, J.; Normant, E.; Grayzel, D.; Demetri, G.D. A Phase I Study of the HSP90 Inhibitor Retaspimycin Hydrochloride (IPI-504) in Patients with Gastrointestinal Stromal Tumors or Soft-Tissue Sarcomas. Clin. Cancer Res. 2013, 19, 6020–6029. [Google Scholar] [CrossRef]
- Jang, W.J.; Jung, S.K.; Kang, J.S.; Jeong, J.W.; Bae, M.K.; Joo, S.H.; Park, G.H.; Kundu, J.K.; Hong, Y.S.; Jeong, C.H. Anti-tumor activity of WK88-1, a novel geldanamycin derivative, in gefitinib-resistant non-small cell lung cancers with Met amplification. Cancer Sci. 2014, 105, 1245–1253. [Google Scholar] [CrossRef]
- Felip, E.; Barlesi, F.; Besse, B.; Chu, Q.; Gandhi, L.; Kim, S.-W.; Carcereny, E.; Sequist, L.V.; Brunsvig, P.; Chouaid, C.; et al. Phase 2 Study of the HSP-90 Inhibitor AUY922 in Previously Treated and Molecularly Defined Patients with Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 576–584. [Google Scholar] [CrossRef]
- Johnson, M.L.; Yu, H.A.; Hart, E.M.; Weitner, B.B.; Rademaker, A.W.; Patel, J.D.; Kris, M.G.; Riely, G.J. Phase I/II Study of HSP90 Inhibitor AUY922 and Erlotinib for EGFR-Mutant Lung Cancer with Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. J. Clin. Oncol. 2015, 33, 1666–1673. [Google Scholar] [CrossRef]
- Ueno, T.; Tsukuda, K.; Toyooka, S.; Ando, M.; Takaoka, M.; Soh, J.; Asano, H.; Maki, Y.; Muraoka, T.; Tanaka, N.; et al. Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on non-small cell lung cancer. Lung Cancer 2012, 76, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.; Curry, J.; Smyth, T.; Fazal, L.; Feltell, R.; Harada, I.; Coyle, J.; Williams, B.; Reule, M.; Angove, H.; et al. The heat shock protein 90 inhibitor, AT13387, displays a long duration of action in vitro and in vivo in non-small cell lung cancer. Cancer Sci. 2012, 103, 522–527. [Google Scholar] [CrossRef]
- Acquaviva, J.; Smith, D.L.; Sang, J.; Friedland, J.C.; He, S.; Sequeira, M.; Zhang, C.; Wada, Y.; Proia, D.A. Targeting KRAS-mutant non-small cell lung cancer with the Hsp90 inhibitor ganetespib. Mol. Cancer Ther. 2012, 11, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Deycmar, S.; Mara, E.; Kerschbaum-Gruber, S.; Waller, V.; Georg, D.; Pruschy, M. Ganetespib selectively sensitizes cancer cells for proximal and distal spread-out Bragg peak proton irradiation. Radiat. Oncol. 2022, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Fennell, D.A.; Danson, S.; Woll, P.J.; Forster, M.; Talbot, D.; Child, J.; Farrelly, L.; Sharkey, A.; Busacca, S.; Ngai, Y.; et al. Ganetespib in Combination with Pemetrexed-Platinum Chemotherapy in Patients with Pleural Mesothelioma (MESO-02): A Phase Ib Trial. Clin. Cancer Res. 2020, 26, 4748–4755. [Google Scholar] [CrossRef]
- Chiosis, G.; Timaul, M.N.; Lucas, B.; Munster, P.N.; Zheng, F.F.; Sepp-Lorenzino, L.; Rosen, N. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem. Biol. 2001, 8, 289–299. [Google Scholar] [CrossRef]
- Allan, R.K.; Mok, D.; Ward, B.K.; Ratajczak, T. Modulation of chaperone function and cochaperone interaction by novobiocin in the C-terminal domain of Hsp90: Evidence that coumarin antibiotics disrupt Hsp90 dimerization. J. Biol. Chem. 2006, 281, 7161–7171. [Google Scholar] [CrossRef]
- Marcu, M.G.; Schulte, T.W.; Neckers, L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J. Natl. Cancer Inst. 2000, 92, 242–248. [Google Scholar] [CrossRef]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef]
