
Phagocytic Activities of Reactive Microglia and Astrocytes Associated with Prion Diseases Are Dysregulated in Opposite Directions


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents, Kits, Antibodies
2.2. Animals
2.3. Isolation and Culturing of Adult Primary Astrocytes
2.4. Isolation and Culturing of Adult Primary Microglia
2.5. Synaptosome Purification
2.6. Transmission Electron Microscopy
2.7. pHrodo Red-Conjugation
2.8. Preparation of Phagocytosis Assay Medium
2.9. Live Imaging Phagocytic Assay with pHrodo Red-Conjugated Synaptosomes or Myelin Debris
2.10. Confocal Microscopy Imaging of Phagocytic Uptake
2.11. Lysotracker Assay
2.12. RT-qPCR
2.13. Protein Extraction and Western Blotting
2.14. Immunocytochemistry and Immunofluorescence of Brain Tissue
2.15. Analysis of Gene Expression by Nanostring
2.16. Statistics
3. Results
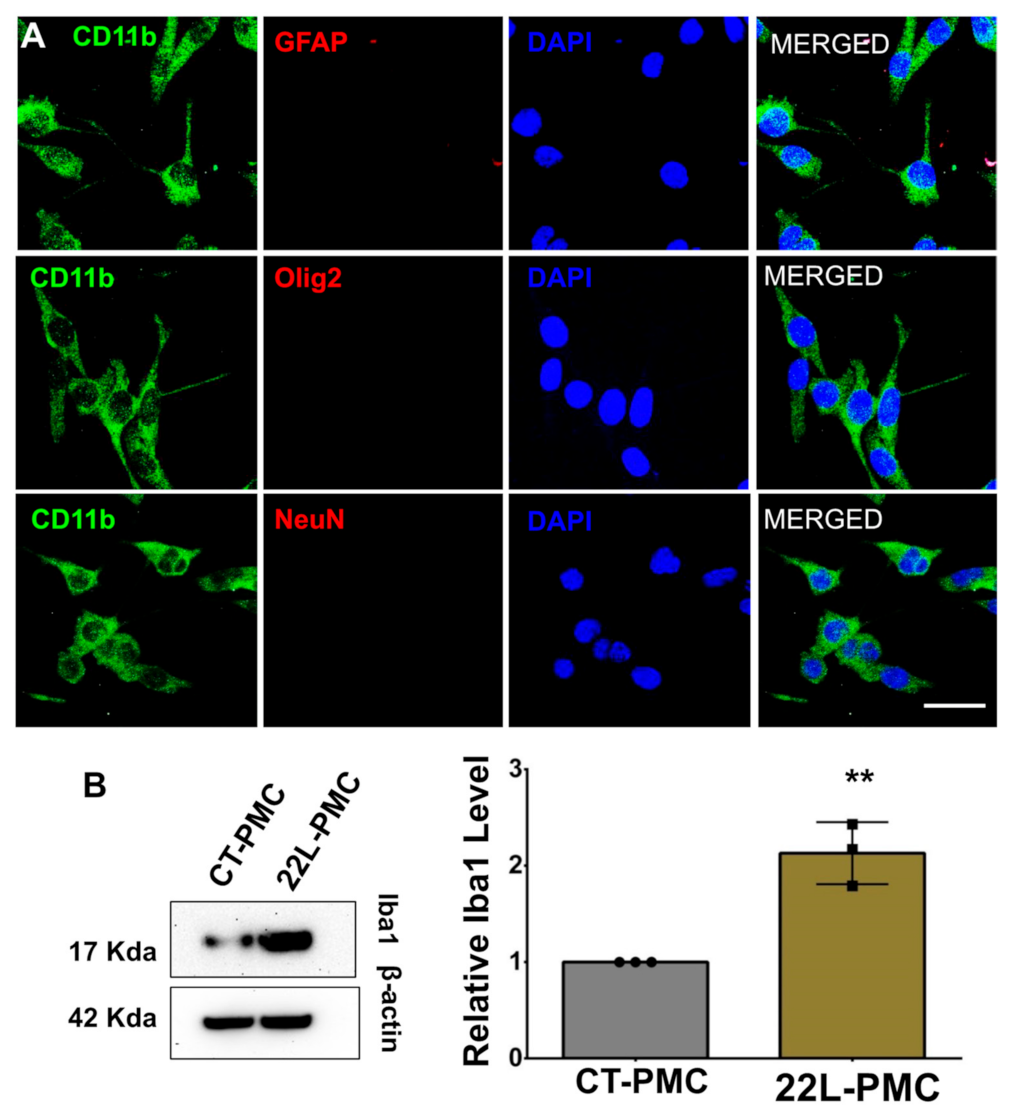
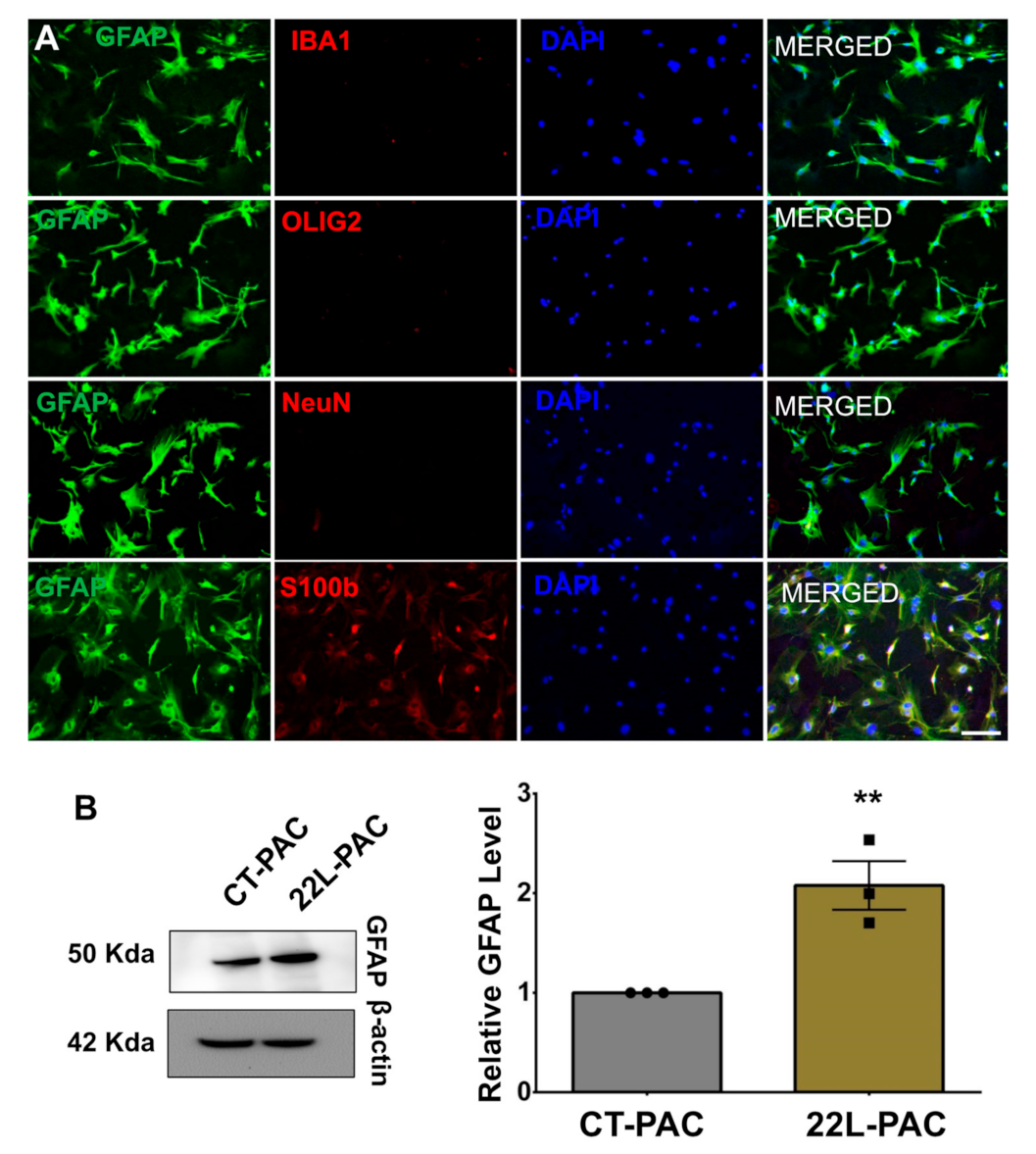
3.1. Isolation of Astrocytes, Microglia, and Synaptosomes from Adult Mouse Brains
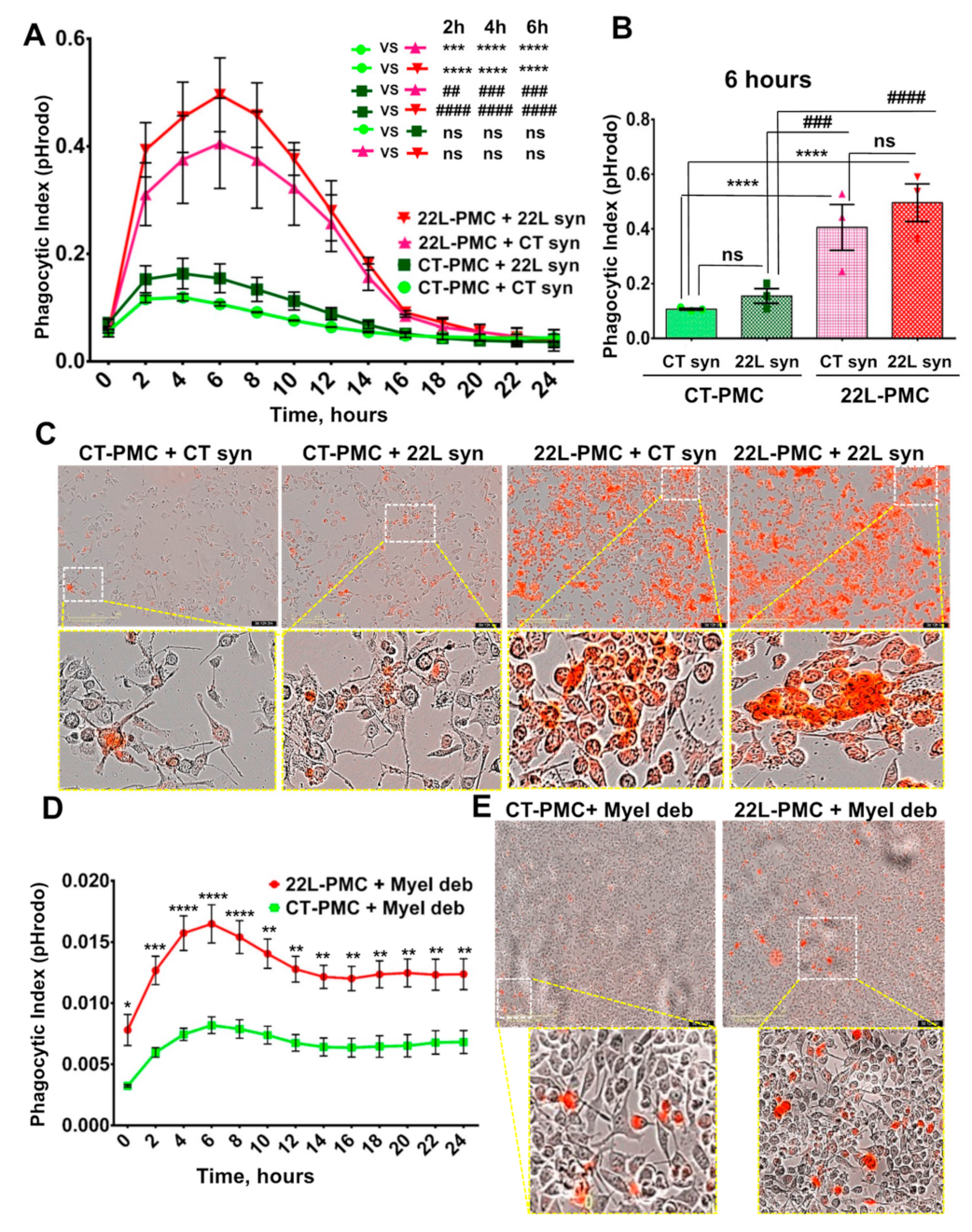
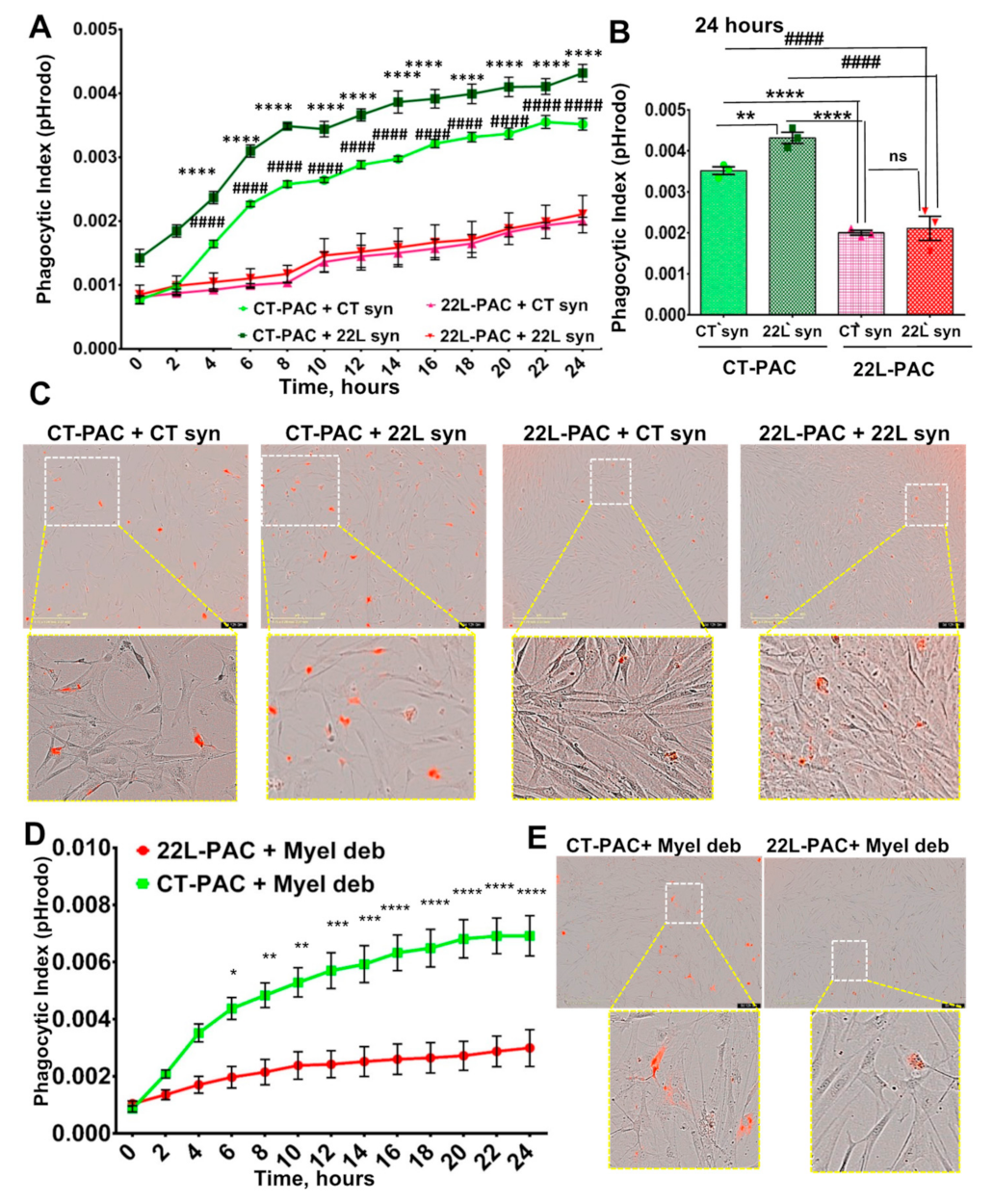
3.2. Phagocytic Uptake of Synaptosomes Is Upregulated in Reactive Microglia
3.3. Phagocytic Uptake of Myelin Debris Is Upregulated in Reactive Microglia
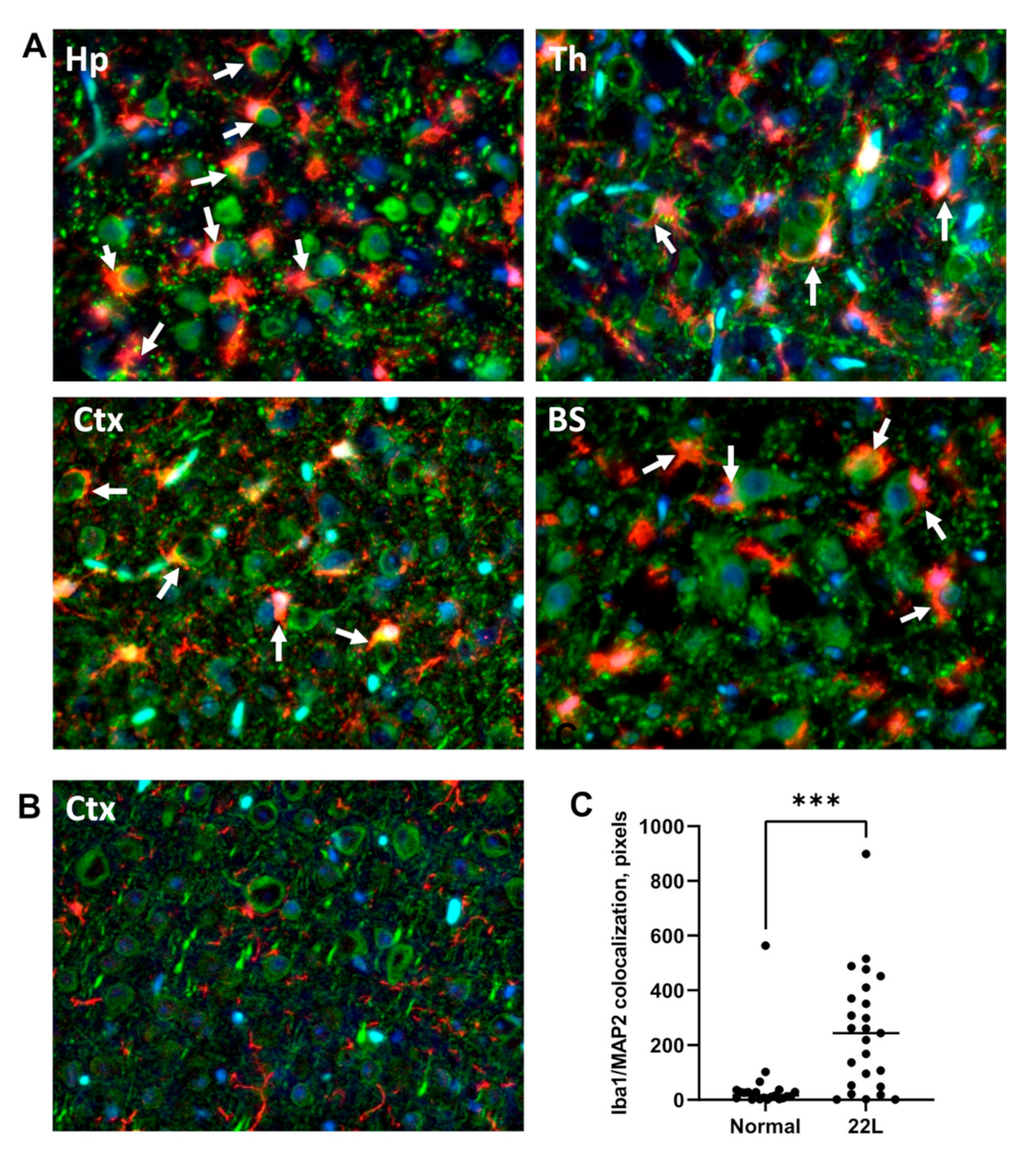
3.4. Engulfment of Neuronal Cells by Reactive Microglia In Vivo
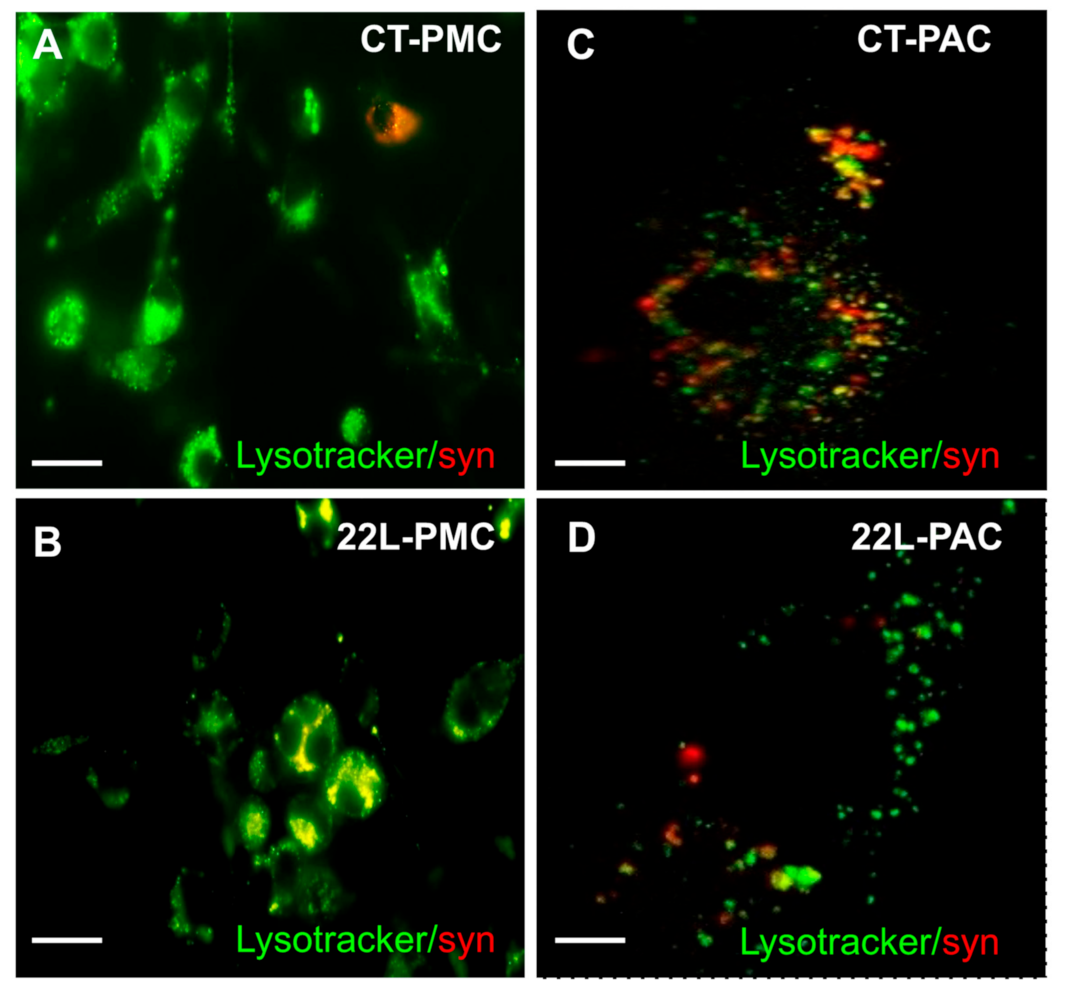
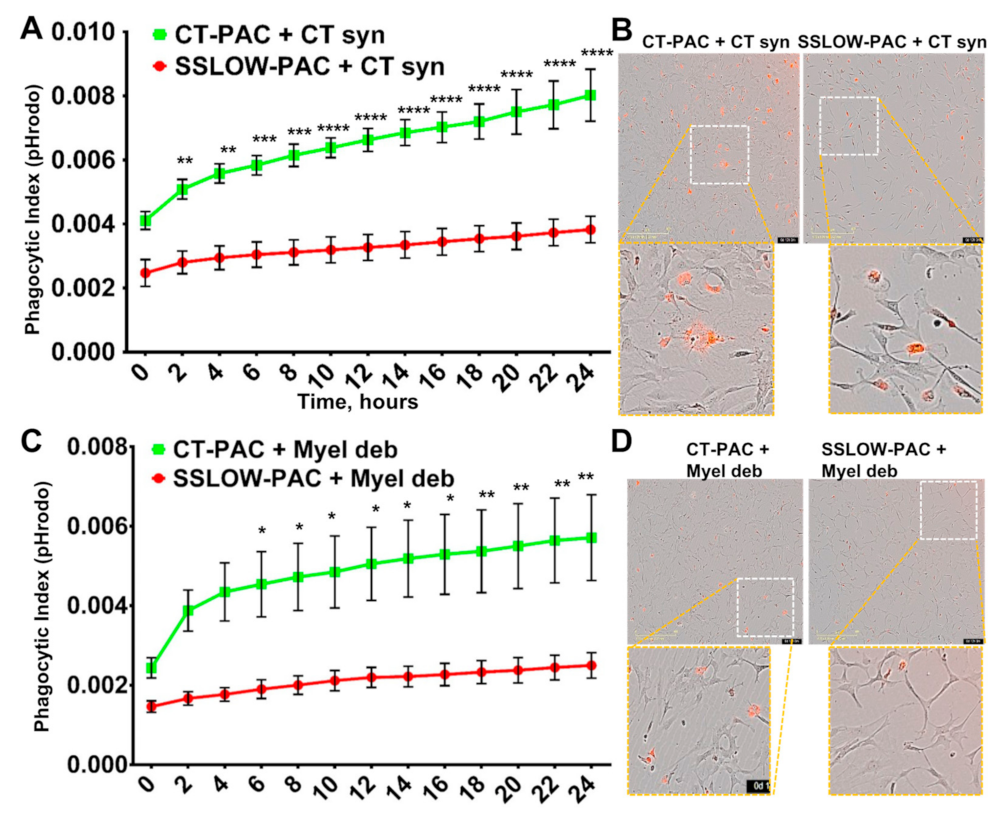
3.5. Phagocytic Activity Is Reduced in Reactive Astrocytes
3.6. Downregulation of Phagocytic Activity Is Common in Reactive Astrocytes Associated with Prion Diseases
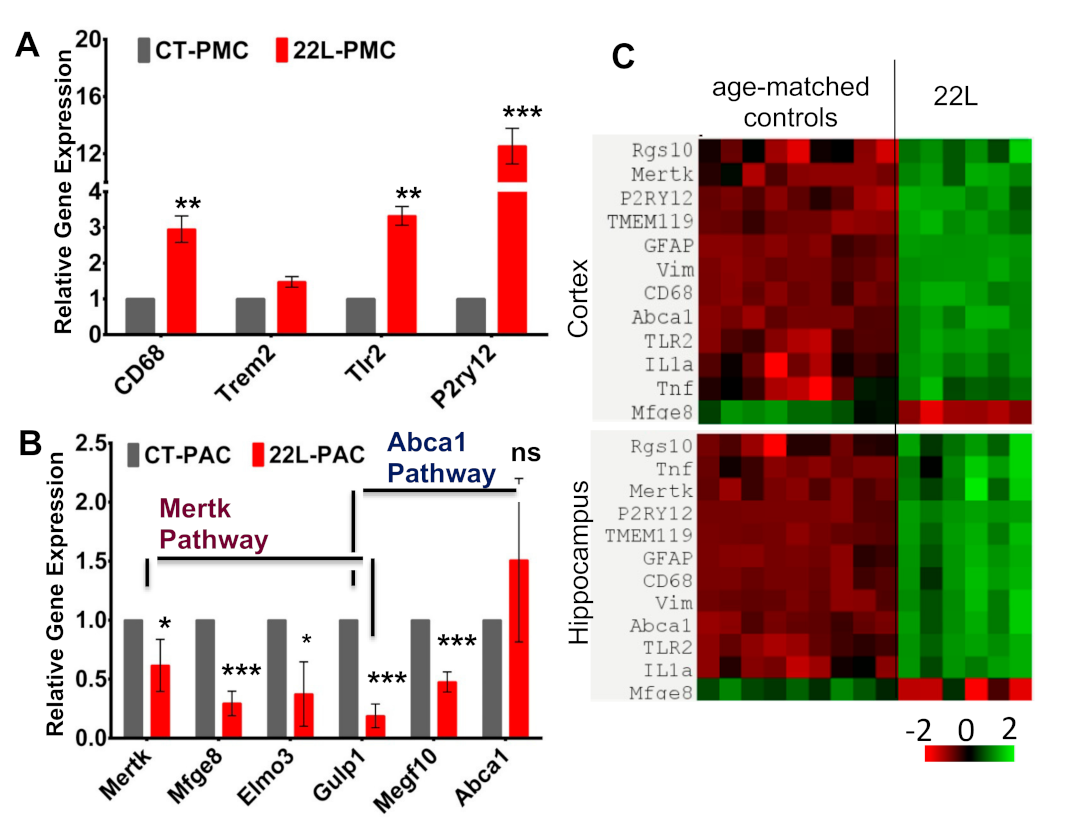
3.7. Analysis of the Expression of Genes That Report on Phagocytic Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarava, N.; Chang, J.C.-Y.; Molesworth, K.; Baskakov, I.V. Region-specific glial homeostatic signature in prion diseases is replaced by a uniform neuroinflammation signature, common for brain regions and prion strains with different cell tropism. Neurobiol. Dis. 2020, 137, e104783. [Google Scholar] [CrossRef]
