
Comprehensive Omics Analysis of a Novel Small-Molecule Inhibitor of Chemoresistant Oncogenic Signatures in Colorectal Cancer Cell with Antitumor Effects


 ,
,  and
and 
Abstract
:1. Introduction
2. Material and Methods
2.1. Identifying Molecular Targets and Therapeutic Classes of RV59
2.2. Gene Expression Microarray Data Extraction
2.3. Validation of MYC/CXCL8/TIMP1 Expression Levels in CRC
2.4. Protein-Protein Interaction (PPI) Network, Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analyses
2.5. Interpretation of Gene Co-Expression in MYC/CXCL8/TIMP1 Genes Network
2.6. Correlation Analysis of MYC/CXCL8/TIMP1 Expressions and Tumor Infiltration Levels
2.7. Drug Sensitivity Analysis of MYC/CXCL8/TIMP1 Oncogenes
2.8. Molecular Docking of Protein-Ligand Interactions
2.9. Statistical Analysis
3. Results
3.1. MYC/CXCL8/TIMP1 Oncogenes Are Potential Drug Targets for RV59
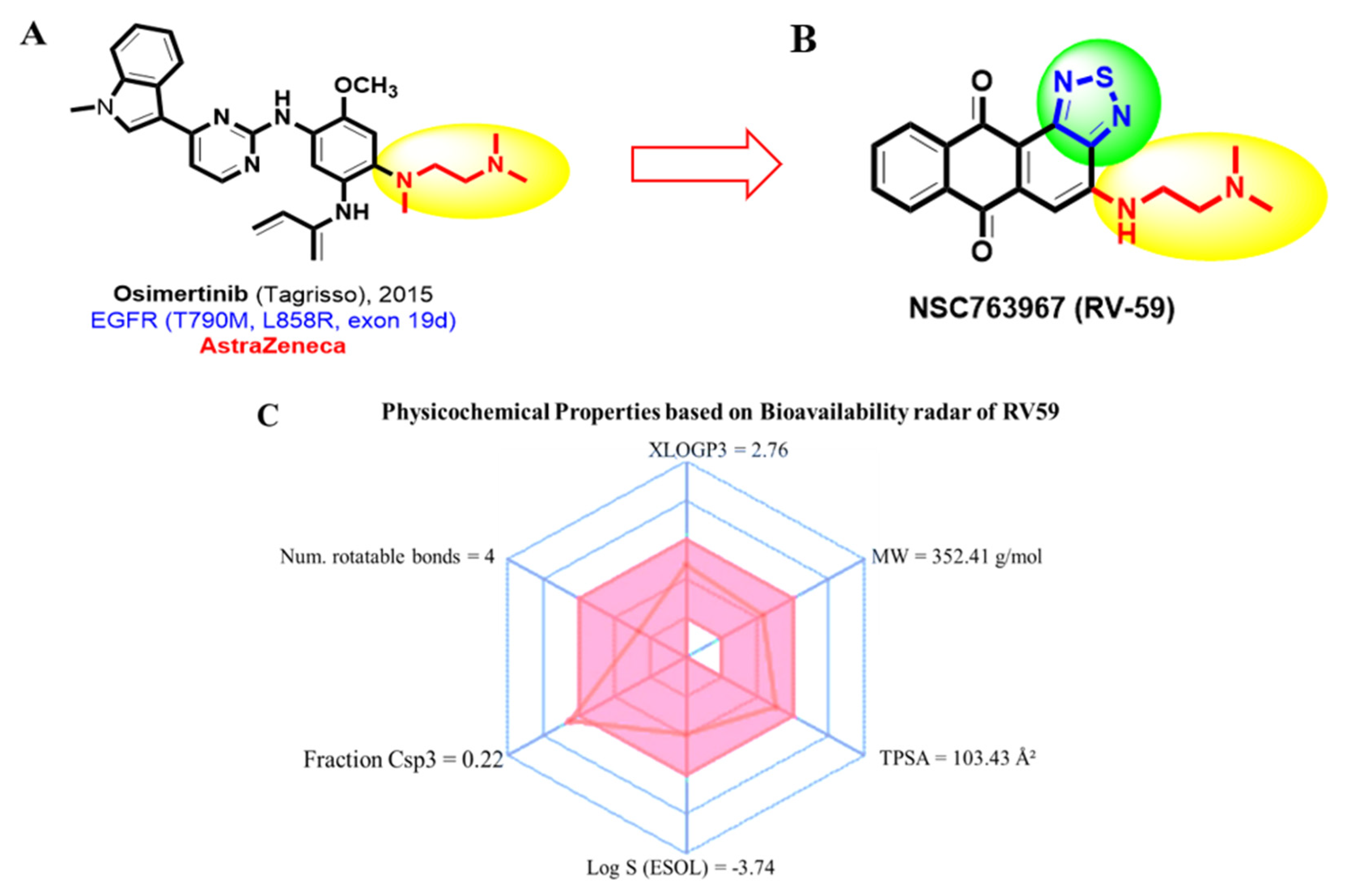
3.2. RV59 Passed the Required Drug-Likeness Criteria
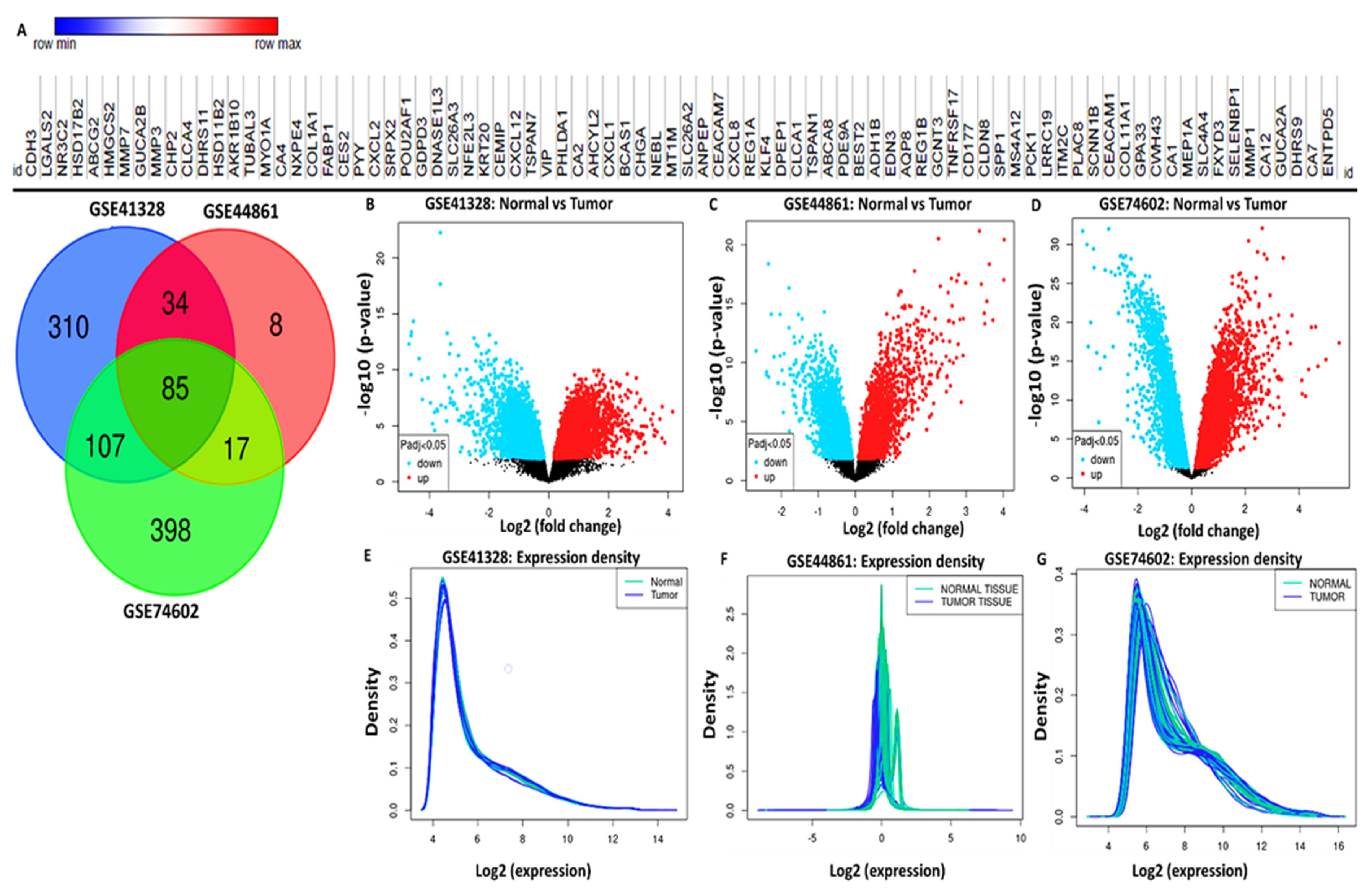
3.3. Identification of Differentially Expressed Genes (DEGs) in CRC
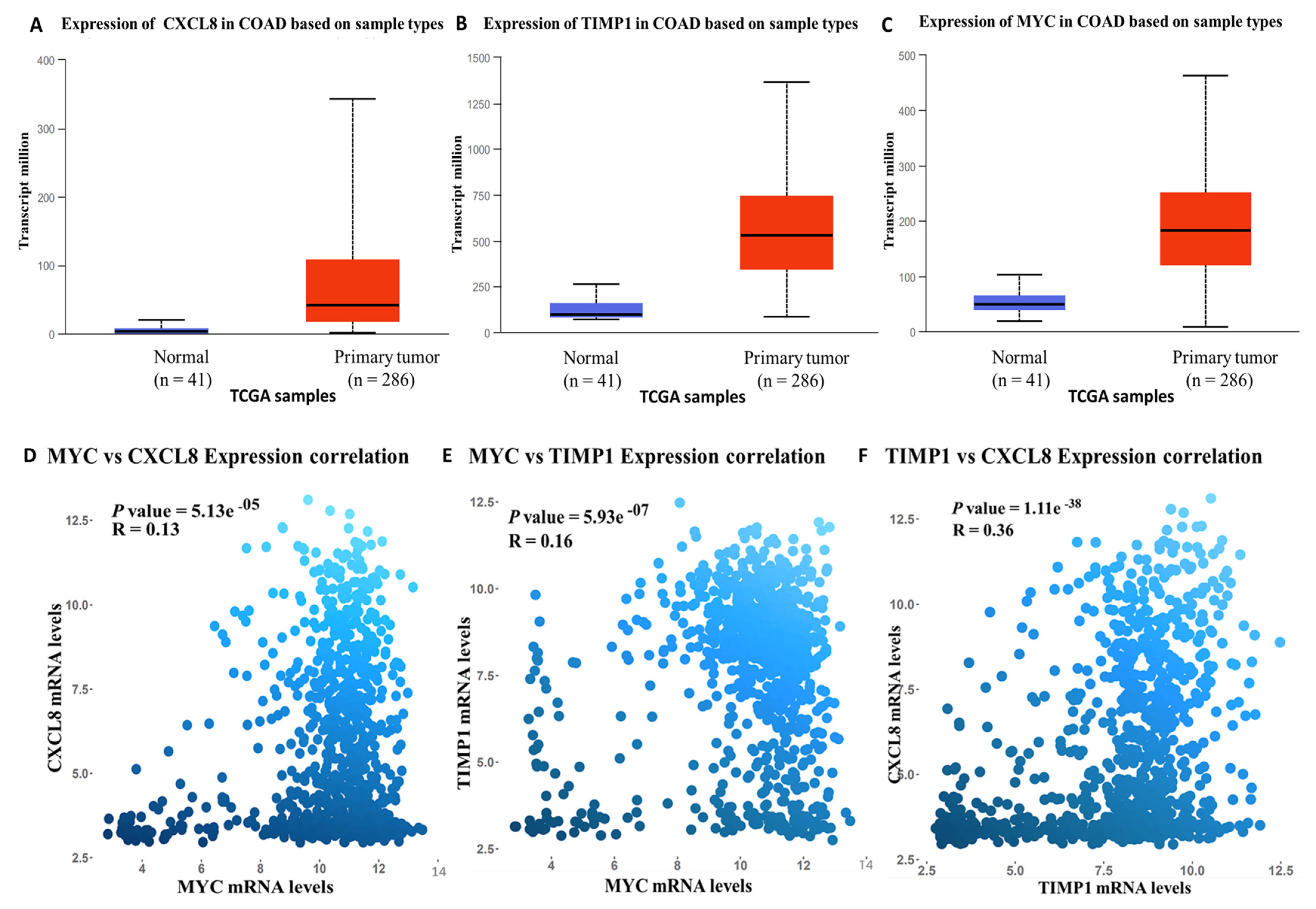
3.4. Validation of MYC/CXCL8/TIMP1 Expression Levels in CRC
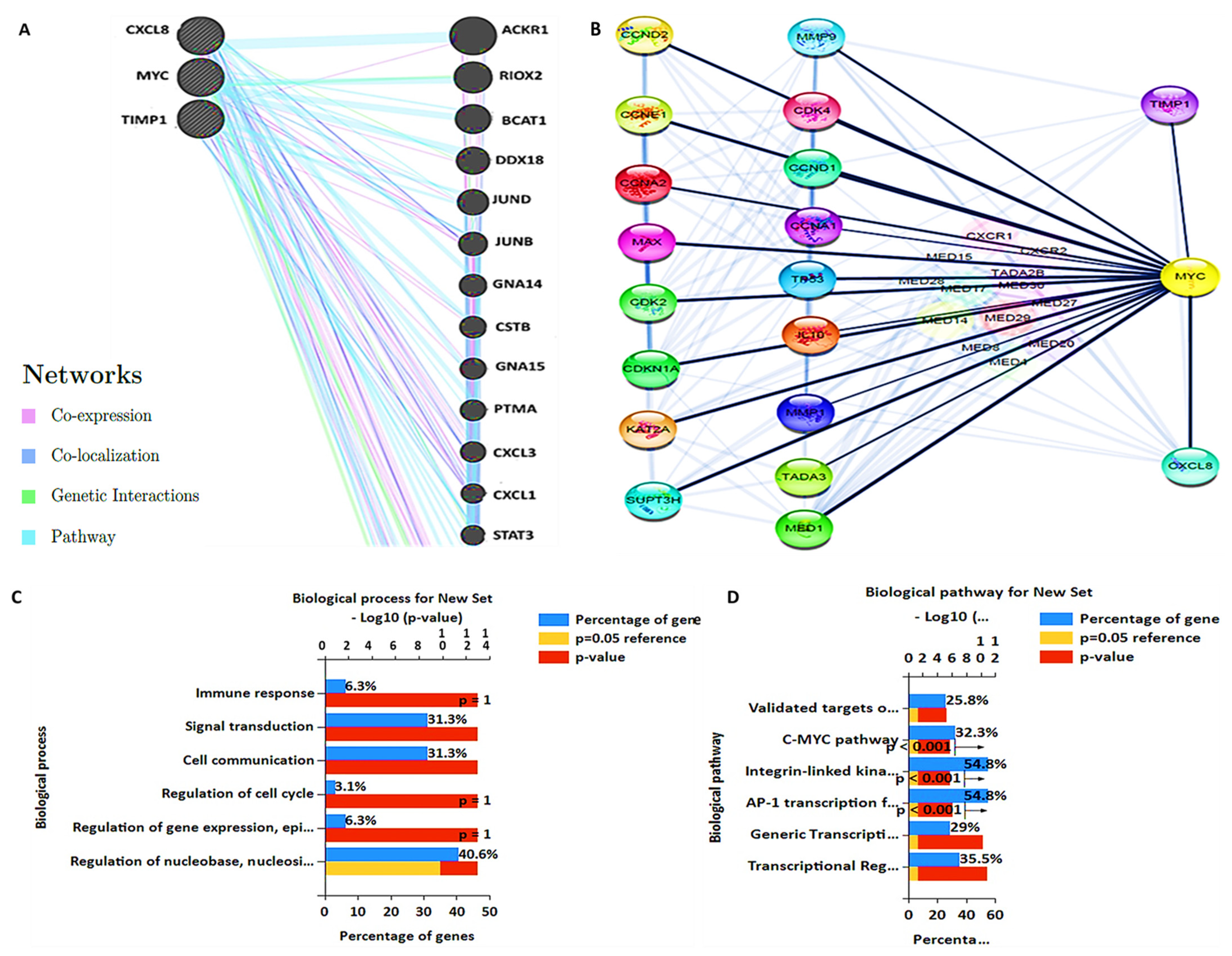
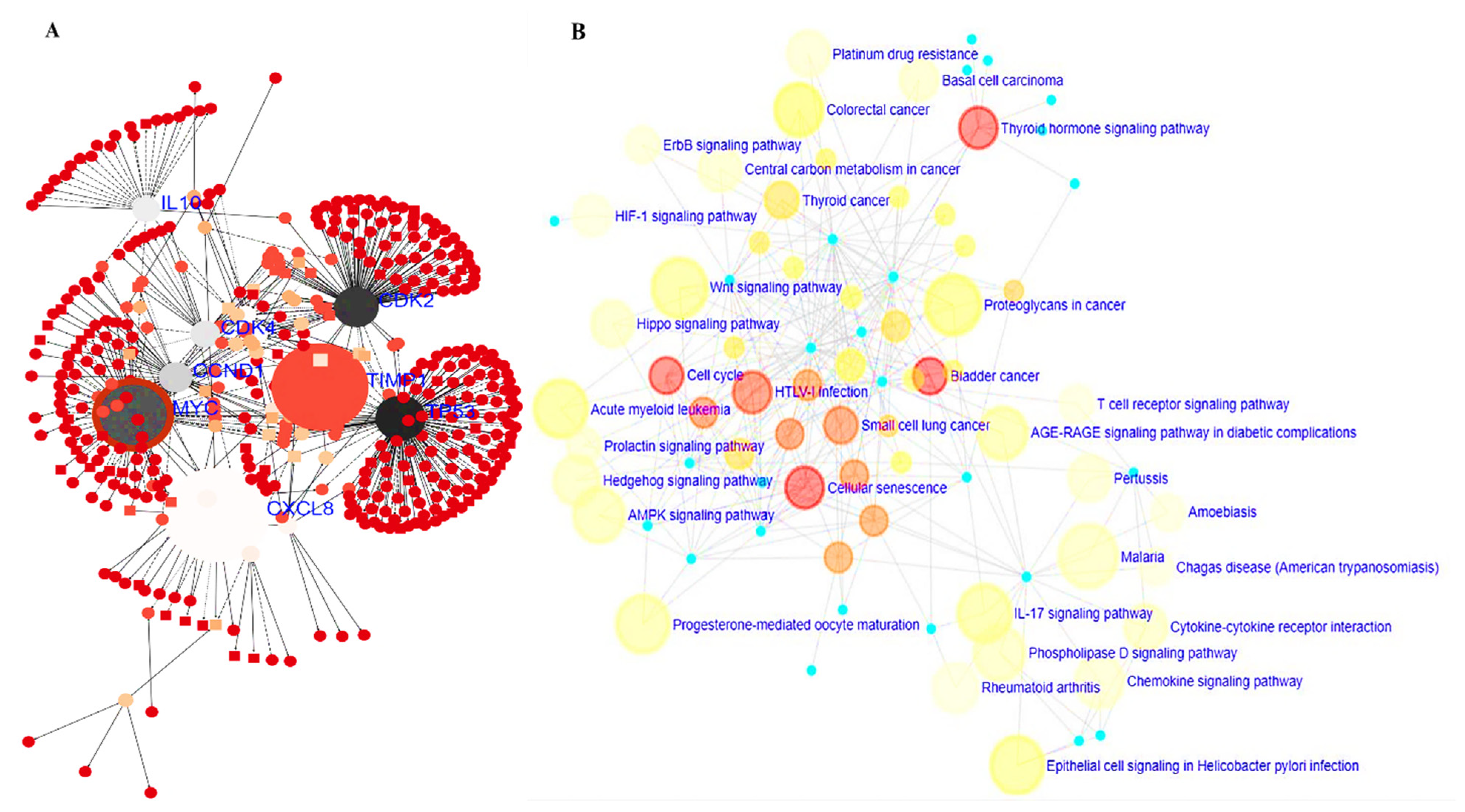
3.5. PPI Network and GO and KEGG Pathway Analysis
3.6. MYC/CXCL8/TIMP1 Gene Co-Expression and Functional Enrichment Analysis
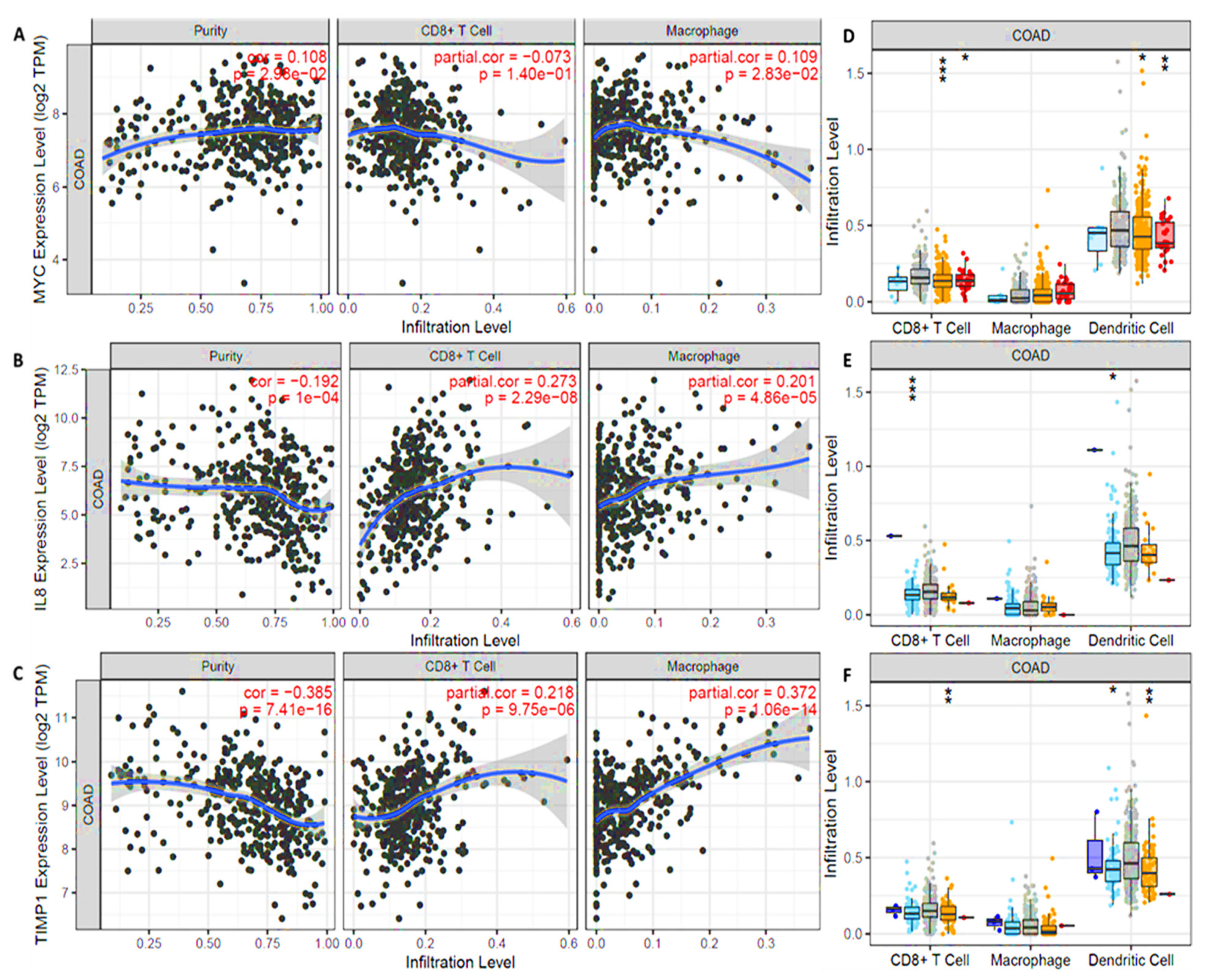
3.7. MYC/CXCL8/TIMP1 Expressions Were Correlated with Immune Cell Infiltration in Both Cancer and Normal Tissues
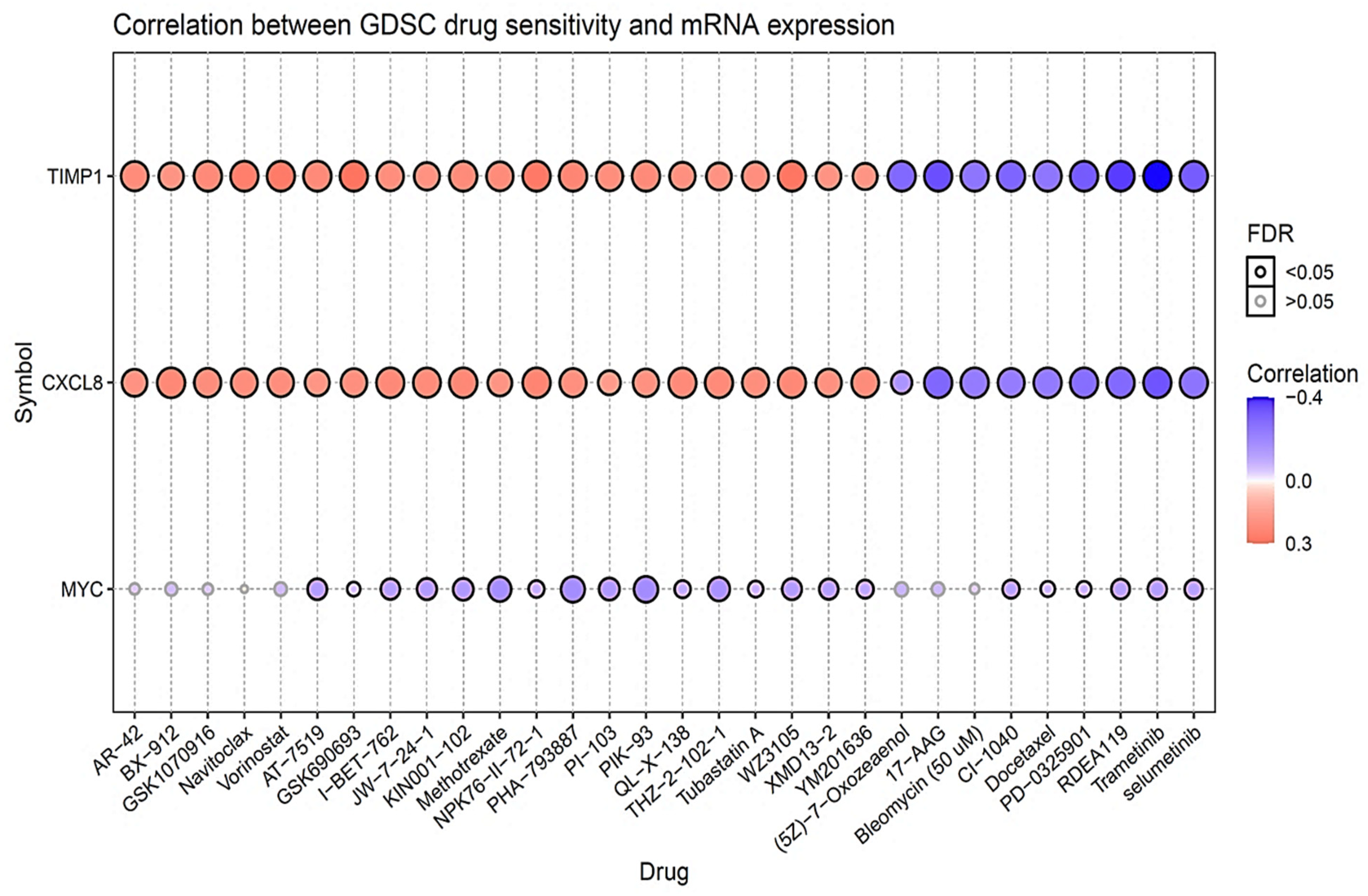
3.8. Drug Sensitivity Analysis of MYC/CXCL8/TIMP1 Oncogenes
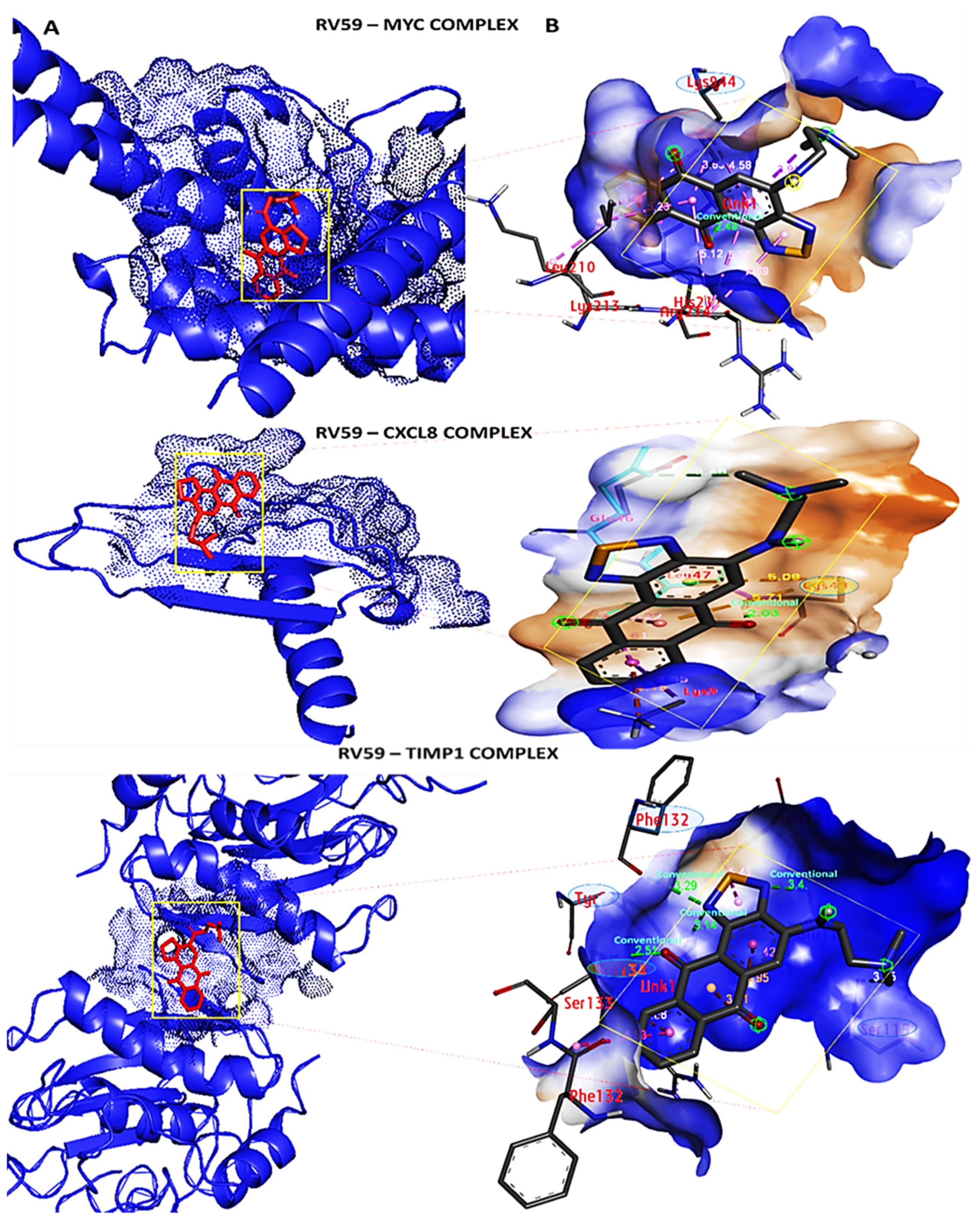
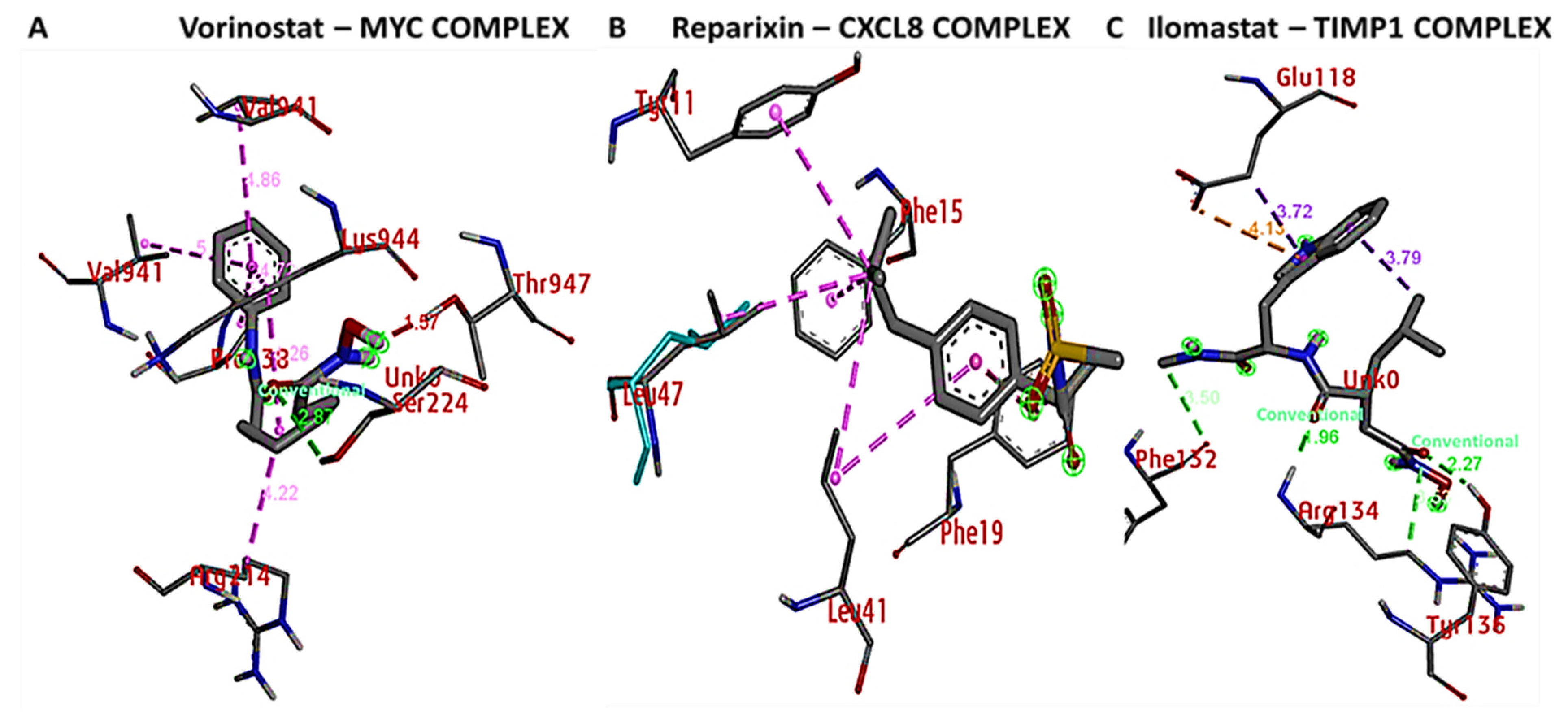
3.9. Docking Results Displayed Strong Binding Energies between RV59 and the MYC/CXCL8/TIMP1 Oncogenes
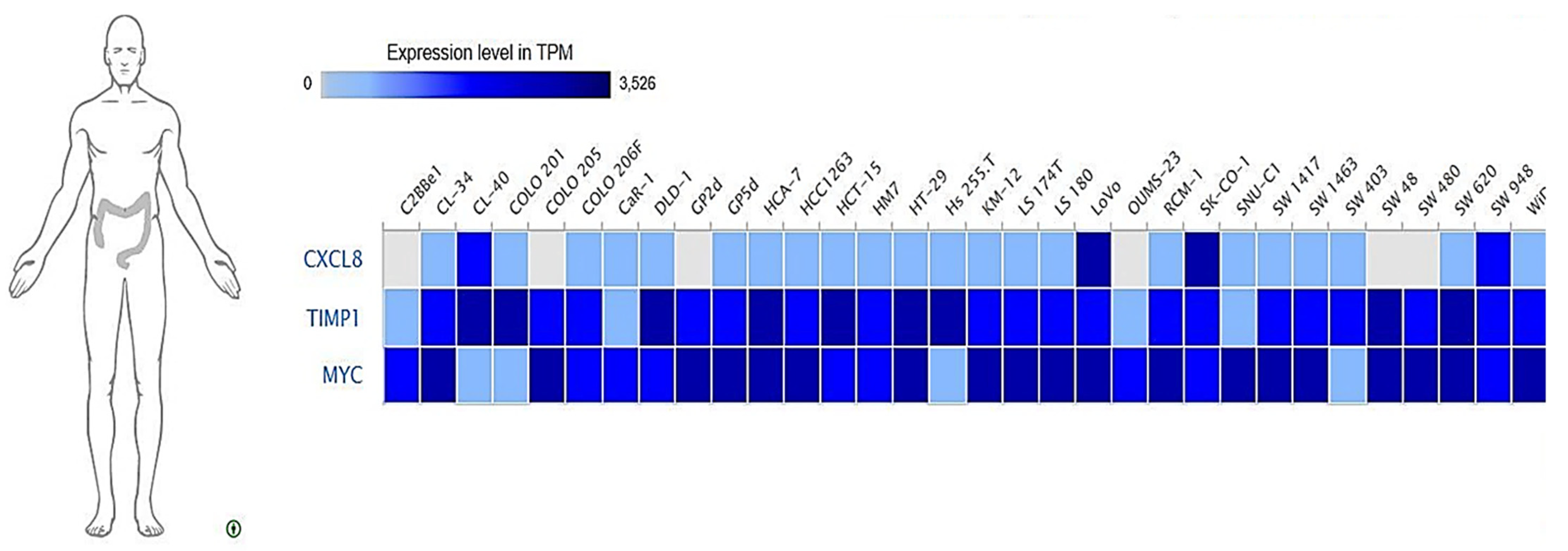
3.10. Expressions of MYC/CXCL8/TIMP1 Oncogenes across Colon Cancer Cell Lines
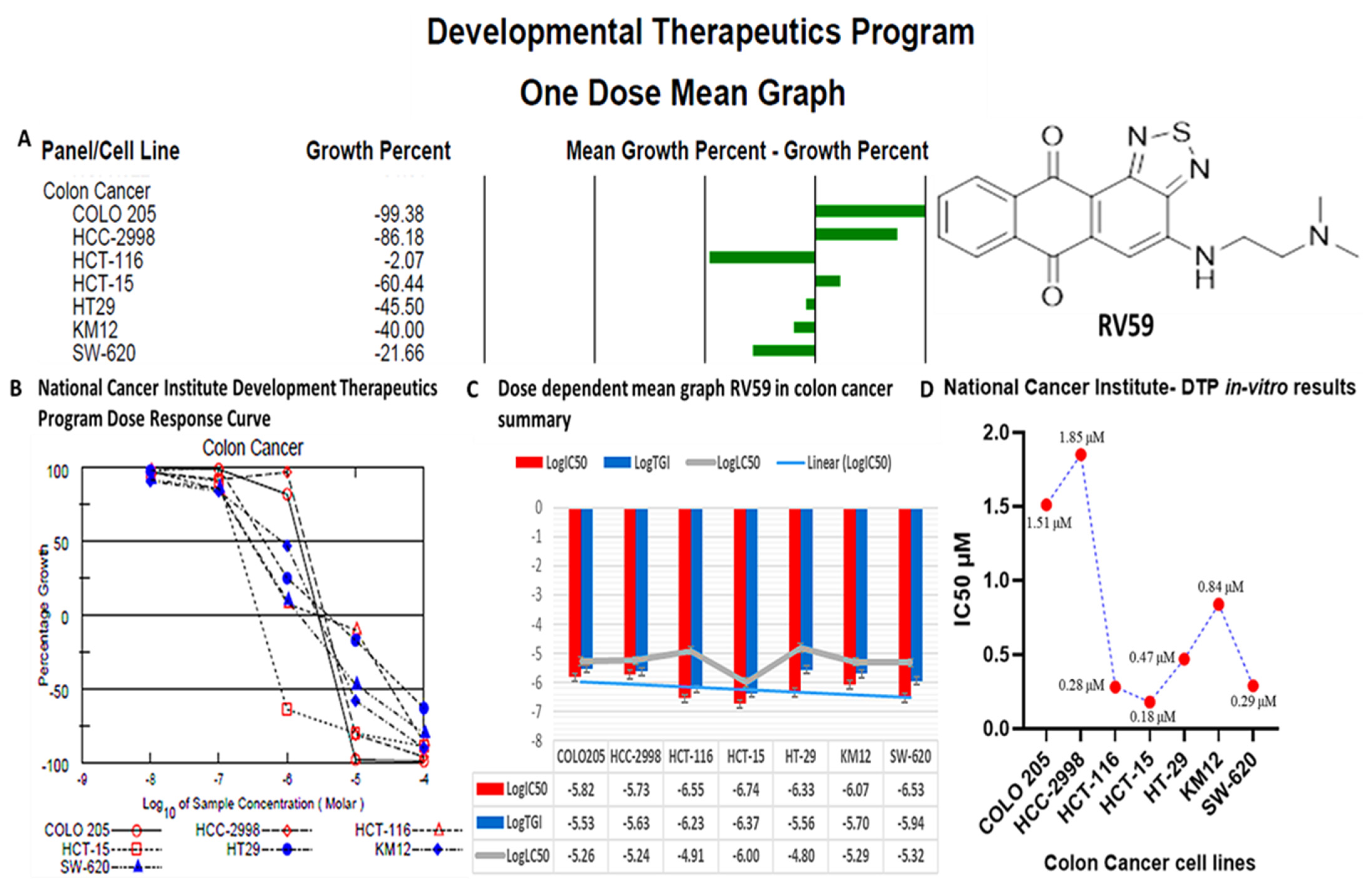
3.11. RV59 Displayed Anti-Proliferative and Cytotoxic Effects in NCI60 Human Colon Cancer Cell Lines
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Böckelman, C.; Beilmann-Lehtonen, I.; Kaprio, T.; Koskensalo, S.; Tervahartiala, T.; Mustonen, H.; Stenman, U.H.; Sorsa, T.; Haglund, C. Serum MMP-8 and TIMP-1 predict prognosis in colorectal cancer. BMC Cancer 2018, 18, 679. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Łukaszewicz-Zając, M.; Mroczko, B. Circulating Biomarkers of Colorectal Cancer (CRC)-Their Utility in Diagnosis and Prognosis. J. Clin. Med. 2021, 10, 2391. