
S1P-Induced TNF-α and IL-6 Release from PBMCs Exacerbates Lung Cancer-Associated Inflammation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. Isolation and Treatment of Human PBMCs
2.3. Cytokine Measurements
2.4. Western Blotting Analysis
2.5. Statistical Analysis
3. Results
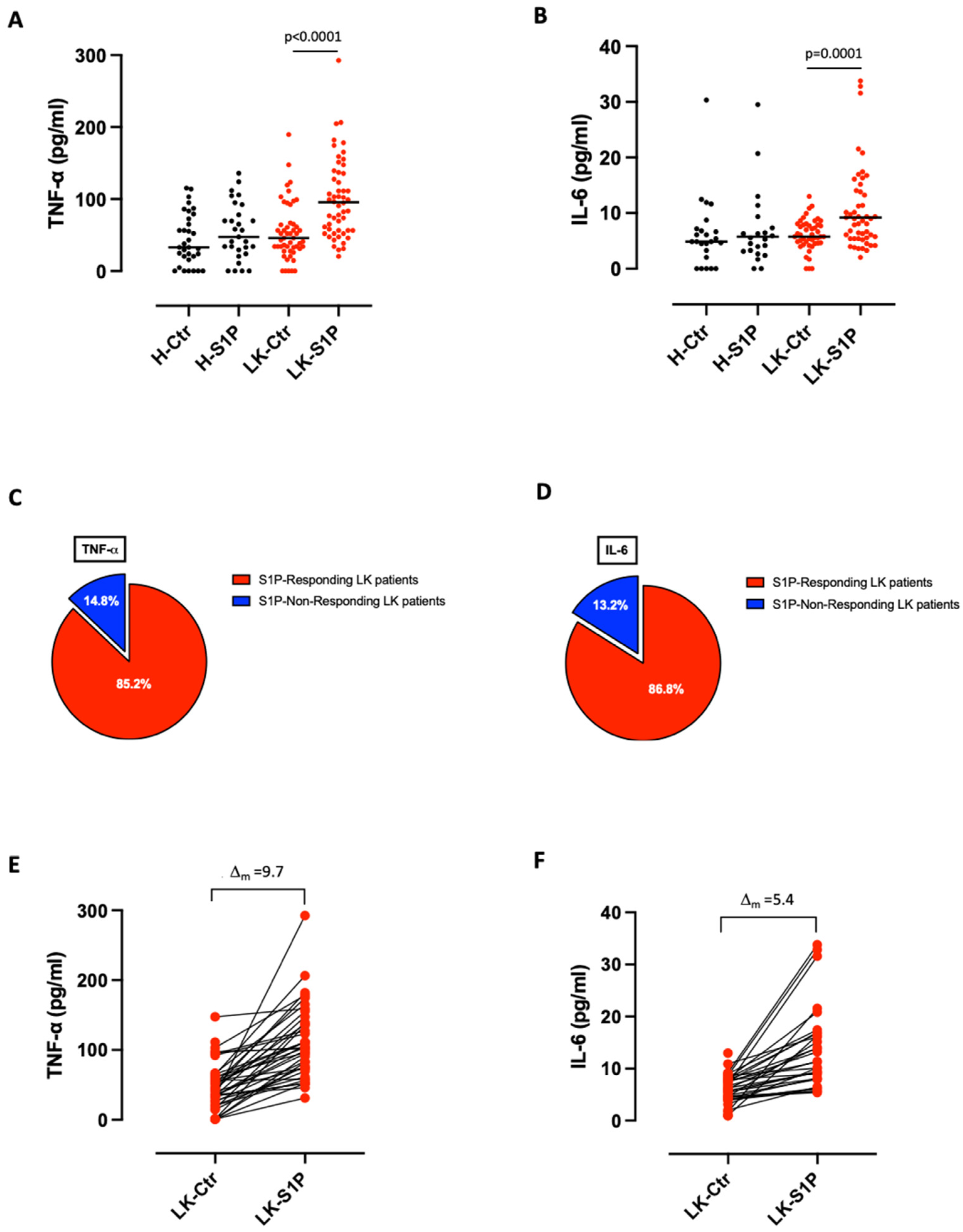
3.1. S1P Induced the Release of Pro-Inflammatory Cytokines from Lung Cancer but Not from Healthy-Derived PBMCs
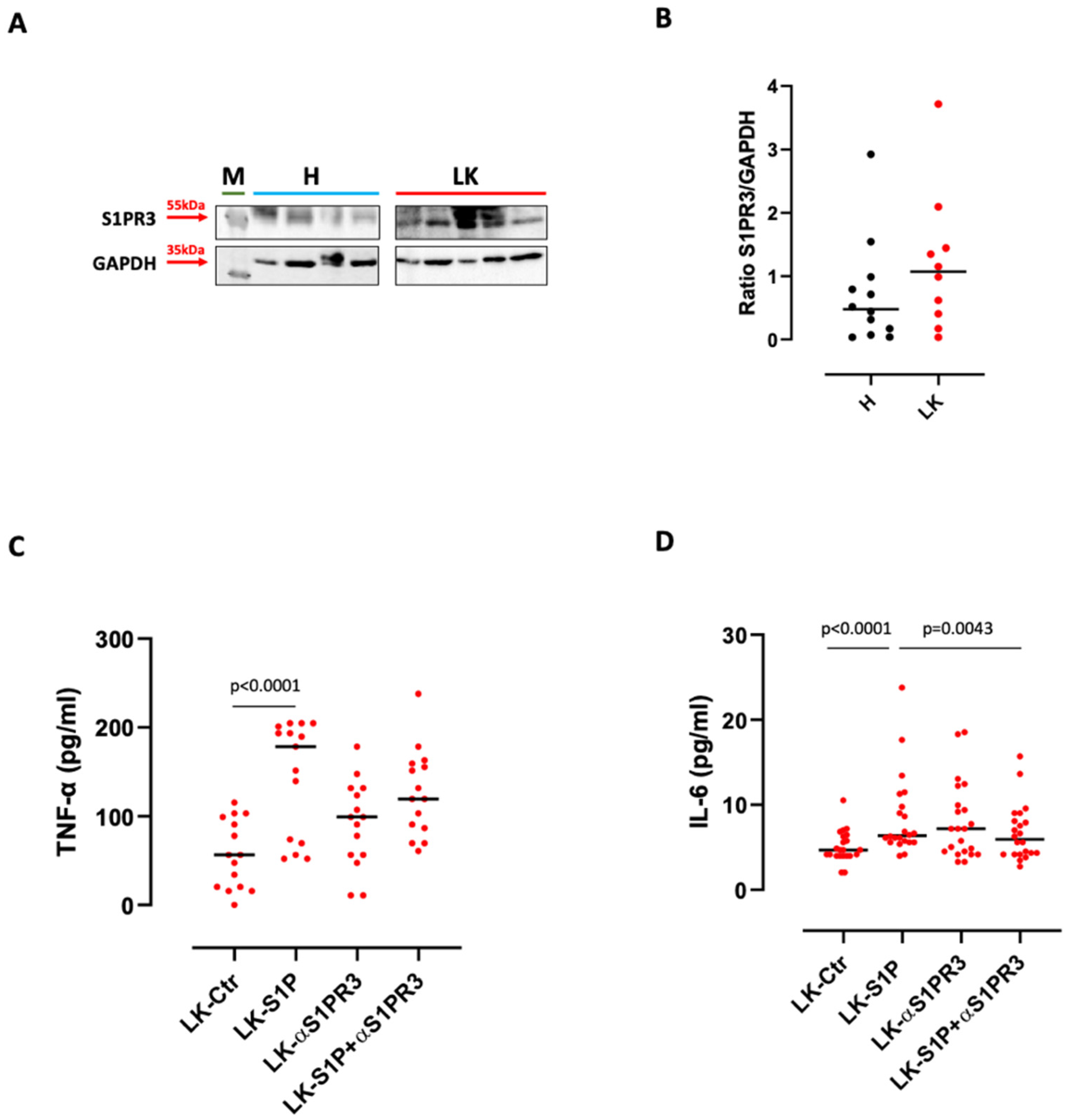
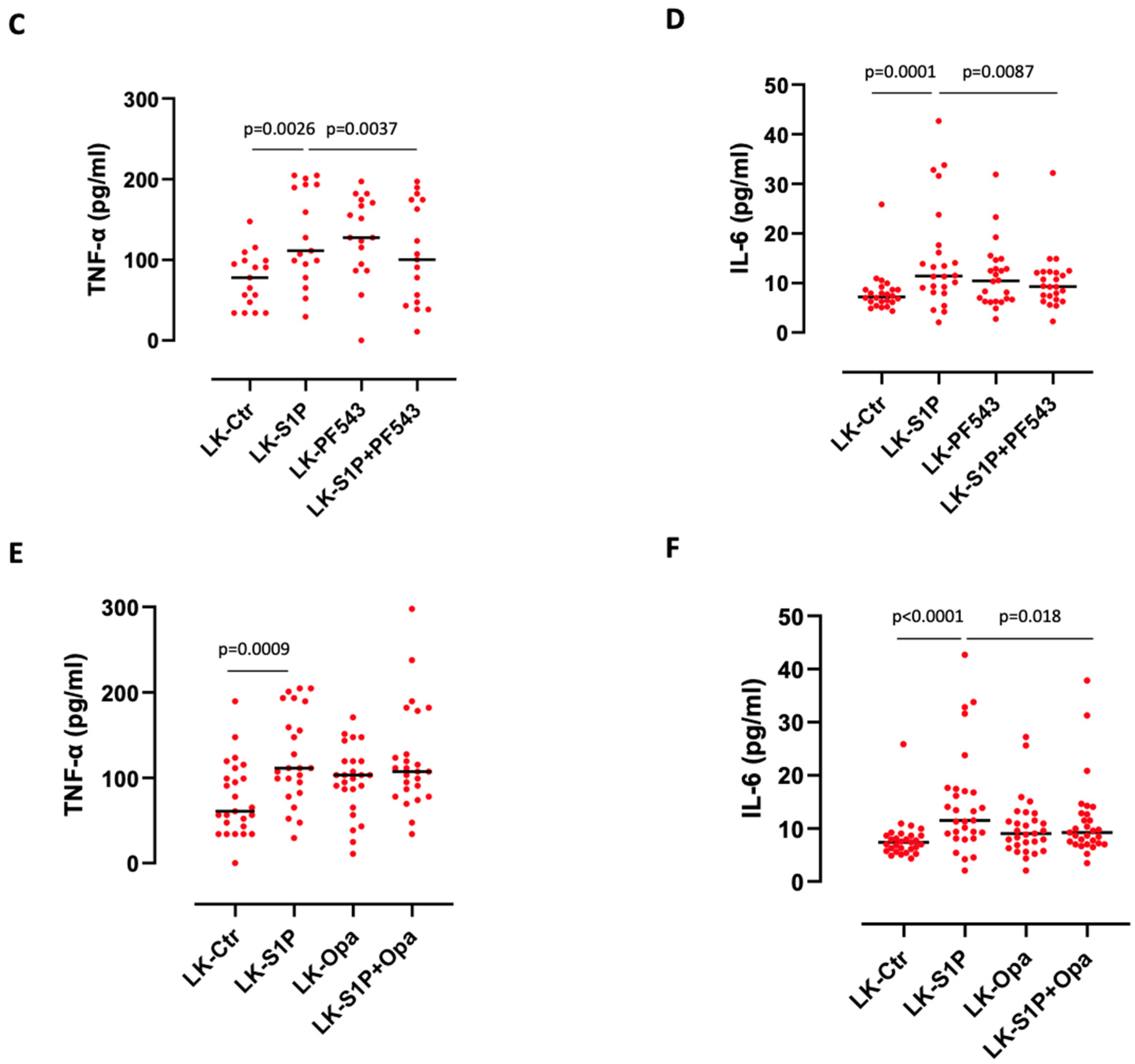