- Makarava, N.; Mychko, O.; Chang, J.C.-Y.; Molesworth, K.; Baskakov, I.V. The degree of astrocyte activation is predictive of the incubation time to prion disease. Acta Neuropathol. Commun. 2021, 9, 87. [Google Scholar] [CrossRef]
- Scheckel, C.; Imeri, M.; Schwarz, P.; Aguzzi, A. Ribosomal profiling during prion disease uncovers progressive translational derangement in glia but not in neurons. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. RNA-seq and network analysis reveal unique glial gene expression signatures during prion infection. Mol. Brain 2020, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, R.; Sinha, A.; Makarava, N.; Molesworth, K.; Baskakov, I.V. Non-cell autonomous astrocyte-mediated neuronal toxicity in prion diseases. Acta Neuropathol. Commun. 2021, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schätzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J.; et al. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 2020, 105, 855–866.e855. [Google Scholar] [CrossRef] [Green Version]
- Baskakov, I.V. On the reactive states of astrocytes in prion diseases. Prion 2021, 15, 87–93. [Google Scholar] [CrossRef]
- Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. Microglia Are Critical in Host Defense against Prion Disease. J. Virol. 2018, 92, e00549-00518. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Herrmann, U.S.; Falsig, J.; Abakumova, I.; Nuvolone, M.; Schwarz, P.; Frauenknecht, K.; Rushing, E.J.; Aguzzi, A. A neuroprotective role for microglia in prion diseases. J. Exp. Med. 2016, 213, 1047–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguzzi, A.; Zhu, C. Microglia in prion diseases. J. Clin. Investig. 2017, 127, 3230–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, K.; Sepulveda-Falla, D.; Rose, I.V.L.; Madore, C.; Muth, C.; Matschke, J.; Butovsky, O.; Liddelow, S.; Glatzel, M.; Krasemann, S. Complement 3+-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathol. Commun. 2019, 7, 83. [Google Scholar] [CrossRef]