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, M.; Maeda, K.; Nagahara, H.; Fukuoka, T.; Iseki, Y.; Matsutani, S.; Kashiwagi, S.; Tanaka, H.; Hirakawa, K.; Ohira, M. Tumor-infiltrating Lymphocytes Predict the Chemotherapeutic Outcomes in Patients with Stage IV Colorectal Cancer. In Vivo 2018, 32, 151–158. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Marcuello, M.; Vymetalkova, V.; Neves, R.P.L.; Duran-Sanchon, S.; Vedeld, H.M.; Tham, E.; van Dalum, G.; Flügen, G.; Garcia-Barberan, V.; Fijneman, R.J.; et al. Circulating biomarkers for early detection and clinical management of colorectal cancer. Mol. Asp. Med. 2019, 69, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Wang, P.; Lu, M.Z.; Zhang, S.D.; Zheng, L. c-Myc maintains the self-renewal and chemoresistance properties of colon cancer stem cells. Oncol. Lett. 2019, 17, 4487–4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, E.C.; Hessman, C.; Levin, T.G.; Monroe, M.M.; Wong, M.H. The role of colorectal cancer stem cells in metastatic disease and therapeutic response. Cancers 2011, 3, 319–339. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Cao, D. The origin of cancer stem cells. Front. Biosci. 2012, 4, 819–830. [Google Scholar] [CrossRef] [Green Version]
- de Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.L.; Yang, M.H.; Tsai, M.L.; Lan, H.Y.; Su, S.H.; Chang, S.C.; Teng, H.W.; Yang, S.H.; Lan, Y.T.; Chiou, S.H.; et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology 2011, 141, 279–291.e5. [Google Scholar] [CrossRef] [Green Version]
- Bommer, G.T.; Fearon, E.R. Role of c-Myc in Apc mutant intestinal phenotype: Case closed or time for a new beginning? Cancer Cell 2007, 11, 391–394. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, P.J. beta-catenin signaling and cancer. Bioessays 1999, 21, 1021–1030. [Google Scholar] [CrossRef]
- de Sousa, E.M.F.; Colak, S.; Buikhuisen, J.; Koster, J.; Cameron, K.; de Jong, J.H.; Tuynman, J.B.; Prasetyanti, P.R.; Fessler, E.; van den Bergh, S.P.; et al. Methylation of cancer-stem-cell-associated Wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell 2011, 9, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Kugimiya, N.; Nishimoto, A.; Hosoyama, T.; Ueno, K.; Enoki, T.; Li, T.S.; Hamano, K. The c-MYC-ABCB5 axis plays a pivotal role in 5-fluorouracil resistance in human colon cancer cells. J. Cell Mol. Med. 2015, 19, 1569–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strippoli, A.; Cocomazzi, A.; Basso, M.; Cenci, T.; Ricci, R.; Pierconti, F.; Cassano, A.; Fiorentino, V.; Barone, C.; Bria, E.; et al. c-MYC Expression Is a Possible Keystone in the Colorectal Cancer Resistance to EGFR Inhibitors. Cancers 2020, 12, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer. Int J. Mol. Sci. 2019, 20, 2340. [Google Scholar] [CrossRef] [Green Version]