3.2. The Inhibition of S1PR3 Reduced S1P-Induced TNF-α and IL-6 Release from PBMCs
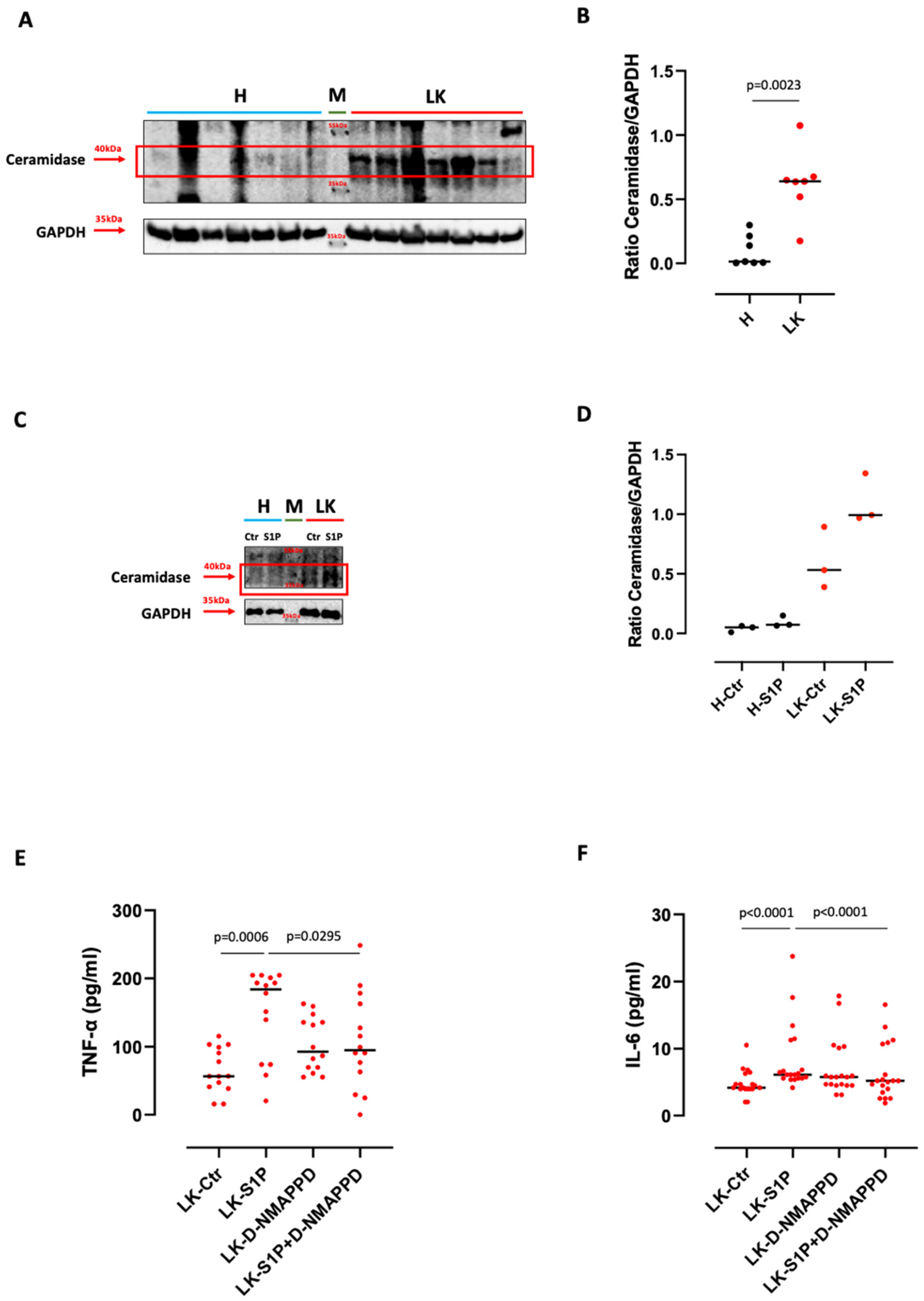
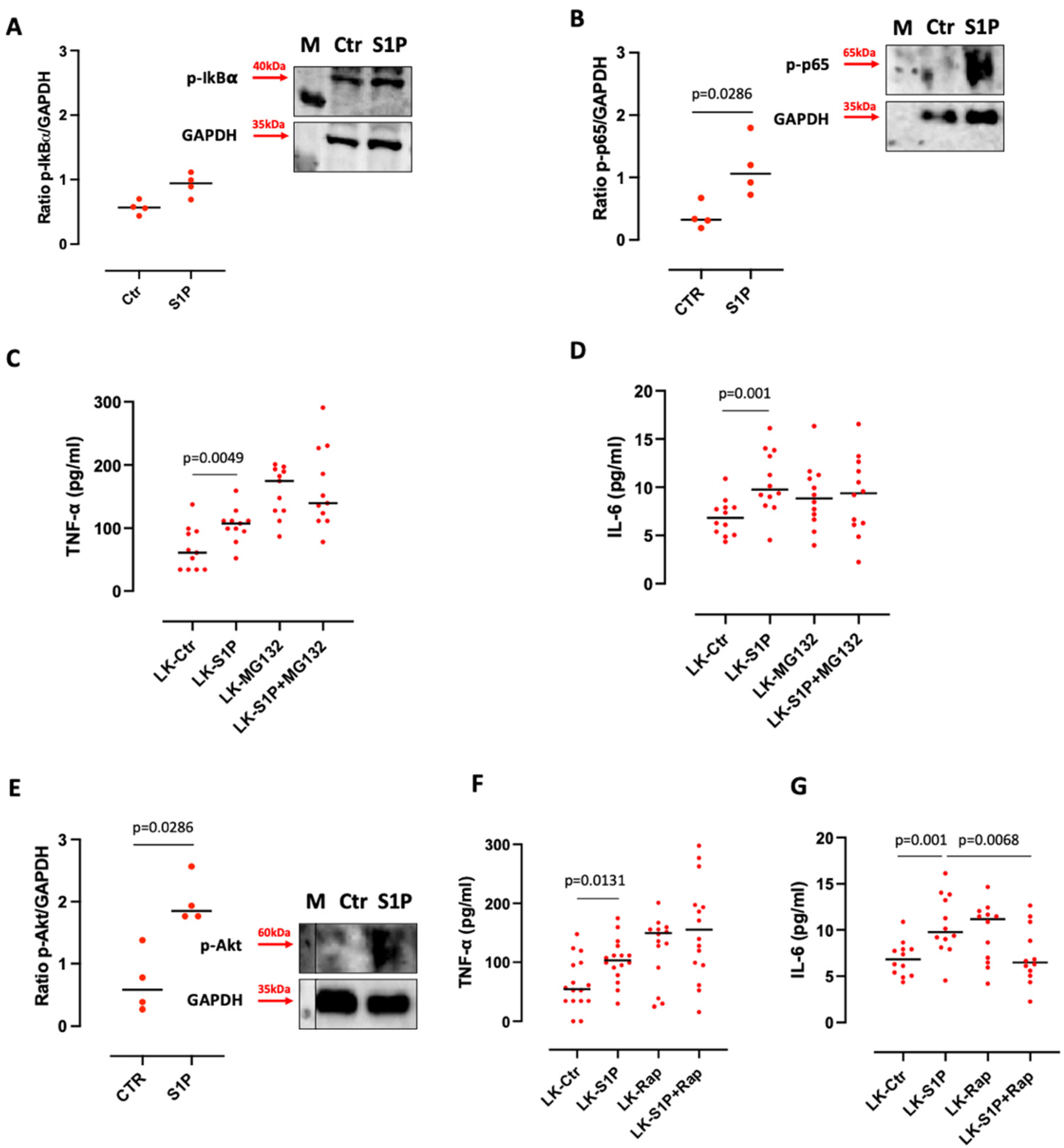
3.3. S1P-Induced TNF-α and IL-6 Release from Lung Cancer-Derived PBMCs Was Ceramidase-Dependent