- Damisah, E.C.; Hill, R.A.; Rai, A.; Chen, F.; Rothlin, C.V.; Ghosh, S.; Grutzendler, J. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci Adv. 2020, 6, eaba3239. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arboledas, A.; Davila, J.C.; Sanchez-Mejias, E.; Navarro, V.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Sanchez-Mico, M.V.; Trujillo-Estrada, L.; Fernandez-Valenzuela, J.J.; Vizuete, M.; et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 2018, 66, 637–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morizawa, Y.M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y.; Nabekura, J.; Sato, K.; Okajima, F.; Takebayashi, H.; et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 2017, 8, 28. [Google Scholar] [CrossRef]
- Hughes, M.M.; Field, R.H.; Perry, V.H.; Murray, C.L.; Cunningham, C. Microglia in the degenerating brain are capable of phagocytosis of beads and of apoptotic cells, but do not efficiently remove PrPSc, even upon LPS stimulation. Glia 2010, 58, 2017–2030. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.P.; Head, M.W.; Ironside, J.W.; Priola, S.A. Uptake and Degradation of Protease-Sensitive and -Resistant Forms of Abnormal Human Prion Protein Aggregates by Human Astrocytes. Am. J. Pathol 2014, 184, 3299–3307. [Google Scholar] [CrossRef] [Green Version]
- Hollister, J.R.; Lee, K.S.; Dorward, D.W.; Baron, G.S. Efficient Uptake and Dissemination of Scrapie Prion Protein by Astrocytes and Fibroblasts from Adult Hamster Brain. PLoS ONE 2015, 10, e0115351. [Google Scholar] [CrossRef] [Green Version]
- Gilch, S.; Schmitz, F.; Aguib, Y.; Kehler, C.; Bulow, S.; Bauer, S.; Kremmer, E.; Schatzl, H.M. CpG and LPS can interfere negatively with prion clearance in macrophage and microglial cells. FEBS J. 2007, 274, 5834–5844. [Google Scholar] [CrossRef]
- Makarava, N.; Chang, J.C.-Y.; Molesworth, K.; Baskakov, I.V. Posttranslational modifications define course of prion strain adaptation and disease phenotype. J. Clin. Investig. 2020, 130, 4382–4395. [Google Scholar] [CrossRef]
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLoS Pathog. 2016, 12, e1005551. [Google Scholar] [CrossRef]