- Hermeking, H.; Eick, D. Mediation of c-Myc-induced apoptosis by p53. Science 1994, 265, 2091–2093. [Google Scholar] [CrossRef]
- Garte, S.J. The c-myc oncogene in tumor progression. Crit. Rev. Oncog. 1993, 4, 435–449. [Google Scholar]
- Jung, J.H.; Jung, D.B.; Kim, H.; Lee, H.; Kang, S.E.; Srivastava, S.K.; Yun, M.; Kim, S.H. Zinc finger protein 746 promotes colorectal cancer progression via c-Myc stability mediated by glycogen synthase kinase 3β and F-box and WD repeat domain-containing 7. Oncogene 2018, 37, 3715–3728. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byron, K.; Gibson, P.R. Interleukin-8 stimulates the migration of human colonic epithelial cells in vitro. Clin. Sci. 1999, 97, 385–390. [Google Scholar] [CrossRef]
- Kitadai, Y.; Haruma, K.; Sumii, K.; Yamamoto, S.; Ue, T.; Yokozaki, H.; Yasui, W.; Ohmoto, Y.; Kajiyama, G.; Fidler, I.J.; et al. Expression of interleukin-8 correlates with vascularity in human gastric carcinomas. Am. J. Pathol. 1998, 152, 93–100. [Google Scholar] [PubMed]
- Setrerrahmane, S.; Xu, H. Tumor-related interleukins: Old validated targets for new anti-cancer drug development. Mol. Cancer 2017, 16, 153. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Q.; Chen, B.; Zhao, Y.; Shen, B.; Wang, H.; Xu, J.; Zhu, M.; Zhao, X.; Xu, C.; et al. Gastric cancer mesenchymal stem cells derived IL-8 induces PD-L1 expression in gastric cancer cells via STAT3/mTOR-c-Myc signal axis. Cell Death Dis. 2018, 9, 928. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 Axis in Cancer and Inflammatory Diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, A.; Tian, Y.; Wu, J.D.; Liu, Y.; Li, T.; Chen, Y.; Han, X.; Wu, K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016, 31, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Dabkeviciene, D.; Jonusiene, V.; Zitkute, V.; Zalyte, E.; Grigaitis, P.; Kirveliene, V.; Sasnauskiene, A. The role of interleukin-8 (CXCL8) and CXCR2 in acquired chemoresistance of human colorectal carcinoma cells HCT116. Med. Oncol. 2015, 32, 258. [Google Scholar] [CrossRef]
- Du, J.; He, Y.; Li, P.; Wu, W.; Chen, Y.; Ruan, H. IL-8 regulates the doxorubicin resistance of colorectal cancer cells via modulation of multidrug resistance 1 (MDR1). Cancer Chemother. Pharm. 2018, 81, 1111–1119. [Google Scholar] [CrossRef]
