
Evolutionary Origins of DNA Repair Pathways: Role of Oxygen Catastrophe in the Emergence of DNA Glycosylases

 , , and
, , and
Abstract
:1. Introduction
2. Types of DNA Damage
2.1. Hydrolytic DNA Damage
2.2. UV Damage
2.3. Ionizing Radiation Damage
2.4. Alkylation Damage
2.5. Oxidative Damage
3. DNA Repair in Reconstructed and Minimal Genomes
4. Spontaneous DNA Damage: To BER or Not to BER?
4.1. DNA Glycosylases: Primordial Players or a Later Adaptation?
4.2. AP Endonucleases: The Archetypal Repair Endonucleases
4.3. Archaeal AP Endonuclease Mth212 in the Repair of Deaminated DNA Bases
4.4. Archaeal Endonuclease Q in the Repair of Deaminated DNA Bases
5. Nucleotide Incision Repair: An Ancient and Versatile Mechanism to Counteract Spontaneous DNA Decay and Ionizing Radiation-Induced DNA Damage
5.1. NIR as a Remnant of the Pre-BER World
5.2. Specialized NIR: UVDE-Initiated Repair of UV Photoproducts and RNAseH2-Catalyzed Ribonucleotide Excision Repair
6. Alternative Excision Repair Pathway: An Unusual Variation on the Endonuclease Theme
7. Oxygen Catastrophe: Back to BER
8. Direct Repair as a Primordial Mechanism to Counteract Alkylation and UV Damage
9. Putative Origins of NER
10. Conclusions: “Nothing in Biology Makes Sense Except in the Light of Evolution”
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eisen, J.A.; Hanawalt, P.C. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat. Res. 1999, 435, 171–213. [Google Scholar] [CrossRef] [Green Version]
- Goosen, N.; Moolenaar, G.F. Repair of UV damage in bacteria. DNA Repair 2008, 7, 353–379. [Google Scholar] [CrossRef]
- Müller, M.; Carell, T. Structural biology of DNA photolyases and cryptochromes. Curr. Opin. Struct. Biol. 2009, 19, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Vechtomova, Y.L.; Telegina, T.A.; Kritsky, M.S. Evolution of proteins of the DNA photolyase/cryptochrome family. Biochemistry 2020, 85, S131–S153. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Wallace, S.S. Hide and seek: How do DNA glycosylases locate oxidatively damaged DNA bases amidst a sea of undamaged bases? Free Radic. Biol. Med. 2017, 107, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Dodd, M.S.; Papineau, D.; Grenne, T.; Slack, J.F.; Rittner, M.; Pirajno, F.; O’Neil, J.; Little, C.T.S. Evidence for early life in Earth’s oldest hydrothermal vent precipitates. Nature 2017, 543, 60–64. [Google Scholar] [CrossRef]
- Betts, H.C.; Puttick, M.N.; Clark, J.W.; Williams, T.A.; Donoghue, P.C.J.; Pisani, D. Integrated genomic and fossil evidence illuminates life’s early evolution and eukaryote origin. Nat. Ecol. Evol. 2018, 2, 1556–1562. [Google Scholar] [CrossRef]
- Pearce, B.K.D.; Tupper, A.S.; Pudritz, R.E.; Higgs, P.G. Constraining the time interval for the origin of life on Earth. Astrobiology 2018, 18, 343–364. [Google Scholar] [CrossRef] [PubMed]
- Kozbial, P.Z.; Mushegian, A.R. Natural history of S-adenosylmethionine-binding proteins. BMC Struct. Biol. 2005, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Weiss, M.C.; Sousa, F.L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 16116. [Google Scholar] [CrossRef]
- Holland, H.D. The oxygenation of the atmosphere and oceans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 903–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, T.W.; Reinhard, C.T.; Planavsky, N.J. The rise of oxygen in Earth’s early ocean and atmosphere. Nature 2014, 506, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Douki, T.; Court, M.; Sauvaigo, S.; Odin, F.; Cadet, J. Formation of the main UV-induced thymine dimeric lesions within isolated and cellular DNA as measured by high performance liquid chromatography-tandem mass spectrometry. J. Biol. Chem. 2000, 275, 11678–11685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westall, F.; de Ronde, C.E.J.; Southam, G.; Grassineau, N.; Colas, M.; Cockell, C.; Lammer, H. Implications of a 3.472–3.333 Gyr-old subaerial microbial mat from the Barberton greenstone belt, South Africa for the UV environmental conditions on the early Earth. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1857–1875. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F. Complexity of damage produced by ionizing radiation. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 377–382. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef]