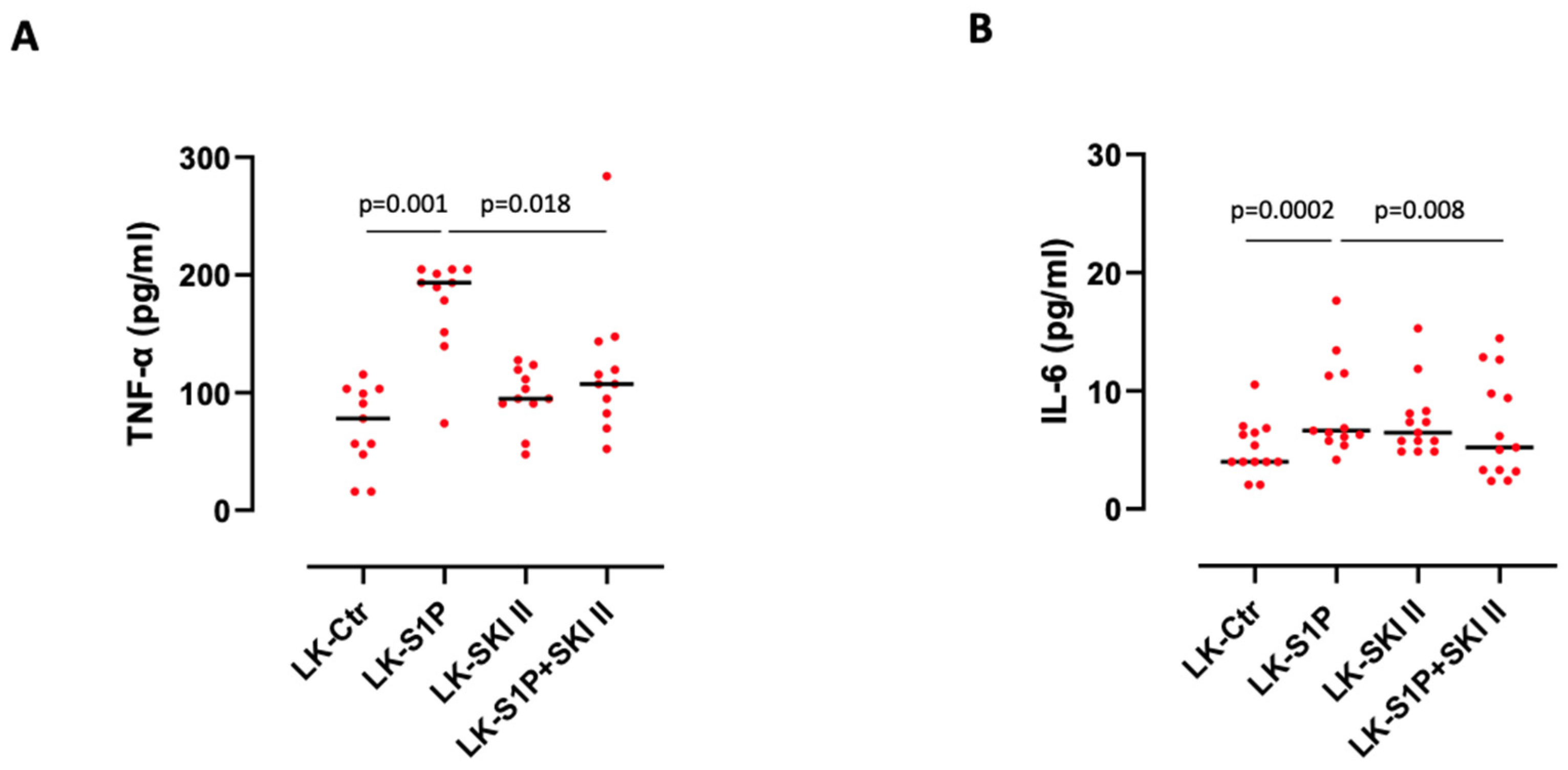
3.4. The Inhibition of SPHKs Reduced the Release of TNF-α and IL-6 after S1P Stimulation of Lung Cancer-Derived PBMCs
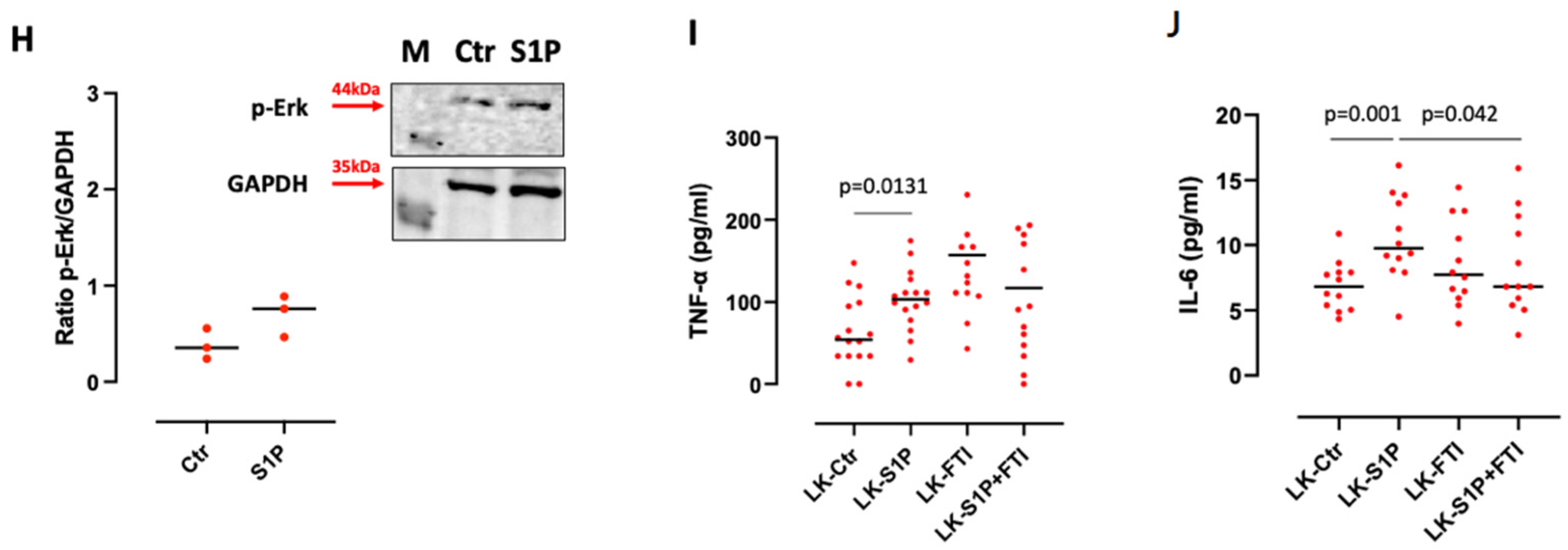