- Mellatyar, H.; Talaei, S.; Pilehvar-Soltanahmadi, Y.; Barzegar, A.; Akbarzadeh, A.; Shahabi, A.; Barekati-Mowahed, M.; Zarghami, N. Targeted cancer therapy through 17-DMAG as an Hsp90 inhibitor: Overview and current state of the art. Biomed. Pharmacother. 2018, 102, 608–617. [Google Scholar] [CrossRef]
- Sidera, K.; Patsavoudi, E. HSP90 inhibitors: Current development and potential in cancer therapy. Recent Pat. Anticancer Drug Discov. 2014, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- McCollum, A.K.; Teneyck, C.J.; Sauer, B.M.; Toft, D.O.; Erlichman, C. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res. 2006, 66, 10967–10975. [Google Scholar] [CrossRef] [PubMed]
- Sawai, A.; Chandarlapaty, S.; Greulich, H.; Gonen, M.; Ye, Q.; Arteaga, C.L.; Sellers, W.; Rosen, N.; Solit, D.B. Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res. 2008, 68, 589–596. [Google Scholar] [CrossRef]
- Agyeman, A.S.; Jun, W.J.; Proia, D.A.; Kim, C.R.; Skor, M.N.; Kocherginsky, M.; Conzen, S.D. Hsp90 Inhibition Results in Glucocorticoid Receptor Degradation in Association with Increased Sensitivity to Paclitaxel in Triple-Negative Breast Cancer. Horm. Cancer 2016, 7, 114–126. [Google Scholar] [CrossRef]
- Ui, T.; Morishima, K.; Saito, S.; Sakuma, Y.; Fujii, H.; Hosoya, Y.; Ishikawa, S.; Aburatani, H.; Fukayama, M.; Niki, T.; et al. The HSP90 inhibitor 17-N-allylamino-17-demethoxy geldanamycin (17-AAG) synergizes with cisplatin and induces apoptosis in cisplatin-resistant esophageal squamous cell carcinoma cell lines via the Akt/XIAP pathway. Oncol. Rep. 2014, 31, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Sonego, M.; Pucci, B.; Addi, L.; Iannelli, F.; Capone, F.; Alfano, L.; Roca, M.S.; Milone, M.R.; Moccia, T.; et al. HSP90 identified by a proteomic approach as druggable target to reverse platinum resistance in ovarian cancer. Mol. Oncol. 2021, 15, 1005–1023. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Issa, I.I.; Haraldsdottir, H.; Hald, J.L.; Schmitz, A.; Due, H.; Dybkaer, K. Hsp90 inhibition sensitizes DLBCL cells to cisplatin. Cancer Chemother. Pharmacol. 2022, 89, 431–440. [Google Scholar] [CrossRef]
- Nagaraju, G.P.; Alese, O.B.; Landry, J.; Diaz, R.; El-Rayes, B.F. HSP90 inhibition downregulates thymidylate synthase and sensitizes colorectal cancer cell lines to the effect of 5FU-based chemotherapy. Oncotarget 2014, 5, 9980–9991. [Google Scholar] [CrossRef]
- Joshi, S.S.; Jiang, S.; Unni, E.; Goding, S.R.; Fan, T.; Antony, P.A.; Hornyak, T.J. 17-AAG inhibits vemurafenib-associated MAP kinase activation and is synergistic with cellular immunotherapy in a murine melanoma model. PLoS ONE 2018, 13, e0191264. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Kroemer, G.; Tang, D. Targeting HSP90 sensitizes pancreas carcinoma to PD-1 blockade. Oncoimmunology 2022, 11, 2068488. [Google Scholar] [CrossRef]
- Liu, K.; Huang, J.; Liu, J.; Li, C.; Kroemer, G.; Tang, D.; Kang, R. HSP90 Mediates IFNγ-Induced Adaptive Resistance to Anti-PD-1 Immunotherapy. Cancer Res. 2022, 82, 2003–2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.M.; Guo, C.L.; Shi, J.J.; Xu, Y.C.; Chen, Y.; Shen, Y.Y.; Su, Y.; Ding, J.; Meng, L.H. HSP90 inhibitor AUY922 abrogates up-regulation of RTKs by mTOR inhibitor AZD8055 and potentiates its antiproliferative activity in human breast cancer. Int. J. Cancer 2014, 135, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L.; Acquaviva, J.; Sequeira, M.; Jimenez, J.P.; Zhang, C.; Sang, J.; Bates, R.C.; Proia, D.A. The HSP90 inhibitor ganetespib potentiates the antitumor activity of EGFR tyrosine kinase inhibition in mutant and wild-type non-small cell lung cancer. Target. Oncol. 