
Age-Dependent Decline in Neuron Growth Potential and Mitochondria Functions in Cortical Neurons

 , ,
, ,  and
and
Abstract
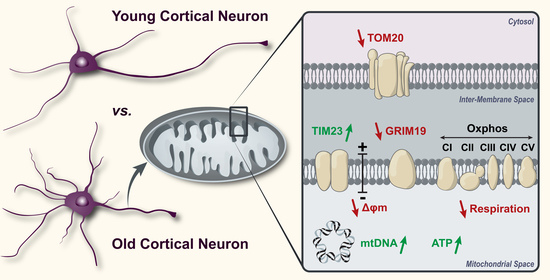
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
3.1. Cell Population Purity of Enriched Cortical Neurons after Isolation and Culture
3.2. Neurite Growth Potential Declines in Mature Cortical Neurons In Vitro with Age
3.3. Expression of Mitochondrial DNA and Mitochondrial DNA Copy Number Changes with Age
3.4. Mitochondrial Respiration of Cultured Mature Cortical Neurons Changes with Age
3.5. Expression of Mitochondrial OXPHOS Complexes in the Electron Transport Chain Is Consistent in Cortical Neurons of Different Ages
3.6. Intracellular ATP Increases in Neurons with Age
3.7. The Mitochondrial Membrane Potential of Isolated Cortical Neurons Changes with Age
3.8. Mitochondrial Membrane-Associated Proteins Are Altered in Aging Neurons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- James, S.L.; Theadom, A.; Ellenbogen, R.G.; Bannick, M.S.; Montjoy-Venning, W.; Lucchesi, L.R.; Abbasi, N.; Abdulkader, R.; Abraha, H.N.; Adsuar, J.C.; et al. Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 56–87. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Tetreault, L.; Kalsi-Ryan, S.; Nouri, A.; Fehlings, M.G. Global prevalence and incidence of traumatic spinal cord injury. Clin. Epidemiol. 2014, 6, 309. [Google Scholar]
- NSCIS. Spinal Cord Injury (SCI) Facts and Figures at a Glance. 2017. Available online: https://www.nscisc.uab.edu/Public/Facts%20and%20Figures%20-%202017.pdf (accessed on 31 July 2020).
- One Degree of Separation: Paralysis and Spinal Cord Injury in the United States. Christopher and Dana Reeve Foundation. 2009, Volume 2019. Available online: http://s3.amazonaws.com/reeve-assets-production/8112REPTFINAL.PDF (accessed on 31 July 2020).
- Fouad, K.; Bixby, J.L.; Callahan, A.; Grethe, J.S.; Jakeman, L.B.; Lemmon, V.P.; Magnuson, D.S.; Martone, M.E.; Nielson, J.L.; Schwab, J.M. FAIR SCI Ahead: The Evolution of the Open Data Commons for Pre-Clinical Spinal Cord Injury Research. J. Neurotrauma 2019, 6, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, C.G.; Meves, J.M.; Zheng, B. The age factor in axonal repair after spinal cord injury: A focus on neuron-intrinsic mechanisms. Neurosci. Lett. 2017, 652, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, T.C.; Geoffroy, C.G. The Influence of Neuron-Extrinsic Factors and Aging on Injury Progression and Axonal Repair in the Central Nervous System. Front. Cell Dev. Biol. 2020, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, C.G.; Hilton, B.J.; Tetzlaff, W.; Zheng, B. Evidence for an age-dependent decline in axon regeneration in the adult mammalian central nervous system. Cell Rep. 2016, 15, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, R.H. Mitochondrial Dysfunction in Aging and Diseases of Aging; Multidisciplinary Digital Publishing Institute: Basel, Switzerland, 2019. [Google Scholar]
- López-Lluch, G.; Irusta, P.M.; Navas, P.; de Cabo, R. Mitochondrial biogenesis and healthy aging. Exp. Gerontol. 2008, 43, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Payne, B.A.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta BBA Bioenerg. 2015, 1847, 1347–1353. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Youle, R.J.; Finkel, T. The mitochondrial basis of aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of brain aging: Adaptive and pathological modification by metabolic states. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.R.; Geddes, J.W.; Sullivan, P.G. Brain Region-Specific, Age-Related, Alterations in Mitochondrial Responses to Elevated Calcium. J. Bioenerg. Biomembr. 2004, 36, 401–406. [Google Scholar] [CrossRef]