- Yamasaki, T.; Suzuki, A.; Hasebe, R.; Horiuchi, M. Flow Cytometric Detection of PrP(Sc) in Neurons and Glial Cells from Prion-Infected Mouse Brains. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffrey, M.; McGovern, G.; Makarava, N.; Gonzalez, L.; Kim, Y.S.; Rohwer, R.G.; Baskakov, I.V. Pathology of SSLOW, a transmissible and fatal synthetic prion protein disorder, and comparison with naturally occurring classical transmissible spongoform encephalopathies. Neuropath. Appl. Neurobiol. 2014, 40, 296–310. [Google Scholar] [CrossRef] [Green Version]
- Makarava, N.; Kovacs, G.G.; Bocharova, O.; Savtchenko, R.; Alexeeva, I.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. Recombinant prion protein induces a new transmissible prion disease in wild type animals. Acta Neuropathol. 2010, 119, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarava, N.; Kovacs, G.G.; Savtchenko, R.; Alexeeva, I.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. Stabilization of a prion strain of synthetic origin requires multiple serial passages. J. Biol. Chem. 2012, 287, 30205–30214. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.H.; Stawski, P.S.; Draycott, A.S.; Udeshi, N.D.; Lehrman, E.K.; Wilton, D.K.; Svinkina, T.; Deerinck, T.J.; Ellisman, M.H.; Stevens, B.; et al. Proteomic Analysis of Unbounded Cellular Compartments: Synaptic Clefts. Cell 2016, 166, 1295–1307.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, Y.G.; Chung, W.S. In Vitro Engulfment Assay to Measure Phagocytic Activity of Astrocytes Using Synaptosomes. Methods Mol. Biol. 2019, 1938, 155–168. [Google Scholar] [PubMed]
- Janda, E.; Boi, L.; Carta, A.R. Microglial Phagocytosis and Its Regulation: A Therapeutic Target in Parkinson’s Disease? Front. Mol. Neusci. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Fekete, R.; Cserép, C.; Lénárt, N.; Tóth, K.; Orsolits, B.; Martinecz, B.; Méhes, E.; Szabó, B.; Németh, V.; Gönci, B.; et al. Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol. 2018, 136, 461–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.-B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Hidetoshi, T.-S.; Makoto, T.; Inoue, K. P2Y receptors in microglia and neuroinflammation. Wiley Interdiscip. Rev. Membr. Transport. Signal. 2012, 1, 493–501. [Google Scholar] [CrossRef]