- Kobayashi, T.; Masaki, T.; Nozaki, E.; Sugiyama, M.; Nagashima, F.; Furuse, J.; Onishi, H.; Watanabe, T.; Ohkura, Y. Microarray Analysis of Gene Expression at the Tumor Front of Colon Cancer. Anticancer Res. 2015, 35, 6577–6581. [Google Scholar] [PubMed]
- Cheng, X.S.; Li, Y.F.; Tan, J.; Sun, B.; Xiao, Y.C.; Fang, X.B.; Zhang, X.F.; Li, Q.; Dong, J.H.; Li, M.; et al. CCL20 and CXCL8 synergize to promote progression and poor survival outcome in patients with colorectal cancer by collaborative induction of the epithelial-mesenchymal transition. Cancer Lett. 2014, 348, 77–87. [Google Scholar] [CrossRef]
- Xu, T.; Jing, C.; Shi, Y.; Miao, R.; Peng, L.; Kong, S.; Ma, Y.; Li, L. microRNA-20a enhances the epithelial-to-mesenchymal transition of colorectal cancer cells by modulating matrix metalloproteinases. Exp. Med. 2015, 10, 683–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egea, V.; Zahler, S.; Rieth, N.; Neth, P.; Popp, T.; Kehe, K.; Jochum, M.; Ries, C. Tissue inhibitor of metalloproteinase-1 (TIMP-1) regulates mesenchymal stem cells through let-7f microRNA and Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E309–E316. [Google Scholar] [CrossRef] [Green Version]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, N.M.; Byström, P.; Christensen, I.J.; Berglund, A.; Nielsen, H.J.; Brünner, N.; Glimelius, B. TIMP-1 is significantly associated with objective response and survival in metastatic colorectal cancer patients receiving combination of irinotecan, 5-fluorouracil, and folinic acid. Clin. Cancer Res. 2007, 13, 4117–4122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirco, R.; Liu, X.W.; Jung, K.K.; Kim, H.R. Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev. 2006, 25, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choi, J.W.; Kim, Y.S. Plasma or serum TIMP-1 is a predictor of survival outcomes in colorectal cancer: A meta-analysis. J. Gastrointest. Liver Dis. 2011, 20, 287–291. [Google Scholar]
- Dong, H.; Yin, H.; Zhao, C.; Cao, J.; Xu, W.; Zhang, Y. Design, Synthesis and Biological Evaluation of Novel Osimertinib-Based HDAC and EGFR Dual Inhibitors. Molecules 2019, 24, 2407. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Chen, G.; Gao, M.; Wu, J. Design, synthesis and evaluation of the osimertinib analogue (C-005) as potent EGFR inhibitor against NSCLC. Bioorg. Med. Chem. 2018, 26, 6135–6145. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Chen, T.C.; Lee, C.C.; Chen, C.L.; Ahmed Ali, A.A.; Tikhomirov, A.; Guh, J.H.; Yu, D.S.; Huang, H.S. Ring fusion strategy for synthesis and lead optimization of sulfur-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivatives as promising scaffold of antitumor agents. Eur. J. Med. Chem. 2015, 102, 661–676. [Google Scholar] [CrossRef] [PubMed]