- Breimer, L.H.; Lindahl, T. Thymine lesions produced by ionizing radiation in double-stranded DNA. Biochemistry 1985, 24, 4018–4022. [Google Scholar] [CrossRef]
- Boorstein, R.J.; Hilbert, T.P.; Cunningham, R.P.; Teebor, G.W. Formation and stability of repairable pyrimidine photohydrates in DNA. Biochemistry 1990, 29, 10455–10460. [Google Scholar] [CrossRef]
- Ganguly, T.; Duker, N.J. Stability of DNA thymine hydrates. Nucleic Acids Res. 1991, 19, 3319–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawley, P.D.; Phillips, D.H. DNA adducts from chemotherapeutic agents. Mutat. Res. 1996, 355, 13–40. [Google Scholar] [CrossRef]
- Nakamura, J.; Mutlu, E.; Sharma, V.; Collins, L.; Bodnar, W.; Yu, R.; Lai, Y.; Moeller, B.; Lu, K.; Swenberg, J. The endogenous exposome. DNA Repair 2014, 19, 3–13. [Google Scholar] [CrossRef]
- Williams, J.S.; Kunkel, T.A. Ribonucleotides in DNA: Origins, repair and consequences. DNA Repair 2014, 19, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Kunkel, T.A.; Bebenek, K. DNA replication fidelity. Annu. Rev. Biochem. 2000, 69, 497–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ischenko, A.A.; Saparbaev, M.K. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature 2002, 415, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Georg, J.; Schomacher, L.; Chong, J.P.J.; Majerník, A.I.; Raabe, M.; Urlaub, H.; Müller, S.; Ciirdaeva, E.; Kramer, W.; Fritz, H.-J. The Methanothermobacter thermautotrophicus ExoIII homologue Mth212 is a DNA uridine endonuclease. Nucleic Acids Res. 2006, 34, 5325–5336. [Google Scholar] [CrossRef]
- Ishchenko, A.A.; Deprez, E.; Maksimenko, A.; Brochon, J.-C.; Tauc, P.; Saparbaev, M.K. Uncoupling of the base excision and nucleotide incision repair pathways reveals their respective biological roles. Proc. Natl. Acad. Sci. USA 2006, 103, 2564–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imlay, J.A.; Linn, S. DNA damage and oxygen radical toxicity. Science 1988, 240, 1302–1309. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Lindahl, T.; Nyberg, B. Rate of depurination of native deoxyribonucleic acid. Biochemistry 1972, 11, 3610–3618. [Google Scholar] [CrossRef]
- Lindahl, T.; Nyberg, B. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry 1974, 13, 3405–3410. [Google Scholar] [CrossRef]
- Boiteux, S.; Laval, J. Coding properties of poly(deoxycytidylic acid) templates containing uracil or apyrimidinic sites: In vitro modulation of mutagenesis by deoxyribonucleic acid repair enzymes. Biochemistry 1982, 21, 6746–6751. [Google Scholar] [CrossRef] [PubMed]
- Strauss, B.S. The ‘A rule’ of mutagen specificity: A consequence of DNA polymerase bypass of non-instructional lesions? Bioessays 1991, 13, 79–84. [Google Scholar] [CrossRef]
- Krokan, H.E.; Drabløs, F.; Slupphaug, G. Uracil in DNA—Occurrence, consequences and repair. Oncogene 2002, 21, 8935–8948. [Google Scholar] [CrossRef] [Green Version]
- Alseth, I.; Dalhus, B.; Bjørås, M. Inosine in DNA and RNA. Curr. Opin. Genet. Dev. 2014, 26, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuenschell, G.E.; O’Connor, T.R.; Termini, J. Stability, miscoding potential, and repair of 2′-deoxyxanthosine in DNA: Implications for nitric oxide-induced mutagenesis. Biochemistry 2003, 42, 3608–3616. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.; Penny, D.; Sjöberg, B.-M. Confounded cytosine! Tinkering and the evolution of DNA. Nat. Rev. Mol. Cell Biol. 2001, 2, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Beukers, R.; Ylstra, J.; Berends, W. The effect of ultraviolet light on some components of the nucleic acids: II: In rapidly frozen solutions. Rec. Trav. Chim. 1958, 77, 729–732. [Google Scholar] [CrossRef]
- Douki, T.; Sage, E. Dewar valence isomers, the third type of environmentally relevant DNA photoproducts induced by solar radiation. Photochem. Photobiol. Sci. 2016, 15, 24–30. [Google Scholar] [CrossRef]