- Schleiff, E.; Turnbull, J.L. Functional and Structural Properties of the Mitochondrial Outer Membrane Receptor Tom20. Biochemistry 1998, 37, 13043–13051. [Google Scholar] [CrossRef]
- Yamamoto, H.; Itoh, N.; Kawano, S.; Yatsukawa, Y.-I.; Momose, T.; Makio, T.; Matsunaga, M.; Yokota, M.; Esaki, M.; Shodai, T.; et al. Dual role of the receptor Tom20 in specificity and efficiency of protein import into mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demishtein-Zohary, K.; Azem, A. The TIM23 mitochondrial protein import complex: Function and dysfunction. Cell Tissue Res 2017, 367, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Mokranjac, D.; Neupert, W. The many faces of the mitochondrial TIM23 complex. Biochim. Et Biophys. Acta BBA Bioenerg. 2010, 1797, 1045–1054. [Google Scholar] [CrossRef] [Green Version]
- Shulga, N.; Pastorino, J.G. GRIM-19-mediated translocation of STAT3 to mitochondria is necessary for TNF-induced necroptosis. J. Cell Sci. 2012, 125, 2995–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lu, H.; Liu, Q.; Huang, G.; Lim, C.P.; Zhang, L.; Hao, A.; Cao, X. Function of GRIM-19, a mitochondrial respiratory chain complex I protein, in innate immunity. J. Biol. Chem. 2012, 287, 27227–27235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanHook, A.M. Delivering STAT3 into Mitochondria. Sci. Signal. 2013, 6, ec50. [Google Scholar] [CrossRef]
- Lu, H.; Cao, X. GRIM-19 Is Essential for Maintenance of Mitochondrial Membrane Potential. Mol. Biol. Cell 2008, 19, 1893–1902. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Lu, H.; Hao, A.; Ng, D.C.H.; Ponniah, S.; Guo, K.; Lufei, C.; Zeng, Q.; Cao, X. GRIM-19, a Cell Death Regulatory Protein, Is Essential for Assembly and Function of Mitochondrial Complex I. Mol. Cell. Biol. 2004, 24, 8447–8456. [Google Scholar] [CrossRef] [Green Version]
- Han, S.M.; Baig, H.S.; Hammarlund, M. Mitochondria localize to injured axons to support regeneration. Neuron 2016, 92, 1308–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiryu-Seo, S.; Kiyama, H. Mitochondrial behavior during axon regeneration/degeneration in vivo. Neurosci. Res. 2019, 139, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Patrón, L.A.; Zinsmaier, K.E. Mitochondria on the road to power axonal regeneration. Neuron 2016, 92, 1152–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaarmann, A.; Mandel, M.; Zeb, A.; Wareski, P.; Liiv, J.; Kuum, M.; Antsov, E.; Liiv, M.; Cagalinec, M.; Choubey, V.; et al. Mitochondrial biogenesis is required for axonal growth. Development 2016, 143, 1981–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G.M.; Gallo, G. The role of mitochondria in axon development and regeneration. Dev. Neurobiol. 2018, 78, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Huang, M.; Shang, D.; Yan, X.; Zhao, B.; Zhang, X. Mitochondrial Behavior in Axon Degeneration and Regeneration. Front. Aging Neurosci. 2021, 13, 103. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Krishnamurthy, S.; Patel, S.P.; Pandya, J.D.; Rabchevsky, A.G. Temporal characterization of mitochondrial bioenergetics after spinal cord injury. J. Neurotrauma 2007, 24, 991–999. [Google Scholar] [CrossRef]
- Visavadiya, N.P.; Patel, S.P.; VanRooyen, J.L.; Sullivan, P.G.; Rabchevsky, A.G. Cellular and subcellular oxidative stress parameters following severe spinal cord injury. Redox Biol. 2016, 8, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.; Rabchevsky, A.; Waldmeier, P.; Springer, J. Mitochondrial permeability transition in CNS trauma: Cause or effect of neuronal cell death? J. Neurosci. Res. 2005, 79, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Tseng, A.M.; Chung, D.D.; Pinson, M.R.; Salem, N.A.; Eaves, S.E.; Miranda, R.C. Ethanol exposure increases miR-140 in extracellular vesicles: Implications for fetal neural stem cell proliferation and maturation. Alcohol. Clin. Exp. Res. 2019, 43, 1414–1426. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the mitochondrial membrane potential using the cationic JC-1 dye as a sensitive fluorescent probe. Bio-Protoc. 2019, 9, 1. [Google Scholar] [CrossRef]