- Pogodin, P.V.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. PASS Targets: Ligand-based multi-target computational system based on a public data and naïve Bayes approach. SAR QSAR Environ. Res. 2015, 26, 783–793. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Lin, G.; He, X.; Ji, H.; Shi, L.; Davis, R.W.; Zhong, S. Reproducibility Probability Score--incorporating measurement variability across laboratories for gene selection. Nat. Biotechnol. 2006, 24, 1476–1477. [Google Scholar] [CrossRef]
- Ryan, B.M.; Zanetti, K.A.; Robles, A.I.; Schetter, A.J.; Goodman, J.; Hayes, R.B.; Huang, W.Y.; Gunter, M.J.; Yeager, M.; Burdette, L.; et al. Germline variation in NCF4, an innate immunity gene, is associated with an increased risk of colorectal cancer. Int. J. Cancer 2014, 134, 1399–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pek, M.; Yatim, S.; Chen, Y.; Li, J.; Gong, M.; Jiang, X.; Zhang, F.; Zheng, J.; Wu, X.; Yu, Q. Oncogenic KRAS-associated gene signature defines co-targeting of CDK4/6 and MEK as a viable therapeutic strategy in colorectal cancer. Oncogene 2017, 36, 4975–4986. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Qiu, H.; Xu, J.; Mo, J.; Liu, Y.; Gui, Y.; Huang, G.; Zhang, S.; Yao, H.; Huang, X.; et al. Expression and prognostic potential of GPX1 in human cancers based on data mining. Ann. Transl. Med. 2020, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Najafov, J.; Najafov, A. GECO: Gene expression correlation analysis after genetic algorithm-driven deconvolution. Bioinformatics 2019, 35, 156–159. [Google Scholar] [CrossRef]
- von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.J.; Hu, F.F.; Xia, M.X.; Han, L.; Zhang, Q.; Guo, A.Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. Methods Mol. Biol. 2008, 443, 365–382. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temml, V.; Kaserer, T.; Kutil, Z.; Landa, P.; Vanek, T.; Schuster, D. Pharmacophore modeling for COX-1 and -2 inhibitors with LigandScout in comparison to Discovery Studio. Future Med. Chem. 2014, 6, 1869–1881. [Google Scholar] [CrossRef]
- Mashukova, A.; Forteza, R.; Wald, F.A.; Salas, P.J. PDK1 in apical signaling endosomes participates in the rescue of the polarity complex atypical PKC by intermediate filaments in intestinal epithelia. Mol. Biol. Cell 2012, 23, 1664–1674. [Google Scholar] [CrossRef]
- Mohamad Anuar, N.N.; Nor Hisam, N.S.; Liew, S.L.; Ugusman, A. Clinical Review: Navitoclax as a Pro-Apoptotic and Anti-Fibrotic Agent. Front. Pharm. 2020, 11, 564108. [Google Scholar] [CrossRef]