- Doyle, S.E.; O’Connell, R.M.; Miranda, G.A.; Vaidya, S.A.; Chow, E.K.; Liu, P.T.; Suzuki, S.; Suzuki, N.; Modlin, R.L.; Yeh, W.C.; et al. Toll-like receptors induce a phagocytic gene program through p38. J. Exp. Med. 2004, 199, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Hanayama, R.; Tanaka, M.; Miwa, K.; Shinohara, A.; Iwamatsu, A.; Nagata, S. Identification of a factor that links apoptotic cells to phagocytes. Nature 2002, 417, 182–187. [Google Scholar] [CrossRef]
- de Oliviera Nascimento, L.; Massari, P.; Wetzler, L. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.A.; Race, B.; Williams, K.; Chesebro, B. Toll-like receptor 2 confers partial neuroprotection during prion disease. PLoS ONE 2018, 13, e0208559. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Lui, H.; Zhang, J.; Makinson, Stefanie R.; Cahill, Michelle K.; Kelley, Kevin W.; Huang, H.-Y.; Shang, Y.; Oldham, Michael C.; Martens, Lauren H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef] [Green Version]
- Stephan, A.H.; Madison, D.V.; Mateos, J.M.; Fraser, D.A.; Lovelett, E.A.; Coutellier, L.; Kim, L.; Tsai, H.-H.; Huang, E.J.; Rowitch, D.H.; et al. A Dramatic Increase of C1q Protein in the CNS during Normal Aging. J. Neurosci. 2013, 33, 13460–13474. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, B.; Allen, N.J.; Zazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincenti, J.E.; Murphy, L.; Grabert, K.; McColl, B.W.; Cancellotti, E.; Freeman, T.C.; Manson, J.C. Defining the Microglia Response during the Time Course of Chronic Neurodegeneration. J. Virol. 2016, 90, 3003–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muth, C.; Schröck, K.; Madore, C.; Hartmann, K.; Fanek, Z.; Butovsky, O.; Glatzel, M.; Krasemann, S. Activation of microglia by retroviral infection correlates with transient clearance of prions from the brain but does not change incubation time. Brain Pathol. 2017, 27, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.; Brown, D.R.; Groschup, M.H.; Feldmann, C.; Haist, I.; Kretzschmar, H.A. Role of microglia in neuronal cell death in prion disease. Brain Pathol. 1998, 8, 449–457. [Google Scholar] [CrossRef]
