
Spectral Library-Based Single-Cell Proteomics Resolves Cellular Heterogeneity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
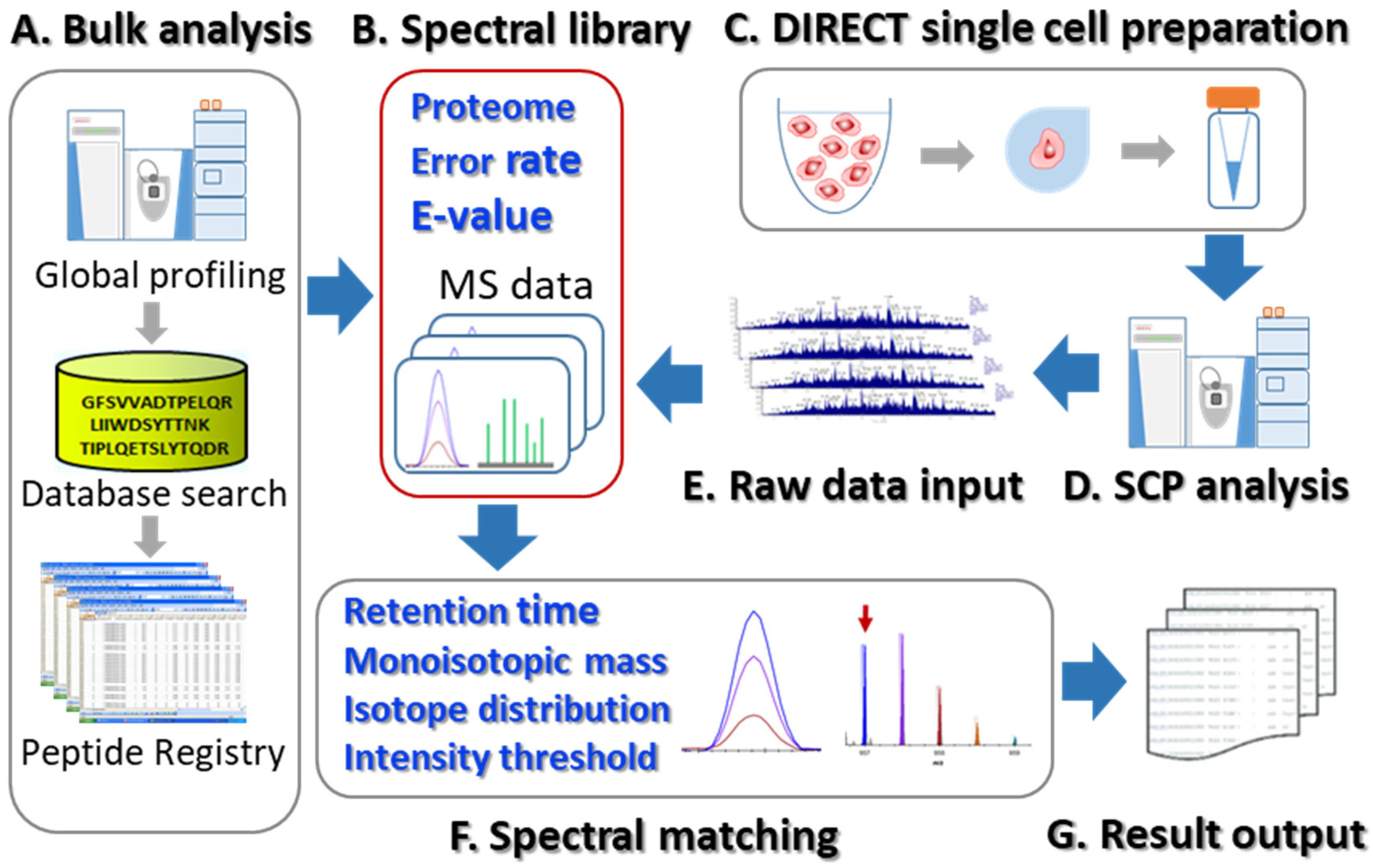
:1. Introduction
2. Materials and Methods
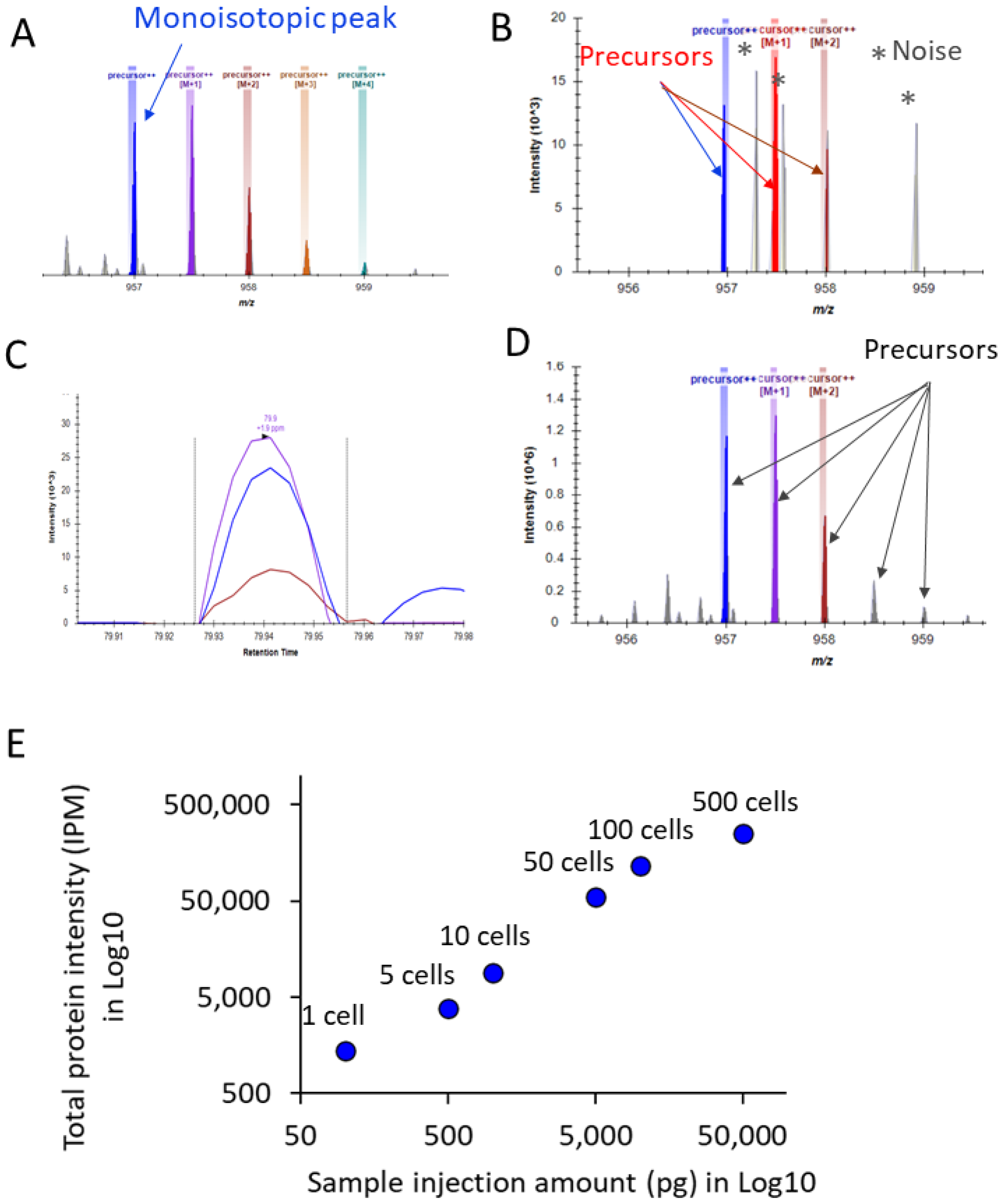
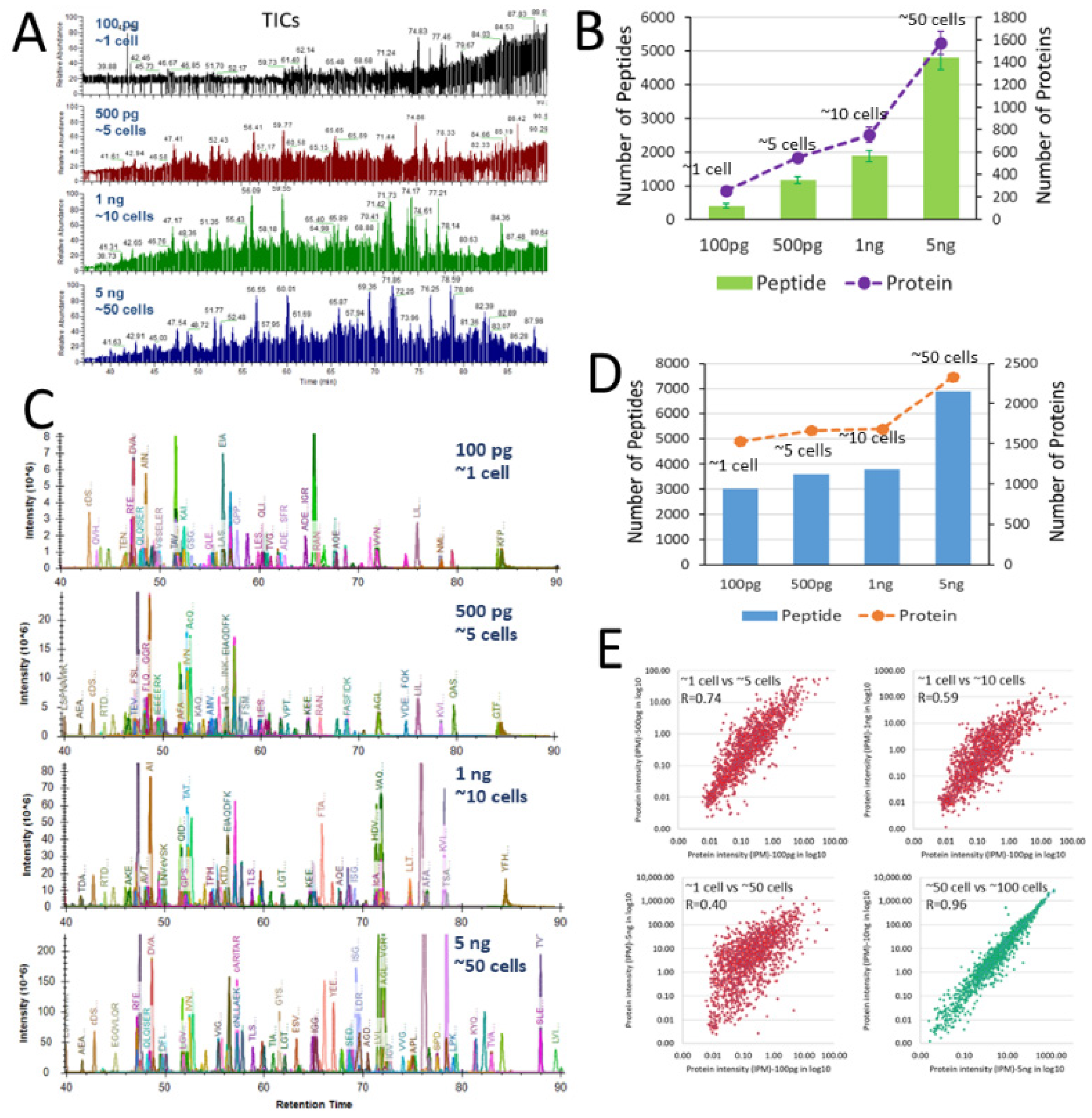
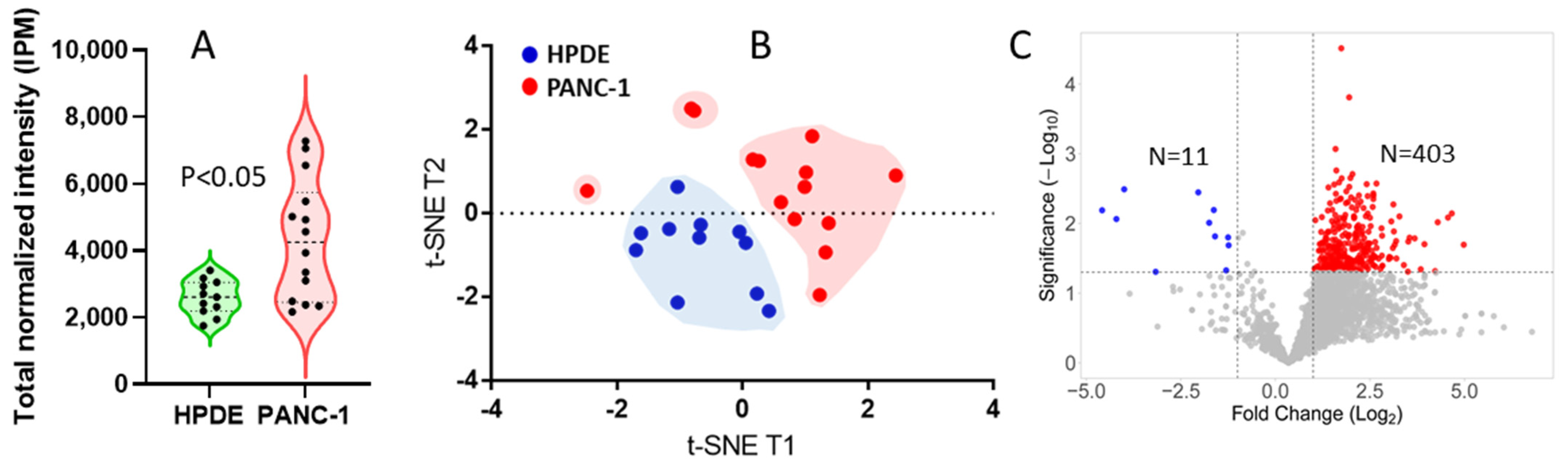
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perkel, J.M. Single-cell proteomics takes centre stage. Nature 2021, 597, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Slavov, N. Driving Single Cell Proteomics Forward with Innovation. J. Proteome Res. 2021, 20, 4915–4918. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Chen, S. Single-cell-type proteomics: Toward a holistic understanding of plant function. Mol. Cell. Proteom. 2012, 11, 1622–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavasso, S.; Gullaksen, S.E.; Skavland, J.; Gjertsen, B.T. Single-cell proteomics: Potential implications for cancer diagnostics. Expert Rev. Mol. Diagn. 2016, 16, 579–589. [Google Scholar] [CrossRef]
- Kelly, R.T. Single-cell Proteomics: Progress and Prospects. Mol. Cell. Proteom. 2020, 19, 1739–1748. [Google Scholar] [CrossRef]
- Li, L.; Yan, S.; Lin, B.; Shi, Q.; Lu, Y. Single-Cell Proteomics for Cancer Immunotherapy. Adv. Cancer Res. 2018, 139, 185–207. [Google Scholar] [CrossRef]
- Liang, Y.; Truong, T.; Zhu, Y.; Kelly, R.T. In-Depth Mass Spectrometry-Based Single-Cell and Nanoscale Proteomics. Methods Mol. Biol. 2021, 2185, 159–179. [Google Scholar] [CrossRef]
- Marx, V. A dream of single-cell proteomics. Nat. Methods 2019, 16, 809–812. [Google Scholar] [CrossRef] [Green Version]
- Paul, I.; White, C.; Turcinovic, I.; Emili, A. Imaging the future: The emerging era of single-cell spatial proteomics. FEBS J. 2020, 288, 6990–7001. [Google Scholar] [CrossRef]