2015, 10, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, N.; Tsutsumi, S.; Sourbier, C.; Beebe, K.; Mollapour, M.; Rivas, C.; Yoshida, S.; Trepel, J.B.; Huang, Y.; Tatokoro, M.; et al. The HSP90 inhibitor ganetespib synergizes with the MET kinase inhibitor crizotinib in both crizotinib-sensitive and -resistant MET-driven tumor models. Cancer Res. 2013, 73, 7022–7033. [Google Scholar] [CrossRef]
- Roue, G.; Perez-Galan, P.; Mozos, A.; Lopez-Guerra, M.; Xargay-Torrent, S.; Rosich, L.; Saborit-Villarroya, I.; Normant, E.; Campo, E.; Colomer, D. The Hsp90 inhibitor IPI-504 overcomes bortezomib resistance in mantle cell lymphoma in vitro and in vivo by down-regulation of the prosurvival ER chaperone BiP/Grp78. Blood 2011, 117, 1270–1279. [Google Scholar] [CrossRef]
- Ambati, S.R.; Lopes, E.C.; Kosugi, K.; Mony, U.; Zehir, A.; Shah, S.K.; Taldone, T.; Moreira, A.L.; Meyers, P.A.; Chiosis, G.; et al. Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol. Oncol. 2014, 8, 323–336. [Google Scholar] [CrossRef]
- George, P.; Bali, P.; Annavarapu, S.; Scuto, A.; Fiskus, W.; Guo, F.; Sigua, C.; Sondarva, G.; Moscinski, L.; Atadja, P.; et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood 2005, 105, 1768–1776. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. The heat shock protein 90 inhibitor SNX5422 has a synergistic activity with histone deacetylase inhibitors in induction of death of anaplastic thyroid carcinoma cells. Endocrine 2016, 51, 274–282. [Google Scholar] [CrossRef]
- Lamoureux, F.; Thomas, C.; Yin, M.J.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Suppression of heat shock protein 27 using OGX-427 induces endoplasmic reticulum stress and potentiates heat shock protein 90 inhibitors to delay castrate-resistant prostate cancer. Eur. Urol. 2014, 66, 145–155. [Google Scholar] [CrossRef]
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in cancer treatment: Activity, chemoresistance and its overcoming. Drug Resist. Updates 2021, 54, 100742. [Google Scholar] [CrossRef]
- Nguyen, D.M.; Lorang, D.; Chen, G.A.; Stewart, J.H.; Tabibi, E.; Schrump, D.S. Enhancement of paclitaxel-mediated cytotoxicity in lung cancer cells by 17-allylamino geldanamycin: In vitro and in vivo analysis. Ann. Thorac. Surg. 2001, 72, 371–379. [Google Scholar] [CrossRef]
- Ramalingam, S.; Goss, G.; Rosell, R.; Schmid-Bindert, G.; Zaric, B.; Andric, Z.; Bondarenko, I.; Komov, D.; Ceric, T.; Khuri, F.; et al. A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second-line therapy of advanced non-small cell lung cancer (GALAXY-1). Ann. Oncol. 2015, 26, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.N.; Fennell, D.A.; Kovcin, V.; Ciuleanu, T.-E.; Ramlau, R.; Kowalski, D.; Schenker, M.; Yalcin, I.; Teofilovici, F.; Vukovic, V.M.; et al. Randomized Phase III Study of Ganetespib, a Heat Shock Protein 90 Inhibitor, with Docetaxel Versus Docetaxel in Advanced Non-Small-Cell Lung Cancer (GALAXY-2). J. Clin. Oncol. 2020, 38, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Huang, E.H.; Christie, I.; Burns, T.F. Reactivation of the p90RSK-CDC25C Pathway Leads to Bypass of the Ganetespib-Induced G2-M Arrest and Mediates Acquired Resistance to Ganetespib in KRAS-Mutant NSCLC. Mol. Cancer Ther. 2017, 16, 1658–1668. [Google Scholar] [CrossRef] [PubMed]