- Montier, L.L.C.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genom. 2009, 36, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Yin, P.H.; Lu, C.Y.; Chi, C.W.; Wei, Y.H. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem. J. 2000, 348 Pt 2, 425–432. [Google Scholar] [CrossRef]
- Wachsmuth, M.; Hübner, A.; Li, M.; Madea, B.; Stoneking, M. Age-related and heteroplasmy-related variation in human mtDNA copy number. PLoS Genet. 2016, 12, e1005939. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Wang, Y.; Ye, K.; Picard, M.; Gu, Z. Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genom. 2017, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Hebert, S.L.; Marquet-de Rougé, P.; Lanza, I.R.; McCrady-Spitzer, S.K.; Levine, J.A.; Middha, S.; Carter, R.E.; Klaus, K.A.; Therneau, T.M.; Highsmith, E.W. Mitochondrial aging and physical decline: Insights from three generations of women. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2015, 70, 1409–1417. [Google Scholar] [CrossRef] [Green Version]
- Rausser, S.; Trumpff, C.; McGill, M.A.; Junker, A.; Wang, W.; Ho, S.-h.; Mitchell, A.; Karan, K.R.; Monk, C.; Segerstrom, S.C.; et al. Mitochondrial phenotypes in purified human immune cell subtypes and cell mixtures. bioRxiv 2021. [Google Scholar] [CrossRef]
- Giordano, C.; Iommarini, L.; Giordano, L.; Maresca, A.; Pisano, A.; Valentino, M.L.; Caporali, L.; Liguori, R.; Deceglie, S.; Roberti, M. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain 2014, 137, 335–353. [Google Scholar] [CrossRef] [Green Version]
- Yu-Wai-Man, P.; Sitarz, K.S.; Samuels, D.C.; Griffiths, P.G.; Reeve, A.K.; Bindoff, L.A.; Horvath, R.; Chinnery, P.F. OPA1 mutations cause cytochrome c oxidase deficiency due to loss of wild-type mtDNA molecules. Hum. Mol. Genet. 2010, 19, 3043–3052. [Google Scholar] [CrossRef] [Green Version]
- Preston, C.C.; Oberlin, A.S.; Holmuhamedov, E.L.; Gupta, A.; Sagar, S.; Syed, R.H.K.; Siddiqui, S.A.; Raghavakaimal, S.; Terzic, A.; Jahangir, A. Aging-induced alterations in gene transcripts and functional activity of mitochondrial oxidative phosphorylation complexes in the heart. Mech. Ageing Dev. 2008, 129, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Yonutas, H.M.; Pandya, J.D.; Sullivan, P.G. Changes in mitochondrial bioenergetics in the brain versus spinal cord become more apparent with age. J. Bioenerg. Biomembr. 2015, 47, 149–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filograna, R.; Koolmeister, C.; Upadhyay, M.; Pajak, A.; Clemente, P.; Wibom, R.; Simard, M.L.; Wredenberg, A.; Freyer, C.; Stewart, J.B.; et al. Modulation of mtDNA copy number ameliorates the pathological consequences of a heteroplasmic mtDNA mutation in the mouse. Sci. Adv. 2019, 5, eaav9824. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Guerra Martinez, C.; Torres-Odio, S.; Bell, S.L.; Birdwell, C.E.; Bryant, J.D.; Tong, C.W.; Watson, R.O.; West, L.C.; West, A.P. Elevated type I interferon responses potentiate metabolic dysfunction, inflammation, and accelerated aging in mtDNA mutator mice. Sci. Adv. 2021, 7, eabe7548. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Whitehall, J.C.; Bradshaw, C.; Gay, D.; Robertson, F.; Blain, A.P.; Hudson, G.; Pyle, A.; Houghton, D.; Hunt, M.; et al. Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer 2020, 1, 976–989. [Google Scholar] [CrossRef]
- Suen, Y.; Fung, K.; Choy, Y.; Lee, C.; Chan, C.; Kong, S. Concanavalin A induced apoptosis in murine macrophage PU5-1.8 cells through clustering of mitochondria and release of cytochrome c. Apoptosis 2000, 5, 369–377. [Google Scholar] [CrossRef]