- Specht, H.; Slavov, N. Transformative Opportunities for Single-Cell Proteomics. J. Proteome Res. 2018, 17, 2565–2571. [Google Scholar] [CrossRef]
- Su, Y.; Shi, Q.; Wei, W. Single cell proteomics in biomedicine: High-dimensional data acquisition, visualization, and analysis. Proteomics 2017, 17, 1600267. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, F. Emerging single-cell technologies for functional proteomics in oncology. Expert Rev. Proteom. 2016, 13, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R. Constellations in a cellular universe. Nature 2003, 422, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. [Google Scholar] [CrossRef] [Green Version]
- Cheung, T.K.; Lee, C.Y.; Bayer, F.P.; McCoy, A.; Kuster, B.; Rose, C.M. Defining the carrier proteome limit for single-cell proteomics. Nat. Methods 2021, 18, 76–83. [Google Scholar] [CrossRef]
- Tsai, C.F.; Zhao, R.; Williams, S.M.; Moore, R.J.; Schultz, K.; Chrisler, W.B.; Pasa-Tolic, L.; Rodland, K.D.; Smith, R.D.; Shi, T.; et al. An Improved Boosting to Amplify Signal with Isobaric Labeling (iBASIL) Strategy for Precise Quantitative Single-cell Proteomics. Mol. Cell. Proteom. 2020, 19, 828–838. [Google Scholar] [CrossRef] [Green Version]
- Schoof, E.M.; Furtwangler, B.; Uresin, N.; Rapin, N.; Savickas, S.; Gentil, C.; Lechman, E.; Keller, U.A.D.; Dick, J.E.; Porse, B.T. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021, 12, 3341. [Google Scholar] [CrossRef]
- Li, Z.Y.; Huang, M.; Wang, X.K.; Zhu, Y.; Li, J.S.; Wong, C.C.L.; Fang, Q. Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis. Anal. Chem. 2018, 90, 5430–5438. [Google Scholar] [CrossRef]
- Williams, S.M.; Liyu, A.V.; Tsai, C.F.; Moore, R.J.; Orton, D.J.; Chrisler, W.B.; Gaffrey, M.J.; Liu, T.; Smith, R.D.; Kelly, R.T.; et al. Automated Coupling of Nanodroplet Sample Preparation with Liquid Chromatography-Mass Spectrometry for High-Throughput Single-Cell Proteomics. Anal. Chem. 2020, 92, 10588–10596. [Google Scholar] [CrossRef]