3.5. S1P-Induced IL-6 Release from Lung Cancer-Derived PBMCs Was mTOR and K-Ras-Dependent
4. Discussion
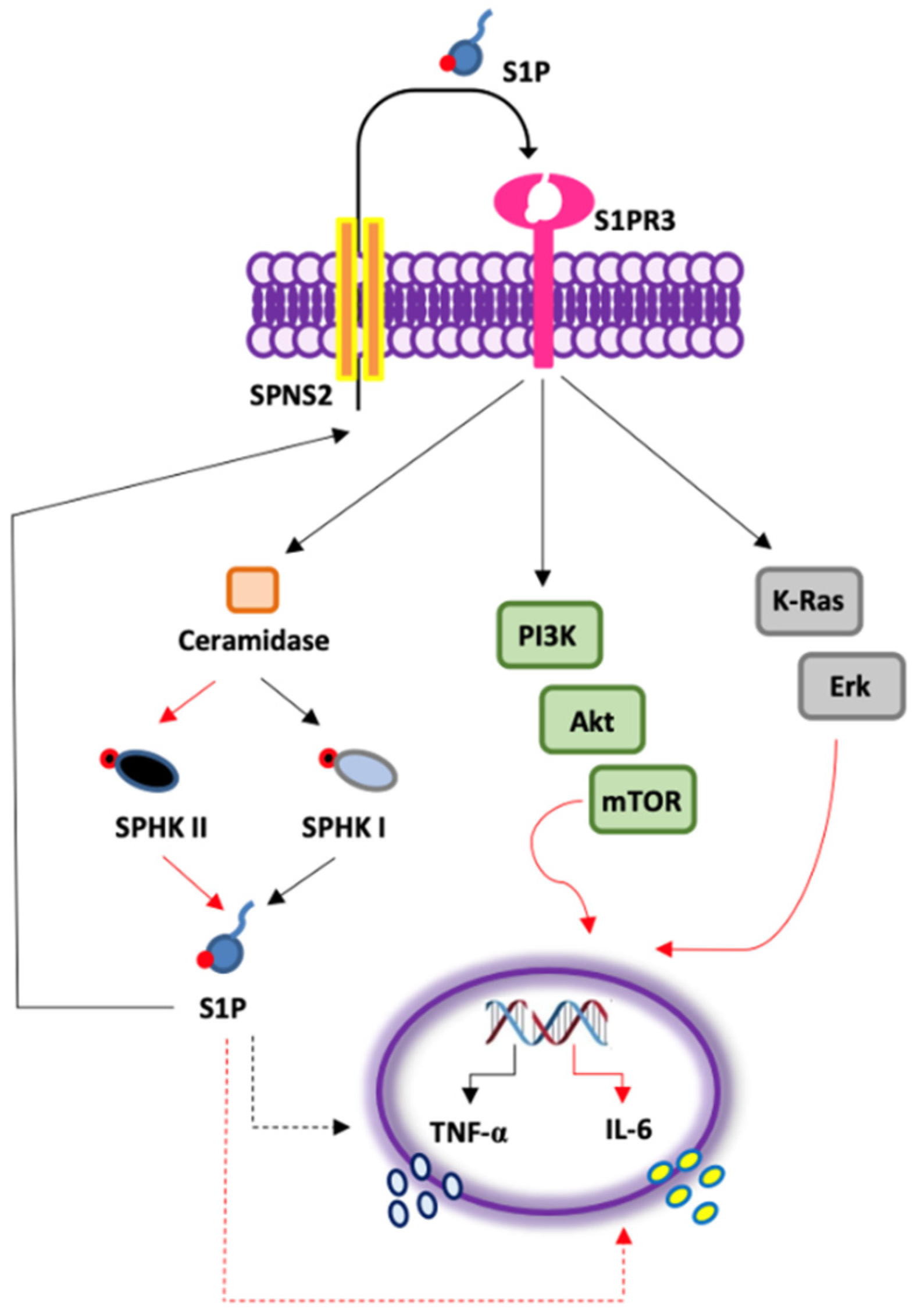
- S1P exacerbates the pro-inflammatory milieu by inducing IL-6 and TNF-α release from LK-derived PBMCs in a S1PR3-dependent manner;
- The activation of S1PR3 by the exogenous S1P induces the release of TNF-α in a SPHK I-dependent manner, and of IL-6 via SPHK I/II;
- S1P-induced IL-6, but not TNF-α, release from PBMCs of lung cancer patients is mTOR- and K-Ras-, but not NF-κB-dependent (Figure 6).