- De Vos, K.; Goossens, V.; Boone, E.; Vercammen, D.; Vancompernolle, K.; Vandenabeele, P.; Haegeman, G.; Fiers, W.; Grooten, J. The 55-kDa Tumor Necrosis Factor Receptor Induces Clustering of Mitochondria through Its Membrane-proximal Region*. J. Biol. Chem. 1998, 273, 9673–9680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, K.S.; Wilkinson, S.; James, J.; Ryan, K.M.; Vousden, K.H. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2009, 16, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Franco-Iborra, S.; Cuadros, T.; Parent, A.; Romero-Gimenez, J.; Vila, M.; Perier, C. Defective mitochondrial protein import contributes to complex I-induced mitochondrial dysfunction and neurodegeneration in Parkinson’s disease. Cell Death Dis. 2018, 9, 1122. [Google Scholar] [CrossRef] [Green Version]
- Jolivet, R.; Magistretti, P.; Weber, B. Deciphering neuron-glia compartmentalization in cortical energy metabolism. Front. Neuroenergetics 2009, 1, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, P.K.; Galeffi, F.; Turner, D.A. Cellular Links between Neuronal Activity and Energy Homeostasis. Front. Pharmacol. 2012, 3, 43. [Google Scholar] [CrossRef] [Green Version]
- Kwong, L.K.; Sohal, R.S. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch. Biochem. Biophys. 2000, 373, 16–22. [Google Scholar] [CrossRef]
- Ojaimi, J.; Masters, C.L.; Opeskin, K.; McKelvie, P.; Byrne, E. Mitochondrial respiratory chain activity in the human brain as a function of age. Mech. Ageing Dev. 1999, 111, 39–47. [Google Scholar] [CrossRef]
- Kokoszka, J.E.; Coskun, P.; Esposito, L.A.; Wallace, D.C. Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. USA 2001, 98, 2278–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathrop, K.L.; Steketee, M.B. Mitochondrial dynamics in retinal ganglion cell axon regeneration and growth cone guidance. J. Ocul. Biol. 2013, 1, 9. [Google Scholar] [PubMed]
- Lenaz, G.; D’Aurelio, M.; Pich, M.M.; Genova, M.; Ventura, B.; Bovina, C.; Formiggini, G.; Castelli, G.P. Mitochondrial bioenergetics in aging. Biochim. Et Biophys. Acta BBA Bioenerg. 2000, 1459, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Pandya, J.D.; Royland, J.E.; MacPhail, R.C.; Sullivan, P.G.; Kodavanti, P.R.S. Age- and brain region-specific differences in mitochondrial bioenergetics in Brown Norway rats. Neurobiol. Aging 2016, 42, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Porras, C.A.-M.; Bai, Y. Respiratory supercomplexes: Plasticity and implications. Front. Biosci. Landmark Ed. 2015, 20, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Shimokata, H.; Kuzuya, F. Aging, basal metabolic rate, and nutrition. Nihon Ronen Igakkai Zasshi. Jpn. J. Geriatr. 1993, 30, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Rubner, M. Machinery of metabolism. JAMA 1916, 66, 10.1111. [Google Scholar]
- Manini, T.M. Energy expenditure and aging. Ageing Res. Rev. 2010, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ravera, S.; Podestà, M.; Sabatini, F.; Dagnino, M.; Cilloni, D.; Fiorini, S.; Barla, A.; Frassoni, F. Discrete Changes in Glucose Metabolism Define Aging. Sci. Rep. 2019, 9, 10347. [Google Scholar] [CrossRef] [Green Version]
- Yaniv, Y.; Juhaszova, M.; Sollott, S.J. Age-related changes of myocardial ATP supply and demand mechanisms. Trends Endocrinol. Metab. 2013, 24, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Nicholls, D.G. Mitochondrial membrane potential and aging. Aging Cell 2004, 3, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Romano, A.D.; Tamborra, R.; Rollo, T.; Altomare, E.; Vendemiale, G. Bioenergetics in aging: Mitochondrial proton leak in aging rat liver, kidney and heart. Redox Rep. 2007, 12, 91–95. [Google Scholar] [CrossRef]