- Herrera-Martínez, M.; Orozco-Samperio, E.; Montaño, S.; Ariza-Ortega, J.A.; Flores-García, Y.; López-Contreras, L. Vorinostat as potential antiparasitic drug. Eur. Rev. Med. Pharm. Sci. 2020, 24, 7412–7419. [Google Scholar] [CrossRef]
- Kretzner, L.; Scuto, A.; Dino, P.M.; Kowolik, C.M.; Wu, J.; Ventura, P.; Jove, R.; Forman, S.J.; Yen, Y.; Kirschbaum, M.H. Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res. 2011, 71, 3912–3920. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Kang, M.H.; Hong, K.; Kim, J.H. Tubastatin A inhibits HDAC and Sirtuin activity rather than being a HDAC6-specific inhibitor in mouse oocytes. Aging 2019, 11, 1759–1777. [Google Scholar] [CrossRef]
- Papatheodorou, I.; Fonseca, N.A.; Keays, M.; Tang, Y.A.; Barrera, E.; Bazant, W.; Burke, M.; Füllgrabe, A.; Fuentes, A.M.; George, N.; et al. Expression Atlas: Gene and protein expression across multiple studies and organisms. Nucleic Acids Res. 2018, 46, D246–D251. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Yaeger, R.; Kotani, D.; Mondaca, S.; Parikh, A.R.; Bando, H.; Van Seventer, E.E.; Taniguchi, H.; Zhao, H.; Thant, C.N.; de Stanchina, E.; et al. Response to Anti-EGFR Therapy in Patients with BRAF non-V600-Mutant Metastatic Colorectal Cancer. Clin. Cancer Res. 2019, 25, 7089–7097. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharm. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Gao, H.; Yang, Z.; Yang, X.; Rao, Y. Synthesis and evaluation of osimertinib derivatives as potent EGFR inhibitors. Bioorg. Med. Chem. 2017, 25, 4553–4559. [Google Scholar] [CrossRef]
- Shen, C.J.; Lin, P.L.; Lin, H.C.; Cheng, Y.W.; Huang, H.S.; Lee, H. RV-59 suppresses cytoplasmic Nrf2-mediated 5-fluorouracil resistance and tumor growth in colorectal cancer. Am. J. Cancer Res. 2019, 9, 2789–2796. [Google Scholar]
- Ali, A.A.A.; Lee, Y.-R.; Wu, A.T.H.; Yadav, V.K.; Yu, D.-S.; Huang, H.-S. Structure-based strategies for synthesis, lead optimization and biological evaluation of N-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivatives as potential multi-target anticancer agents. Arab. J. Chem. 2021, 14, 102884. [Google Scholar] [CrossRef]