- Bagatell, R.; Beliakoff, J.; David, C.L.; Marron, M.T.; Whitesell, L. Hsp90 inhibitors deplete key anti-apoptotic proteins in pediatric solid tumor cells and demonstrate synergistic anticancer activity with cisplatin. Int. J. Cancer 2005, 113, 179–188. [Google Scholar] [CrossRef]
- Jacquemont, C.; Simon, J.A.; D’Andrea, A.D.; Taniguchi, T. Non-specific chemical inhibition of the Fanconi anemia pathway sensitizes cancer cells to cisplatin. Mol. Cancer 2012, 11, 26. [Google Scholar] [CrossRef]
- Ewers, K.M.; Patil, S.; Kopp, W.; Thomale, J.; Quilitz, T.; Magerhans, A.; Wang, X.; Hessmann, E.; Dobbelstein, M. HSP90 Inhibition Synergizes with Cisplatin to Eliminate Basal-like Pancreatic Ductal Adenocarcinoma Cells. Cancers 2021, 13, 6163. [Google Scholar] [CrossRef]
- Gutierrez, M.; Guo, R.; Giaccone, G.; Liu, S.V.; Hao, Z.; Hilton, C.; Hinson, J.M.; Kris, M.G.; Orlemans, E.O.; Drilon, A. Phase 1 multicenter study of the HSP90 inhibitor SNX-5422 plus carboplatin and paclitaxel in patients with lung cancers. Lung Cancer 2021, 162, 23–28. [Google Scholar] [CrossRef]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef]
- Sharma, K.; Vabulas, R.M.; Macek, B.; Pinkert, S.; Cox, J.; Mann, M.; Hartl, F.U. Quantitative proteomics reveals that Hsp90 inhibition preferentially targets kinases and the DNA damage response. Mol. Cell Proteom. 2012, 11, M111.014654. [Google Scholar] [CrossRef]
- Yin, X.; Zhang, H.; Lundgren, K.; Wilson, L.; Burrows, F.; Shores, C.G. BIIB021, a novel Hsp90 inhibitor, sensitizes head and neck squamous cell carcinoma to radiotherapy. Int. J. Cancer 2010, 126, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gong, Y.; Ma, Y.; Thompson, R.C.; Wang, J.; Cheng, Z.; Xue, L. A Brain-Penetrating Hsp90 Inhibitor NXD30001 Inhibits Glioblastoma as a Monotherapy or in Combination with Radiation. Front. Pharm. 2020, 11, 974. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.K.; Pal, S.; Kondapi, K.; Sitto, M.; Dewar, C.; Devasia, T.; Schipper, M.J.; Thomas, D.G.; Basrur, V.; Pai, M.P.; et al. Low-Dose Hsp90 Inhibitor Selectively Radiosensitizes HNSCC and Pancreatic Xenografts. Clin. Cancer Res. 2020, 26, 5246–5257. [Google Scholar] [CrossRef] [PubMed]
- Proia, D.A.; Kaufmann, G.F. Targeting Heat-Shock Protein 90 (HSP90) as a Complementary Strategy to Immune Checkpoint Blockade for Cancer Therapy. Cancer Immunol. Res. 2015, 3, 583–589. [Google Scholar] [CrossRef]
- Mbofung, R.M.; McKenzie, J.A.; Malu, S.; Zhang, M.; Peng, W.; Liu, C.; Kuiatse, I.; Tieu, T.; Williams, L.; Devi, S.; et al. HSP90 inhibition enhances cancer immunotherapy by upregulating interferon response genes. Nat. Commun. 2017, 8, 451. [Google Scholar] [CrossRef]
- Zavareh, R.B.; Spangenberg, S.H.; Woods, A.; Martinez-Pena, F.; Lairson, L.L. HSP90 Inhibition Enhances Cancer Immunotherapy by Modulating the Surface Expression of Multiple Immune Checkpoint Proteins. Cell Chem. Biol. 2021, 28, 158–168.e155. [Google Scholar] [CrossRef]
- Zhang, Y.; Ware, M.B.; Zaidi, M.Y.; Ruggieri, A.N.; Olson, B.M.; Komar, H.; Farren, M.R.; Nagaraju, G.P.; Zhang, C.; Chen, Z.; et al. Heat Shock Protein-90 Inhibition Alters Activation of Pancreatic Stellate Cells and Enhances the Efficacy of PD-1 Blockade in Pancreatic Cancer. Mol. Cancer Ther. 2021, 20, 150–160. [Google Scholar] [CrossRef]
- Vaishampayan, U.N.; Burger, A.M.; Sausville, E.A.; Heilbrun, L.K.; Li, J.; Horiba, M.N.; Egorin, M.J.; Ivy, P.; Pacey, S.; Lorusso, P.M. Safety, efficacy, pharmacokinetics, and pharmacodynamics of the combination of sorafenib and tanespimycin. Clin. Cancer Res. 2010, 16, 3795–3804. [Google Scholar] [CrossRef]