- Murchison, D.; Zawieja, D.C.; Griffith, W.H. Reduced mitochondrial buffering of voltage-gated calcium influx in aged rat basal forebrain neurons. Cell Calcium 2004, 36, 61–75. [Google Scholar] [CrossRef]
- Murchison, D.; Griffith, W.H. Calcium buffering systems and calcium signaling in aged rat basal forebrain neurons. Aging Cell 2007, 6, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The mitochondrial membrane potential (Δψ m) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Meijer, M. Mitochondrial biogenesis: The Tom and Tim machine. Curr. Biol. 1997, 7, R100–R103. [Google Scholar] [CrossRef] [Green Version]
- Formosa, L.E.; Ryan, M.T. Mitochondrial OXPHOS complex assembly lines. Nat. Cell Biol. 2018, 20, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.M.; Adhihetty, P.J.; Buford, T.W.; Wohlgemuth, S.E.; Lees, H.A.; Nguyen, L.M.D.; Aranda, J.M.; Sandesara, B.D.; Pahor, M.; Manini, T.M. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012, 11, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.-M.; Adhihetty, P.J.; Wawrzyniak, N.R.; Wohlgemuth, S.E.; Picca, A.; Kujoth, G.C.; Prolla, T.A.; Leeuwenburgh, C. Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS ONE 2013, 8, e69327. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.-M.; Ljubicic, V.; Adhihetty, P.J.; Hood, D.A. Biogenesis of the mitochondrial Tom40 channel in skeletal muscle from aged animals and its adaptability to chronic contractile activity. Am. J. Physiol. -Cell Physiol. 2010, 298, C1308–C1314. [Google Scholar] [CrossRef]
- Dukanovic, J.; Rapaport, D. Multiple pathways in the integration of proteins into the mitochondrial outer membrane. Biochim. Et Biophys. Acta BBA Biomembr. 2011, 1808, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Yano, H.; Baranov, S.V.; Baranova, O.V.; Kim, J.; Pan, Y.; Yablonska, S.; Carlisle, D.L.; Ferrante, R.J.; Kim, A.H.; Friedlander, R.M. Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci. 2014, 17, 822–831. [Google Scholar] [CrossRef]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B.V. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, D.; LeVault, K.R.; Barnett, A.J.; Brewer, G.J. A reversible early oxidized redox state that precedes macromolecular ROS damage in aging nontransgenic and 3xTg-AD mouse neurons. J. Neurosci. 2012, 32, 5821–5832. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.-T.; Wu, J.; Holstein, D.; Upadhyay, G.; Rourk, W.; Muller, E.; Lechleiter, J.D. Ca2+ signaling, mitochondria and sensitivity to oxidative stress in aging astrocytes. Neurobiol. Aging 2007, 28, 99–111. [Google Scholar] [CrossRef]
- Han, Q.; Xie, Y.; Ordaz, J.D.; Huh, A.J.; Huang, N.; Wu, W.; Liu, N.; Chamberlain, K.A.; Sheng, Z.-H.; Xu, X.-M. Restoring cellular energetics promotes axonal regeneration and functional recovery after spinal cord injury. Cell Metab. 2020, 31, 623–641.e628. [Google Scholar] [CrossRef] [PubMed]
- Vanhauwaert, R.; Bharat, V.; Wang, X. Surveillance and transportation of mitochondria in neurons. Curr. Opin. Neurobiol. 2019, 57, 87–93. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutherland, T.C.; Sefiani, A.; Horvat, D.; Huntington, T.E.; Lei, Y.; West, A.P.; Geoffroy, C.G. Age-Dependent Decline in Neuron Growth Potential and Mitochondria Functions in Cortical Neurons. Cells 2021, 10, 1625. https://doi.org/10.3390/cells10071625
Sutherland TC, Sefiani A, Horvat D, Huntington TE, Lei Y, West AP, Geoffroy CG. Age-Dependent Decline in Neuron Growth Potential and Mitochondria Functions in Cortical Neurons. Cells. 2021; 10(7):1625. https://doi.org/10.3390/cells10071625
Chicago/Turabian StyleSutherland, Theresa C., Arthur Sefiani, Darijana Horvat, Taylor E. Huntington, Yuanjiu Lei, A. Phillip West, and Cédric G. Geoffroy. 2021. "Age-Dependent Decline in Neuron Growth Potential and Mitochondria Functions in Cortical Neurons" Cells 10, no. 7: 1625. https://doi.org/10.3390/cells10071625