- Grizenkova, J.; Akhtar, S.; Brandner, S.; Collinge, J.; Lloyd, S.E. Microglial Cx3cr1 knockout reduces prion disease incubation time in mice. BMC Neurosci. 2014, 15, 15–44. [Google Scholar] [CrossRef] [Green Version]
- Baik, S.H.; Kang, S.; Son, S.M.; Mook-Jung, I. Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer’s disease mouse model. Glia 2016, 64, 2274–2290. [Google Scholar] [CrossRef]
- Huang, Y.; Happonen, K.E.; Burrola, P.G.; O’Connor, C.; Hah, N.; Huang, L.; Nimmerjahn, A.; Lemke, G. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 2021. [Google Scholar] [CrossRef]
- Safaiyan, S.; Kannaiyan, N.; Snaidero, N.; Brioschi, S.; Biber, K.; Yona, S.; Edinger, A.L.; Jung, S.; Rossner, M.J.; Simons, M. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 2016, 19, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Wendt, S.; Maricos, M.; Vana, N.; Meyer, N.; Guneykaya, D.; Semtner, M.; Kettenmann, H. Changes in phagocytosis and potassium channel activity in microglia of 5xFAD mice indicate alterations in purinergic signaling in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2017, 58, 41–53. [Google Scholar] [CrossRef]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef]
- Baskakov, I.V.; Katorcha, E. Multifaceted role of sialylation in prion diseases. Front. Neurosci. 2016, 10, e358. [Google Scholar] [CrossRef] [Green Version]
- Baskakov, I.V.; Katorcha, E.; Makarava, N. Prion Strain-Specific Structure and Pathology: A View from the Perspective of Glycobiology. Viruses 2018, 10, 723. [Google Scholar] [CrossRef] [Green Version]
- Varki, A. Sialic acids in human health and disease. Trends Mol. Med. 2008, 14, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Katorcha, E.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci. Rep. 2015, 5, 16912. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Makarava, N.; Katorcha, E.; Savtchenko, R.; Brossmer, R.; Baskakov, I.V. Post-conversion sialylation of prions in lymphoid tissues. Proc. Acad. Natl. Sci. USA 2015, 112, E6654–E6662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katorcha, E.; Makarava, N.; Savtchenko, R.; D’Azzo, A.; Baskakov, I.V. Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLoS Pathog. 2014, 10, e1004366. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Katorcha, E.; Daus, M.L.; Lasch, P.; Beekes, M.; Baskakov, I.V. Sialylation controls prion fate in vivo. J. Biol. Chem. 2017, 292, 2359–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katorcha, E.; Daus, M.L.; Gonzalez-Montalban, N.; Makarava, N.; Lasch, P.; Beekes, M.; Baskakov, I.V. Reversible off and on switching of prion infectivity via removing and reinstalling prion sialylation. Sci. Rep. 2016, 6, 33119. [Google Scholar] [CrossRef]
- Srivastava, S.; Katorcha, E.; Makarava, N.; Barrett, J.P.; Loane, D.J.; Baskakov, I.V. Inflammatory response of microglia to prions is controlled by sialylation of PrPSc. Sci. Rep. 2018, 8, e11326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranich, J.; Krautler, N.J.; Falsig, J.; Ballmer, B.; Li, S.; Hutter, G.; Schwarz, P.; Moos, R.; Julius, C.; Miele, G.; et al. Engulfment of cerebral apoptotic bodies controls the course of prion disease in a mouse strain-dependent manner. J. Exp. Med. 2010, 207, 2271–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Reitboeck, P.; Phillips, A.; Piers, T.M.; Villegas-Llerena, C.; Butler, M.; Mallach, A.; Rodrigues, C.; Arber, C.E.; Heslegrave, A.; Zetterberg, H.; et al. Human Induced Pluripotent Stem Cell-Derived Microglia-Like Cells Harboring TREM2 Missense Mutations Show Specific Deficits in Phagocytosis. Cell Rep. 2018, 24, 2300–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarava, N.; Chang, J.C.-Y.; Baskakov, I.V. Region-Specific Sialylation Pattern of Prion Strains Provides Novel Insight into Prion Neurotropism. Int. J. Mol. Sci. 2020, 21, 828. [Google Scholar] [CrossRef] [Green Version]
- Karapetyan, Y.E.; Saá, P.; Mahal, S.P.; Sferrazza, G.F.; Sherman, A.; Salès, N.; Weissmann, C.; Lasmézas, C.I. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS ONE 2009, 4, e5730. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Chin, G.; Cho, F.S.; Kelley, K.W.; Miller, J.G.; Chien, E.C.; Liddelow, S.A.; Nguyen, P.T.; Nakao-Inoue, H.; Dorman, L.C.; et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018, 359, 1269–1273. [Google Scholar] [CrossRef] [Green Version]