- Sahin, I.H.; Akce, M.; Alese, O.; Shaib, W.; Lesinski, G.B.; El-Rayes, B.; Wu, C. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br. J. Cancer 2019, 121, 809–818. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Huang, L.; Ding, G.; Huang, H.; Cao, G.; Sun, X.; Lou, N.; Wei, Q.; Shen, T.; Xu, X.; et al. Interferon gamma inhibits CXCL8-CXCR2 axis mediated tumor-associated macrophages tumor trafficking and enhances anti-PD1 efficacy in pancreatic cancer. J. Immunother. Cancer 2020, 8, e000308. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, E.; Sato, Y.; Minagawa, A.; Okuyama, R. Pathogenesis of psoriasis and development of treatment. J. Derm. 2018, 45, 264–272. [Google Scholar] [CrossRef] [Green Version]
- Seubert, B.; Grünwald, B.; Kobuch, J.; Cui, H.; Schelter, F.; Schaten, S.; Siveke, J.T.; Lim, N.H.; Nagase, H.; Simonavicius, N.; et al. Tissue inhibitor of metalloproteinases (TIMP)-1 creates a premetastatic niche in the liver through SDF-1/CXCR4-dependent neutrophil recruitment in mice. Hepatology 2015, 61, 238–248. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCI-Synthetic Compounds | NCI-Standard Agents | ArrayCGH-Gray | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Rank | r | CCLC | Target Descriptor | r | CCLC | Target Descriptor | r | CCLC | Target Descriptor |
| 1 | 0.6 | 50 | 3-Nitro-5-Formylisoxazole | 0.51 | 58 | Trimetrexate | 0.27 | 57 | MET |
| 2 | 0.58 | 58 | CI.49700 | 0.49 | 59 | Methotrexate | 0.27 | 57 | CDK4 |
| 3 | 0.56 | 57 | Tryptanthrin | 0.46 | 58 | Dichloroali lawsone | 0.2 | 55 | YES1 |
| 4 | 0.55 | 58 | Tolonium Chloride | 0.46 | 58 | 5HP | 0.18 | 53 | DRD3 |
| 5 | 0.54 | 57 | Metoprine (USAN) | 0.46 | 59 | Cyclocytidine | 0.13 | 55 | TP53 |
| 6 | 0.54 | 45 | Bafilomycin Deriv | 0.42 | 59 | Thioguanine | 0.13 | 55 | WNT1 |
| 7 | 0.53 | 53 | Brilliant cresyl blue | 0.41 | 58 | Hycanthone | 0.11 | 55 | MYC |
| 8 | 0.52 | 46 | Bafilomycin Antibiotic | 0.4 | 45 | Tetraplatin | 0.11 | 57 | AKT1 |
| 9 | 0.52 | 41 | Lapachol | 0.4 | 58 | Cytosine arabinoside | 0.1 | 56 | ABR |
| 10 | 0.51 | 58 | Piroctone olamine | 0.38 | 58 | Pyrazofurin | 0.1 | 56 | CCND1 |
| Pa | Pi | Activity |
|---|---|---|
| 0.477 | 0.029 | MYC inhibitor |
| 0.375 | 0.166 | Catenin beta inhibitor |
| 0.302 | 0.139 | MAP kinase stimulant |
| 0.238 | 0.086 | Antineoplastic (colon cancer) |
| 0.113 | 0.058 | Protein kinase (CK1) inhibitor |
| 0.268 | 0.227 | MAP kinase 8 inhibitor |
| 0.210 | 0.174 | Transcription factor NF kappa A inhibitor |
| 0.143 | 0.110 | Protein kinase (CK1) delta inhibitor |
| 0.136 | 0.109 | Acetylcholine M2 receptor antagonist |
| 0.146 | 0.134 | Chemokine (C-X-C motif) ligand 8 (CXCL8) antagonist |
| Pharmacokinetics | GI Absorption (High) | BBB (Low) |
|---|---|---|
| Drug-likeness (Yes to all) | Lipinski, Ghose, Veber, Egan, Muegge | |
| Bioavailability score | 55% | |
| Medical Chemistry | Synthetic accessibility: | 2.95 |
| RV59 | Standard Inhibitotrs | ||