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef]
- Brinkmann, V. Sphingosine 1-phosphate receptors in health and disease: Mechanistic insights from gene deletion studies and reverse pharmacology. Pharm. Ther. 2007, 115, 84–105. [Google Scholar] [CrossRef]
- Mohammed, S.; Harikumar, K.B. Sphingosine 1-Phosphate: A Novel Target for Lung Disorders. Front. Immunol. 2017, 8, 296. [Google Scholar] [CrossRef]
- Terlizzi, M.; Colarusso, C.; Ferraro, G.; Monti, M.C.; Cerqua, I.; Roviezzo, F.; Pinto, A.; Sorrentino, R. Sphingosine-1-Phosphate Contributes to TLR9-Induced TNF-α Release in Lung Tumor Cells. Cell Physiol. Biochem. 2021, 55, 222–234. [Google Scholar] [PubMed]
- Terlizzi, M.; Colarusso, C.; Ferraro, G.; Monti, M.C.; Rosa, I.; Somma, P.; Salvi, R.; Pinto, A.; Sorrentino, R. Intracellular Sphingosine-1-Phosphate Receptor 3 Contributes to Lung Tumor Cell Proliferation. Cell Physiol. Biochem. 2021, 55, 539–552. [Google Scholar]
- Riboni, L.; Abdel Hadi, L.; Navone, S.E.; Guarnaccia, L.; Campanella, R.; Marfia, G. Sphingosine-1-Phosphate in the Tumor Microenvironment: A Signaling Hub Regulating Cancer Hallmarks. Cells 2020, 9, 337. [Google Scholar] [CrossRef]
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295. [Google Scholar] [CrossRef]
- Pyne, S.; Edwards, J.; Ohotski, J.; Pyne, N.J. Sphingosine 1-phosphate receptors and sphingosine kinase 1: Novel biomarkers for clinical prognosis in breast, prostate, and hematological cancers. Front. Oncol. 2012, 2, 168. [Google Scholar] [CrossRef]
- Alshaker, H.; Thrower, H.; Pchejetski, D. Sphingosine Kinase 1 in Breast Cancer-A New Molecular Marker and a Therapy Target. Front. Oncol. 2020, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Han, M.H. Sphingosine-1-Phosphate (S1P) and S1P Signaling Pathway: Therapeutic Targets in Autoimmunity and Inflammation. Drugs 2016, 76, 1067–1079. [Google Scholar] [CrossRef]
- Rodriguez, Y.I.; Campos, L.E.; Castro, M.G.; Aladhami, A.; Oskeritzian, C.A.; Alvarez, S.E. Sphingosine-1 Phosphate: A New Modulator of Immune Plasticity in the Tumor Microenvironment. Front. Oncol. 2016, 6, 218. [Google Scholar] [CrossRef]
- Colarusso, C.; Terlizzi, M.; Molino, A.; Imitazione, P.; Somma, P.; Rega, R.; Saccomanno, A.; Aquino, R.P.; Pinto, A.; Sorrentino, R. AIM2 Inflammasome Activation Leads to IL-1α and TGF-β Release from Exacerbated Chronic Obstructive Pulmonary Disease-Derived Peripheral Blood Mononuclear Cells. Front. Pharm. 2019, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Goliwas, K.F.; Deshane, J.S. Sphingolipids in Lung Pathology in the Coronavirus Disease Era: A Review of Sphingolipid Involvement in the Pathogenesis of Lung Damage. Front. Physiol. 2021, 12, 760638. [Google Scholar] [CrossRef]
- Wang, X.; Sun, Y.; Peng, X.; Naqvi, S.M.A.S.; Yang, Y.; Zhang, J.; Chen, M.; Chen, Y.; Chen, H.; Yan, H.; et al. The Tumorigenic Effect of Sphingosine Kinase 1 and Its Potential Therapeutic Target. Cancer Control. 2020, 27, 1073274820976664. [Google Scholar] [CrossRef]
- Cao, M.; Ji, C.; Zhou, Y.; Huang, W.; Ni, W.; Tong, X.; Wei, J.F. Sphingosine kinase inhibitors: A patent review. Int. J. Mol. Med. 2018, 41, 2450–2460. [Google Scholar] [CrossRef]
- Blom, T.; Bergelin, N.; Meinander, A.; Löf, C.; Slotte, J.P.; Eriksson, J.E.; Törnquist, K. An autocrine sphingosine-1-phosphate signaling loop enhances NF-kappaB-activation and survival. BMC Cell Biol. 2010, 11, 45. [Google Scholar] [CrossRef]
- Beckham, T.H.; Cheng, J.C.; Lu, P.; Shao, Y.; Troyer, D.; Lance, R.; Marrison, S.T.; Norris, J.S.; Liu, X. Acid ceramidase induces sphingosine kinase 1/S1P receptor 2-mediated activation of oncogenic Akt signaling. Oncogenesis 2013, 2, e49. [Google Scholar] [CrossRef]