- Fu, W.; Sharma, S.S.; Ma, L.; Chu, B.; Bui, M.M.; Reed, D.; Pledger, W.J. Apoptosis of osteosarcoma cultures by the combination of the cyclin-dependent kinase inhibitor SCH727965 and a heat shock protein 90 inhibitor. Cell Death Dis. 2013, 4, e566. [Google Scholar] [CrossRef]
- Abdalla, A.N.; Abdallah, M.E.; Aslam, A.; Bader, A.; Vassallo, A.; Tommasi, N.; Malki, W.H.; Gouda, A.M.; Mukhtar, M.H.; El-Readi, M.Z.; et al. Synergistic Anti Leukemia Effect of a Novel Hsp90 and a Pan Cyclin Dependent Kinase Inhibitors. Molecules 2020, 25, 2220. [Google Scholar] [CrossRef]
- Sasame, J.; Ikegaya, N.; Kawazu, M.; Natsumeda, M.; Hayashi, T.; Isoda, M.; Satomi, K.; Tomiyama, A.; Oshima, A.; Honma, H.; et al. HSP90 Inhibition Overcomes Resistance to Molecular Targeted Therapy in BRAFV600E-mutant High-grade Glioma. Clin. Cancer Res. 2022, 28, 2425–2439. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Shin, S.; Kang, J.; Han, K.C.; Kim, Y.H.; Bae, J.W.; Park, K.H. HSP90 Inhibitor, 17-DMAG, Alone and in Combination with Lapatinib Attenuates Acquired Lapatinib-Resistance in ER-positive, HER2-Overexpressing Breast Cancer Cell Line. Cancers 2020, 12, 2630. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Stopeck, A.T.; Gordon, M.S.; Mendelson, D.; Solit, D.B.; Bagatell, R.; Ma, W.; Wheler, J.; Rosen, N.; Norton, L.; et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: A phase I dose-escalation study. J. Clin. Oncol. 2007, 25, 5410–5417. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D’Andrea, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. HSP90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Kung, A.L.; Davies, F.E.; Morgan, G.; Akiyama, M.; Shringarpure, R.; Munshi, N.C.; et al. Antimyeloma activity of heat shock protein-90 inhibition. Blood 2006, 107, 1092–1100. [Google Scholar] [CrossRef]
- Richardson, P.G.; Chanan-Khan, A.A.; Lonial, S.; Krishnan, A.Y.; Carroll, M.P.; Alsina, M.; Albitar, M.; Berman, D.; Messina, M.; Anderson, K.C. Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: Results of a phase 1/2 study. Br. J. Haematol. 2011, 153, 729–740. [Google Scholar] [CrossRef]
- Ishii, T.; Seike, T.; Nakashima, T.; Juliger, S.; Maharaj, L.; Soga, S.; Akinaga, S.; Cavenagh, J.; Joel, S.; Shiotsu, Y. Anti-tumor activity against multiple myeloma by combination of KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J. 2012, 2, e68. [Google Scholar] [CrossRef]
- Cavenagh, J.; Oakervee, H.; Baetiong-Caguioa, P.; Davies, F.; Gharibo, M.; Rabin, N.; Kurman, M.; Novak, B.; Shiraishi, N.; Nakashima, D.; et al. A phase I/II study of KW-2478, an Hsp90 inhibitor, in combination with bortezomib in patients with relapsed/refractory multiple myeloma. Br. J. Cancer 2017, 117, 1295–1302. [Google Scholar] [CrossRef]
- Peron, M.; Bonvini, P.; Rosolen, A. Effect of inhibition of the ubiquitin-proteasome system and Hsp90 on growth and survival of rhabdomyosarcoma cells in vitro. BMC Cancer 2012, 12, 233. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar] [CrossRef] [PubMed]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-L.; Tu, H.-J.; Pan, S.-L.; Liou, J.-P.; Yang, C.-R. Anti-metastatic activity of MPT0G211, a novel HDAC6 inhibitor, in human breast cancer cells in vitro and in vivo. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-W.; Chao, M.-W.; Tu, H.-J.; Chen, L.-C.; Hsu, K.-C.; Liou, J.-P.; Yang, C.-R.; Yen, S.-C.; HuangFu, W.-C.; Pan, S.-L. A novel dual HDAC and HSP90 inhibitor, MPT0G449, downregulates oncogenic pathways in human acute leukemia in vitro and in vivo. Oncogenesis 2021, 10, 39. [Google Scholar] [CrossRef]