|---|---|---|---|
| RV59-MYC Complex (ΔG = −7.6 Kcal/mol) | Vorinostat-MYC Complex (ΔG = −6.3 Kcal/mol) | ||
| Type of interactions and number of bonds | distance of interacting Amino acids | Type of interactions and number of bonds | distance of interacting Amino acid |
| Conventional Hydrogen bond (1) | LYS944 (2.49 Å) | Conventional Hydrogen bond (1) | SER (2.87Å) |
| Pi sigma | LEU217 | Van der Waals forces | LEU94, SER221, LUE225, LYS213, ASP220, VAL940 |
| Pi-pi stacked | HIS217 | Alkyl | ARG214, LYS944 |
| Pi-Alkyl | ARG214,LYS213 | ||
| RV59-CXCL8 Complex (ΔG = −7.7 Kcal/mol) | Reparixinx-CXCL8 Complex (ΔG = −6.3 Kcal/mol) | ||
| Type of interactions and number of bonds | distance of interacting Amino acids | Type of interactions and number of bonds | distance of interacting Amino acids |
| Conventional Hydrogen bond (1) | CYS47 (2.03 Å) | Van der Waals forces | ASP43, ARG45 |
| Carbon hydrogen bond | GLU46 | Pi-pi stacked | PHE19 |
| Pi-cation | LYS9 | Alkyl | LEU41, TYR11 |
| Pi-Alkyl | LEU47 | Pi-Alkyl | LEU47, PHE15 |
| RV59-TIMP1 Complex (ΔG = −6.9 Kcal/mol) | Ilomastat-TIMP1 Complex (ΔG = −7.4 Kcal/mol) | ||
| Type of interactions and number of bonds | distance of interacting Amino acids | Type of interactions and number of bonds | distance of interacting Amino acids |
| Conventional Hydrogen bond (4) | PHE132 (3.29 Å), TRY99 (3.41 Å), GLU118 (3.4 Å), ARG134 (2.51 Å) | Conventional Hydrogen bond (2) | ARG134, TYR136 |
| Carbon hydrogen bond | SER133 | Van der Waals | THR131, TYR99, GLU118, ASP114, GLU125, LYS122 |
| Pi-cation | SER115 | Carbon hydrogen bond | PHE132 |
| Pi-Alkyl | PHE132 | Pi-Anion | GLU118 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, T.-H.; Mokgautsi, N.; Huang, Y.-J.; Wu, A.T.H.; Huang, H.-S. Comprehensive Omics Analysis of a Novel Small-Molecule Inhibitor of Chemoresistant Oncogenic Signatures in Colorectal Cancer Cell with Antitumor Effects. Cells 2021, 10, 1970. https://doi.org/10.3390/cells10081970
Huang T-H, Mokgautsi N, Huang Y-J, Wu ATH, Huang H-S. Comprehensive Omics Analysis of a Novel Small-Molecule Inhibitor of Chemoresistant Oncogenic Signatures in Colorectal Cancer Cell with Antitumor Effects. Cells. 2021; 10(8):1970. https://doi.org/10.3390/cells10081970
Chicago/Turabian StyleHuang, Tse-Hung, Ntlotlang Mokgautsi, Yan-Jiun Huang, Alexander T. H. Wu, and Hsu-Shan Huang. 2021. "Comprehensive Omics Analysis of a Novel Small-Molecule Inhibitor of Chemoresistant Oncogenic Signatures in Colorectal Cancer Cell with Antitumor Effects" Cells 10, no. 8: 1970. https://doi.org/10.3390/cells10081970