- Gault, C.R.; Eblen, S.T.; Neumann, C.A.; Hannun, Y.A.; Obeid, L.M. Oncogenic K-Ras regulates bioactive sphingolipids in a sphingosine kinase 1-dependent manner. J. Biol. Chem. 2012, 287, 31794–31803. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Recent advances in the role of sphingosine 1-phosphate in cancer. FEBS Lett. 2020, 594, 3583–3601. [Google Scholar] [CrossRef]
- Tang, X.; Chen, H.; Chen, G.; Duan, C.; Fan, Q.; Li, H.; Wang, Y.; Li, Z.; Shi, W.; Liu, Y. Validated LC-MS/MS method of Sphingosine 1-phosphate quantification in human serum for evaluation of response to radiotherapy in lung cancer. Thorac. Cancer 2020, 11, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-ras-Mutant Lung Cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Hsiao, L.D.; Su, M.H.; Yang, C.M. Sphingosine 1-Phosphate Induces Cyclooxygenase-2/Prostaglandin E2Expression via PKCα-dependent Mitogen-Activated Protein Kinases and NF-κB Cascade in Human Cardiac Fibroblasts. Front. Pharm. 2020, 11, 569802. [Google Scholar] [CrossRef]
- Wang, C.; Xu, T.; Lachance, B.B.; Zhong, X.; Shen, G.; Xu, T.; Tang, C.; Jia, X. Critical roles of sphingosine kinase 1 in the regulation of neuroinflammation and neuronal injury after spinal cord injury. J. Neuroinflamm. 2021, 18, 50. [Google Scholar] [CrossRef]
- Berndt, N.; Patel, R.; Yang, H.; Balasis, M.E.; Sebti, S.M. Akt2 and acid ceramidase cooperate to induce cell invasion and resistance to apoptosis. Cell Cycle 2013, 12, 2024–2032. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.M.; Kwon, H.J. Acid ceramidase, an emerging target for anti-cancer and anti-angiogenesis. Arch. Pharm. Res. 2019, 42, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Monick, M.M.; Mallampalli, R.K.; Bradford, M.; McCoy, D.; Gross, T.J.; Flaherty, D.M.; Powers, L.S.; Cameron, K.; Kelly, S.; Merrill, A.H., Jr.; et al. Cooperative prosurvival activity by ERK and Akt in human alveolar macrophages is dependent on high levels of acid ceramidase activity. J. Immunol. 2004, 173, 123–135. [Google Scholar] [CrossRef]
- Yan, H.Q.; Huang, X.B.; Ke, S.Z.; Jiang, Y.N.; Zhang, Y.H.; Wang, Y.N.; Li, J.; Gao, F.G. Interleukin 6 augments lung cancer chemotherapeutic resistance via ataxia-telangiectasia mutated/NF-kappaB pathway activation. Cancer Sci. 2014, 105, 1220–1227. [Google Scholar] [CrossRef]
- Duan, S.; Tsai, Y.; Keng, P.; Chen, Y.; Lee, S.O.; Chen, Y. IL-6 signaling contributes to cisplatin resistance in non-small cell lung cancer via the up-regulation of anti-apoptotic and DNA repair associated molecules. Oncotarget 2015, 6, 27651–27660. [Google Scholar] [CrossRef]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terlizzi, M.; Colarusso, C.; Somma, P.; De Rosa, I.; Panico, L.; Pinto, A.; Sorrentino, R. S1P-Induced TNF-α and IL-6 Release from PBMCs Exacerbates Lung Cancer-Associated Inflammation. Cells 2022, 11, 2524. https://doi.org/10.3390/cells11162524
Terlizzi M, Colarusso C, Somma P, De Rosa I, Panico L, Pinto A, Sorrentino R. S1P-Induced TNF-α and IL-6 Release from PBMCs Exacerbates Lung Cancer-Associated Inflammation. Cells. 2022; 11(16):2524. https://doi.org/10.3390/cells11162524
Chicago/Turabian StyleTerlizzi, Michela, Chiara Colarusso, Pasquale Somma, Ilaria De Rosa, Luigi Panico, Aldo Pinto, and Rosalinda Sorrentino. 2022. "S1P-Induced TNF-α and IL-6 Release from PBMCs Exacerbates Lung Cancer-Associated Inflammation" Cells 11, no. 16: 2524. https://doi.org/10.3390/cells11162524