- Lee, C.H.; Hong, H.M.; Chang, Y.Y.; Chang, W.W. Inhibition of heat shock protein (Hsp) 27 potentiates the suppressive effect of Hsp90 inhibitors in targeting breast cancer stem-like cells. Biochimie 2012, 94, 1382–1389. [Google Scholar] [CrossRef]
- Guo, F.; Rocha, K.; Bali, P.; Pranpat, M.; Fiskus, W.; Boyapalle, S.; Kumaraswamy, S.; Balasis, M.; Greedy, B.; Armitage, E.S.; et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res. 2005, 65, 10536–10544. [Google Scholar] [CrossRef]
- Lin, T.-y.; Guo, W.; Long, Q.; Ma, A.; Liu, Q.; Zhang, H.; Huang, Y.; Chandrasekaran, S.; Pan, C.; Lam, K.S.; et al. HSP90 Inhibitor Encapsulated Photo-Theranostic Nanoparticles for Synergistic Combination Cancer Therapy. Theranostics 2016, 6, 1324–1335. [Google Scholar] [CrossRef]
- Moon, S.J.; Jeong, B.C.; Kim, H.J.; Lim, J.E.; Kim, H.J.; Kwon, G.Y.; Jackman, J.A.; Kim, J.H. Bruceantin targets HSP90 to overcome resistance to hormone therapy in castration-resistant prostate cancer. Theranostics 2021, 11, 958–973. [Google Scholar] [CrossRef]
- Bai, J.; Zhou, G.; Qiu, Y.; Hu, Y.; Liu, J.; Zhao, J.; Zhang, S.; Zhang, J. HSP90 inhibitor AUY922 can reverse Fulvestrant induced feedback reaction in human breast cancer cells. Cancer Sci. 2017, 108, 1177–1184. [Google Scholar] [CrossRef]
- Whitesell, L.; Santagata, S.; Mendillo, M.L.; Lin, N.U.; Proia, D.A.; Lindquist, S. HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc. Natl. Acad. Sci. USA 2014, 111, 18297–18302. [Google Scholar] [CrossRef]
- Heske, C.M.; Mendoza, A.; Edessa, L.D.; Baumgart, J.T.; Lee, S.; Trepel, J.; Proia, D.A.; Neckers, L.; Helman, L.J. STA-8666, a novel HSP90 inhibitor/SN-38 drug conjugate, causes complete tumor regression in preclinical mouse models of pediatric sarcoma. Oncotarget 2016, 7, 65540–65552. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhu, H.-P.; Xie, X.; Mao, Q.; Liu, Y.-Q.; He, X.-H.; Peng, C.; Jiang, Q.-L.; Huang, W. Novel HSP90-PI3K Dual Inhibitor Suppresses Melanoma Cell Proliferation by Interfering with HSP90-EGFR Interaction and Downstream Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 1845. [Google Scholar] [CrossRef]
- Oh, Y.J.; Park, S.Y.; Seo, Y.H. Selective targeting of cancer cells using a hydrogen peroxide-activated Hsp90 inhibitor. Bioorg. Chem. 2021, 115, 105195. [Google Scholar] [CrossRef]
- Rachidi, S.; Sun, S.; Wu, B.X.; Jones, E.; Drake, R.R.; Ogretmen, B.; Cowart, L.A.; Clarke, C.J.; Hannun, Y.A.; Chiosis, G.; et al. Endoplasmic reticulum heat shock protein gp96 maintains liver homeostasis and promotes hepatocellular carcinogenesis. J. Hepatol. 2015, 62, 879–888. [Google Scholar] [CrossRef]
- Hua, Y.; White-Gilbertson, S.; Kellner, J.; Rachidi, S.; Usmani, S.Z.; Chiosis, G.; Depinho, R.; Li, Z.; Liu, B. Molecular chaperone gp96 is a novel therapeutic target of multiple myeloma. Clin. Cancer Res. 2013, 19, 6242–6251. [Google Scholar] [CrossRef]
- Morales, C.; Rachidi, S.; Hong, F.; Sun, S.; Ouyang, X.; Wallace, C.; Zhang, Y.; Garret-Mayer, E.; Wu, J.; Liu, B.; et al. Immune chaperone gp96 drives the contributions of macrophages to inflammatory colon tumorigenesis. Cancer Res. 2014, 74, 446–459. [Google Scholar] [CrossRef]