- Kalxdorf, M.; Muller, T.; Stegle, O.; Krijgsveld, J. IceR improves proteome coverage and data completeness in global and single-cell proteomics. Nat. Commun. 2021, 12, 4787. [Google Scholar] [CrossRef] [PubMed]
- Gebreyesus, S.T.; Siyal, A.A.; Kitata, R.B.; Chen, E.S.; Enkhbayar, B.; Angata, T.; Lin, K.I.; Chen, Y.J.; Tu, H.L. Streamlined single-cell proteomics by an integrated microfluidic chip and data-independent acquisition mass spectrometry. Nat. Commun. 2022, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Motamedchaboki, K.; Misal, S.A.; Liang, Y.; Guise, A.J.; Truong, T.; Huguet, R.; Plowey, E.D.; Zhu, Y.; Lopez-Ferrer, D.; et al. Ultrasensitive single-cell proteomics workflow identifies >1000 protein groups per mammalian cell. Chem. Sci. 2020, 12, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.D.; Thielert, M.; Vasilopoulou, C.; Ammar, C.; Coscia, F.; Mund, A.; Hoerning, O.B.; Bache, N.; Apalategui, A.; Lubeck, M.; et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Biol. 2022, 18, e10798. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Wang, X.; Guan, S.; Lin, H.; Yan, G.; Gao, M.; Deng, C.; Zhang, X. Integrated Proteome Analysis Device for Fast Single-Cell Protein Profiling. Anal. Chem. 2018, 90, 14003–14010. [Google Scholar] [CrossRef]
- Zhu, Y.; Piehowski, P.D.; Zhao, R.; Chen, J.; Shen, Y.; Moore, R.J.; Shukla, A.K.; Petyuk, V.A.; Campbell-Thompson, M.; Mathews, C.E.; et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun. 2018, 9, 882. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, P.; Rose, C.M. Understanding the effect of carrier proteomes in single cell proteomic studies—Key lessons. Expert Rev. Proteom. 2022, 19, 5–15. [Google Scholar] [CrossRef]