- Boddaert, J.; Kinugawa, K.; Lambert, J.C.; Boukhtouche, F.; Zoll, J.; Merval, R.; Blanc-Brude, O.; Mann, D.; Berr, C.; Vilar, J.; et al. Evidence of a role for lactadherin in Alzheimer’s disease. Am. J. Pathol. 2007, 170, 921–929. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Accession Number | Sequence |
|---|---|---|
| CD68 | NM_001291058.1 | F 5′- CTGCCAGTCCGAAAATGGAAC-3′ R 5′- CTTCATCCACCGGGGCTATC-3′ |
| P2ry12 | NM_027571.4 | F 5′- AACACCACCTCAGCCAATAC-3′ R 5′- ACAGCAATGGGAAGAGAACC-3′ |
| Trem2 | NM_031254.3 | F 5′- TGGGACCTCTCCACCAGTT-3′ R 5′- GTGGTGTTGAGGGCTTGG-3′ |
| Tlr2 | NM_011905.3 | F 5′- CACTATCCGGAGGTTGCATATC-3′ R 5′- GGAAGACCTTGCTGTTCTCTAC-3′ |
| Abca1 | NM_013454.3 | F 5′- ATGGAGCAGGGAAGACCAC-3′ R 5′- GTAGGCCGTGCCAGAAGTT-3′ |
| Megf10 | NM_001001979.2 | F 5′- CTACAGACACAAGCAGAAGAGG-3′ R 5′- CAGGGTTTCTGCGATGGTATAG-3′ |
| Mfge8 | NM_008594.2 | F 5′- GTGCCCTGTGGGCTACTC-3′ R 5′- GTATTGGGGACGGCTGTG-3′ |
| Mertk | NM_008587.2 | F 5′- GATGGTTCTGGCCCCACT-3′ R 5′- CTGATCTAGCTCGGTCTCTTCC-3′ |
| Elmo3 | NM_17‘2760.3 | F 5′- CTCAGCACTGCCCCAGAT-3′ R 5′- GCACCCTTCACGACTTGTATT-3′ |
| Gulp1 | NM_028450.3 | F 5′- GGATTAGAAGGAGGGAGAGGA-3′ R 5′- ACGATCGCGTGTTCGTATC-3′ |
| Gapdh | NM_001289726.1 | F 5′- AACAGCAACTCCCACTCTTC-3′ R 5′- CCTGTTGCTGTAGCCGTATT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, A.; Kushwaha, R.; Molesworth, K.; Mychko, O.; Makarava, N.; Baskakov, I.V. Phagocytic Activities of Reactive Microglia and Astrocytes Associated with Prion Diseases Are Dysregulated in Opposite Directions. Cells 2021, 10, 1728. https://doi.org/10.3390/cells10071728
Sinha A, Kushwaha R, Molesworth K, Mychko O, Makarava N, Baskakov IV. Phagocytic Activities of Reactive Microglia and Astrocytes Associated with Prion Diseases Are Dysregulated in Opposite Directions. Cells. 2021; 10(7):1728. https://doi.org/10.3390/cells10071728
Chicago/Turabian StyleSinha, Anshuman, Rajesh Kushwaha, Kara Molesworth, Olga Mychko, Natallia Makarava, and Ilia V. Baskakov. 2021. "Phagocytic Activities of Reactive Microglia and Astrocytes Associated with Prion Diseases Are Dysregulated in Opposite Directions" Cells 10, no. 7: 1728. https://doi.org/10.3390/cells10071728