- Wu, B.X.; Hong, F.; Zhang, Y.; Ansa-Addo, E.; Li, Z. GRP94/gp96 in Cancer: Biology, Structure, Immunology, and Drug Development. Adv. Cancer Res. 2016, 129, 165–190. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Client | Function | Cancer |
|---|---|---|
| Transcription factors | ||
| p53 | Tumor-suppressor protein | Mutated in cancer |
| HIF-1α | Heat-shock response | Angiogenesis |
| Estrogen receptors | Response to estrogens | Breast cancer |
| Androgen receptor | Response to androgens | Prostate cancer [20] |
| OCT4 | Embryonic development and induction of pluripotent cells | Chemotherapy resistance and tumor differentiation [21] |
| STAT3 | JAK–STAT3 signal pathway | Proliferation and survival [22] |
| Kinase | ||
| AKT (PKB) | PI3K/AKT pathway | impaired apoptosis |
| CDK4(Cyclin D-dependent kinases 4) | Cell cycling | Tumor proliferation [23] |
| SRC | Nonreceptor tyrosine kinase | Tumor growth and metastasis [24] |
| BRAF | Mitogen signaling | Proliferation and invasion |
| JAK (Janus kinases) | JAK–STAT signal pathway | Proliferation and survival |
| BCR–ABL | Fusion tyrosine kinase | Hallmarks of CML cells |
| HCK (hematopoietic cell kinase) | Immune response | Distant metastasis |
| IκB | Activation of NF-κB pathway | Proliferation, antiapoptotic and angiogenesis [25] |
| MMP2 (matrix metalloproteinase 2) | Decomposition of extracellular matrix (ECM) components and basement membrane (BM) | Invasion/metastasis |
| Others | ||
| TERT | Telomere maintenance | Immortalization |
| RAD51 and/or RAD52 | DNA repair | Radiotherapy resistance |
| Other Anticancer Treatment | Hsp90 Inhibitor | Cancer Cell Type | Synergistic Mechanism | Conditions | Refs |
|---|---|---|---|---|---|
| Chemotherapy | |||||
| Taxanes | 17-AAG | EGFR mutant non–small-cell lung cancers (NSCLC) | Degradation of epidermal growth factor receptor (EGFR) | In vitro, in vivo | [63] |
| Ganetespib, NVP-AUY922 | Triple-negative breast cancer (TNBC) | Degradation of Glucocorticoid receptor (GR), apoptosis↑proliferation↓ | In vitro, in vivo | [64] | |
| Cisplatin | 17-AAG | Cisplatin-resistant esophageal squamous cell carcinoma (ESCC) | Apoptosis↑by Akt/XIAP pathway | In vitro | [65] |
| 17AAG, ganetespib | Platinum-resistant ovarian cancer | Apoptosis↑, DNA damage↑ | In vitro, in vivo | [66] | |
| 17AAG | Relapsed diffuse large B-Cell lymphoma (DLBCL) | Apoptosis↑, DNA damage↑ | In vitro | [67] | |
| 5-flfluorouracil (5-FU) | ganetespib | Colorectal cancer (CRC) | Inducing G0/G1 cell cycle arrest; downregulating thymidylate synthase | In vitro, in vivo | [68] |
| Radiotherapy | |||||
| Radiotherapy | Ganetespib | Pancreatic ductal adenocarcinoma (PDAC) | Proliferation↓, angiogenesis↓, apoptosis↑, HIF-1α expression↓ | In vitro, in vivo | [35] |
| Fractionated, conebeam CT (CBCT)-based irradiation | NW457 | Glioblastoma | Disrupting DNA damage response (DDR) | In vitro, in vivo | [43] |
| Immunotherapy | |||||
| Cellular immunotherapy | 17-AAG | Wild-type BRAF, NRAS mutant melanoma cells | ERK signaling↓, CRAF↓ | In vitro, in vivo | [69] |
| Anti-PD-1 antibody | Ganetespib | PDAC | Downregulating STAT1; indoleamine 2,3-dioxygenase 1 (IDO1) ↓, PD-L1↓ | In vitro, in vivo | [70,71] |
| Protein kinase inhibitors | |||||
| mTOR inhibitor AZD8055 | AUY922 | breast cancer | Enhancing cell cycle arrest; destabilizing multiple tyrosine kinases; abrogating activation of AKT induced by AZD8055 | In vitro, in vivo | [72] |
| EGFR inhibitor (erlotinib, gefitinib) | Ganetespib | NSCLC | Stabilizing EGFR protein levels in an inactive state; completely abrogating ERK and AKT signaling activity | In vitro, in vivo | [73] |