- Rosenberger, G.; Koh, C.C.; Guo, T.; Rost, H.L.; Kouvonen, P.; Collins, B.C.; Heusel, M.; Liu, Y.; Caron, E.; Vichalkovski, A.; et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data 2014, 1, 140031. [Google Scholar] [CrossRef]
- Schubert, O.T.; Gillet, L.C.; Collins, B.C.; Navarro, P.; Rosenberger, G.; Wolski, W.E.; Lam, H.; Amodei, D.; Mallick, P.; Maclean, B.; et al. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat. Protoc. 2015, 10, 426–441. [Google Scholar] [CrossRef]
- Pan, S.; Brand, R.E.; Lai, L.A.; Dawson, D.W.; Donahue, T.R.; Kim, S.; Khalaf, N.I.; Othman, M.O.; Fisher, W.E.; Bronner, M.P.; et al. Proteome heterogeneity and malignancy detection in pancreatic cyst fluids. Clin. Transl. Med. 2021, 11, e506. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Chen, R.; Brentnall, T.A.; Eng, J.K.; Picozzi, V.J.; Pan, S. Predictive proteomic signatures for response of pancreatic cancer patients receiving chemotherapy. Clin. Proteom. 2019, 16, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet: An open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Mendoza, L.; Shteynberg, D.; Slagel, J.; Sun, Z.; Moritz, R.L. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteom. Clin. Appl. 2015, 9, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef] [PubMed]
- Maclean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; Maccoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, J.K.; Fischer, B.; Grossmann, J.; Maccoss, M.J. A fast SEQUEST cross correlation algorithm. J. Proteome Res. 2008, 7, 4598–4602. [Google Scholar] [CrossRef] [PubMed]
- Leys, C.; Ley, C.; Klein, O.; Bernard, P.; Licata, L. Detecting outliers: Do not use standard deviation around the mean, use absolute deviation around the median. J. Exp. Soc. Psychol. 2013, 49, 764–766. [Google Scholar] [CrossRef] [Green Version]
- Belkina, A.C.; Ciccolella, C.O.; Anno, R.; Halpert, R.; Spidlen, J.; Snyder-Cappione, J.E. Automated optimized parameters for T-distributed stochastic neighbor embedding improve visualization and analysis of large datasets. Nat. Commun. 2019, 10, 5415. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Kovatch, J.R.; Badiong, A.; Merbouh, N. Optimization and Modeling of Quadrupole Orbitrap Parameters for Sensitive Analysis toward Single-Cell Proteomics. J. Proteome Res. 2017, 16, 3711–3721. [Google Scholar] [CrossRef] [PubMed]
- Sperling, E.; Bunner, A.E.; Sykes, M.T.; Williamson, J.R. Quantitative analysis of isotope distributions in proteomic mass spectrometry using least-squares Fourier transform convolution. Anal. Chem. 2008, 80, 4906–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valkenborg, D.; Jansen, I.; Burzykowski, T. A model-based method for the prediction of the isotopic distribution of peptides. J. Am. Soc. Mass Spectrom. 2008, 19, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Xiao, K.; Yu, F.; Fang, H.; Xue, B.; Liu, Y.; Tian, Z. Accurate and Efficient Resolution of Overlapping Isotopic Envelopes in Protein Tandem Mass Spectra. Sci. Rep. 2015, 5, 14755. [Google Scholar] [CrossRef] [Green Version]
- May, D.; Fitzgibbon, M.; Liu, Y.; Holzman, T.; Eng, J.; Kemp, C.J.; Whiteaker, J.; Paulovich, A.; McIntosh, M. A platform for accurate mass and time analyses of mass spectrometry data. J. Proteome Res. 2007, 6, 2685–2694. [Google Scholar] [CrossRef]
- May, D.; Liu, Y.; Law, W.; Fitzgibbon, M.; Wang, H.; Hanash, S.; McIntosh, M. Peptide sequence confidence in accurate mass and time analysis and its use in complex proteomics experiments. J. Proteome Res. 2008, 7, 5148–5156. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, J.S.; Monroe, M.E.; Qian, W.J.; Smith, R.D. Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom. Rev. 2006, 25, 450–482. [Google Scholar] [CrossRef] [Green Version]