| MET kinase inhibitor Crizotinib | Ganetespib | MET-driven cancers | Synergistically inhibiting MET and its downstream signaling pathways | In vitro, in vivo | [74] |
| Proteasome inhibitors | |||||
| Bortezomib | IPI-504 | Mantle cell lymphoma (MCL) | Inducing depletion of BiP/Grp78, inhibiting unfolded protein response, promoting NOXA-mediated mitochondrial depolarization | In vitro, in vivo | [75] |
| Bortezomib | PU-H71 | Ewing sarcoma | G2/M phase arrest; depletion of proteins including AKT, pERK, RAF-1, c-MYC, c-KIT, IGF1R, hTERT and EWS-FLI1 | In vitro, in vivo | [76] |
| Histone deacetylases inhibitors | |||||
| LBH589 | 17-AAG | CML, AML | Degradation of FLT-3 and Bcr-Abl↑ | In vitro | [77] |
| PXD101, suberoylanilide hydroxamic acid (SAHA), trichostatin A (TSA) | SNX5422 | Anaplastic thyroid carcinoma (ATC) | Inducing cell death by suppressing PI3K/Akt/mTOR signaling | In vitro | [78] |
| Other HSP inhibitors | |||||
| HSP27 inhibitor OGX-427 | 17-AAG | Castration-resistant prostate cancer (CRPC) | OGX-427 attenuates Hsp27 expression induced by HSP90 inhibitor; ER stress↑ apoptosis↑ | In vitro, in vivo | [79] |
| Other therapies | |||||
| Fulvestrant (hormone therapy) | AUY922 | ER positive breast cancer | Downregulation of ErbB receptors and downstream PI3K/AKT and ERK pathway; reversing Fulvestrant resistance | In vitro | [79] |
| Anticancer Therapy | Hsp90 Inhibitor | Cancer Type | Phase | Status | NCT Number |
|---|---|---|---|---|---|
| Abiraterone acetate | AT13387 | Prostate Cancer | 1/2 | Completed | NCT01685268 |
| Crizotinib | AT13387 | Non-small-cell lung cancer (NSCLC) | 1/2 | Completed | NCT01712217 |
| Erlotinib hydrochloride | AUY922 | Non-small-cell lung cancer (NSCLC) | 1/2 | Completed | NCT01259089 |
| Niraparib, carboplatin | Ganetespib | Ovarian Cancer | 2 | Active, not recruiting | NCT03783949 |
| Paclitaxel | Ganetespib | Epithelial ovarian cancer (EOC) | 2 | Terminated | NCT02012192 |
| Fulvestrant | Ganetespib | HR+ breast cancer | 2 | Completed | NCT01560416 |
| Paclitaxel | Ganetespib | Recurrent fallopian tube cancer; Recurrent ovarian epithelial cancer; Recurrent primary peritoneal cavity cancer | 1/2 | Terminated | NCT01962948 |
| Trastuzumab | AUY922 | Advanced gastric cancer | 2 | Terminated | NCT01402401 |
| Trastuzumab | AUY922 | Advanced HER2-positive breast cancer | 1/2 | Completed | NCT01271920 |
| Bortezomib | KW-2478 | Multiple Myeloma | 1/2 | Completed | NCT01063907 |
| Sirolimus | Ganetespib | Malignant peripheral nerve sheath tumors (MPNST); Sarcoma | 1/2 | Completed | NCT02008877 |
| Bortezomib | AUY922 | Relapsed or refractory multiple myeloma | 1/2 | Completed | NCT00708292 |
| Bortezomib | 17-AAG | Multiple Myeloma | 2/3 | Completed | NCT00514371 |
| Bortezomib | 17-AAG | Multiple Myeloma | 3 | Completed | NCT00546780 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, X.; Li, T.; Zhang, W.; Yang, X. Targeting Heat-Shock Protein 90 in Cancer: An Update on Combination Therapy. Cells 2022, 11, 2556. https://doi.org/10.3390/cells11162556
Ren X, Li T, Zhang W, Yang X. Targeting Heat-Shock Protein 90 in Cancer: An Update on Combination Therapy. Cells. 2022; 11(16):2556. https://doi.org/10.3390/cells11162556
Chicago/Turabian StyleRen, Xiude, Tao Li, Wei Zhang, and Xuejun Yang. 2022. "Targeting Heat-Shock Protein 90 in Cancer: An Update on Combination Therapy" Cells 11, no. 16: 2556. https://doi.org/10.3390/cells11162556