- May, D.; Pan, S.; Crispin, D.A.; Lai, K.; Bronner, M.P.; Hogan, J.; Hockenbery, D.M.; McIntosh, M.; Brentnall, T.A.; Chen, R. Investigating neoplastic progression of ulcerative colitis with label-free comparative proteomics. J. Proteome Res. 2011, 10, 200–209. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Batth, T.S.; Ruther, P.; Olsen, J.V. A deeper look at carrier proteome effects for single-cell proteomics. Commun. Biol. 2022, 5, 150. [Google Scholar] [CrossRef]
- Bielow, C.; Mastrobuoni, G.; Kempa, S. Proteomics Quality Control: Quality Control Software for MaxQuant Results. J. Proteome Res. 2016, 15, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.Y.; Paulo, J.A.; Gygi, S.P. Evaluating False Transfer Rates from the Match-between-Runs Algorithm with a Two-Proteome Model. J. Proteome Res. 2019, 18, 4020–4026. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickaert, A.; Saiselet, M.; Dom, G.; De Deken, X.; Dumont, J.E.; Feron, O.; Sonveaux, P.; Maenhaut, C. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene 2017, 36, 2637–2642. [Google Scholar] [CrossRef] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Senga, S.S.; Grose, R.P. Hallmarks of cancer-the new testament. Open Biol. 2021, 11, 200358. [Google Scholar] [CrossRef]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senavirathna, L.; Ma, C.; Chen, R.; Pan, S. Spectral Library-Based Single-Cell Proteomics Resolves Cellular Heterogeneity. Cells 2022, 11, 2450. https://doi.org/10.3390/cells11152450
Senavirathna L, Ma C, Chen R, Pan S. Spectral Library-Based Single-Cell Proteomics Resolves Cellular Heterogeneity. Cells. 2022; 11(15):2450. https://doi.org/10.3390/cells11152450
Chicago/Turabian StyleSenavirathna, Lakmini, Cheng Ma, Ru Chen, and Sheng Pan. 2022. "Spectral Library-Based Single-Cell Proteomics Resolves Cellular Heterogeneity" Cells 11, no. 15: 2450. https://doi.org/10.3390/cells11152450