- Boorstein, R.J.; Hilbert, T.P.; Cadet, J.; Cunningham, R.P.; Teebor, G.W. UV-induced pyrimidine hydrates in DNA are repaired by bacterial and mammalian DNA glycosylase activities. Biochemistry 1989, 28, 6164–6170. [Google Scholar] [CrossRef]
- Ganguly, T.; Weems, K.M.; Duker, N.J. Ultraviolet-induced thymine hydrates in DNA are excised by bacterial and human DNA glycosylase activities. Biochemistry 1990, 29, 7222–7228. [Google Scholar] [CrossRef]
- Tarduno, J.A.; Cottrell, R.D.; Davis, W.J.; Nimmo, F.; Bono, R.K. A Hadean to Paleoarchean geodynamo recorded by single zircon crystals. Science 2015, 349, 521–524. [Google Scholar] [CrossRef] [Green Version]
- Doglioni, C.; Pignatti, J.; Coleman, M. Why did life develop on the surface of the Earth in the Cambrian? Geosci. Front. 2016, 7, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Pogozelski, W.K.; Tullius, T.D. Oxidative strand scission of nucleic acids: Routes initiated by hydrogen abstraction from the sugar moiety. Chem. Rev. 1998, 98, 1089–1107. [Google Scholar] [CrossRef] [PubMed]
- Teoule, R.; Bert, C.; Bonicel, A. Thymine fragment damage retained in the DNA polynucleotide chain after gamma irradiation in aerated solutions. II. Radiat. Res. 1977, 72, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Teoule, R.; Bonicel, A.; Bert, C.; Fouque, B. Isolation and identification of thymine products from DNA γ irradiated in oxygen-free aqueous solutions. J. Am. Chem. Soc. 1978, 100, 6749–6750. [Google Scholar] [CrossRef]
- Bonicel, A.; Mariaggi, N.; Hughes, E.; Teoule, R. In vitro γ irradiation of DNA: Identification of radioinduced chemical modifications of the adenine moiety. Radiat. Res. 1980, 83, 19–26. [Google Scholar] [CrossRef]
- Furlong, E.A.; Jorgensen, T.J.; Henner, W.D. Production of dihydrothymidine stereoisomers in DNA by γ-irradiation. Biochemistry 1986, 25, 4344–4349. [Google Scholar] [CrossRef]
- Akhlaq, M.S.; Schuchmann, H.-P.; von Sonntag, C. The reverse of the ‘repair’ reaction of thiols: H-abstraction at carbon by thiyl radicals. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1987, 51, 91–102. [Google Scholar] [CrossRef]
- Lesiak, K.B.; Wheeler, K.T. Formation of α-deoxyadenosine in polydeoxynucleotides exposed to ionizing radiation under anoxic conditions. Radiat. Res. 1990, 121, 328–337. [Google Scholar] [CrossRef]
- Von Sonntag, C. Free-Radical-Induced DNA Damage and Its Repair: A Chemical Perspective; Springer: Berlin/Heidelberg, Germany, 2006; p. 523. [Google Scholar]
- Lawley, P.D. Effects of some chemical mutagens and carcinogens on nucleic acids. Prog. Nucleic Acid Res. Mol. Biol. 1966, 5, 89–131. [Google Scholar] [CrossRef] [PubMed]
- Singer, B.; Kuśmierek, J.T. Chemical mutagenesis. Annu. Rev. Biochem. 1982, 51, 655–691. [Google Scholar] [CrossRef] [PubMed]
- Singer, B. All oxygens in nucleic acids react with carcinogenic ethylating agents. Nature 1976, 264, 333–339. [Google Scholar] [CrossRef]
- Pegg, A.E. Methylation of the O6 position of guanine in DNA is the most likely initiating event in carcinogenesis by methylating agents. Cancer Investig. 1984, 2, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Bodell, W.J.; Singer, B. Influence of hydrogen bonding in DNA and polynucleotides on reaction of nitrogens and oxygens toward ethylnitrosourea. Biochemistry 1979, 18, 2860–2863. [Google Scholar] [CrossRef]
- Boiteux, S.; Laval, J. Mutagenesis by alkylating agents: Coding properties for DNA polymerase of poly (dC) template containing 3-methylcytosine. Biochimie 1982, 64, 637–641. [Google Scholar] [CrossRef]
- Larson, K.; Sahm, J.; Shenkar, R.; Strauss, B. Methylation-induced blocks to in vitro DNA replication. Mutat. Res. 1985, 150, 77–84. [Google Scholar] [CrossRef]
- Swann, P.F. Why do O6-alkylguanine and O4-alkylthymine miscode? The relationship between the structure of DNA containing O6-alkylguanine and O4-alkylthymine and the mutagenic properties of these bases. Mutat. Res. 1990, 233, 81–94. [Google Scholar] [CrossRef]
- Taverna, P.; Sedgwick, B. Generation of an endogenous DNA-methylating agent by nitrosation in Escherichia coli. J. Bacteriol. 1996, 178, 5105–5111. [Google Scholar] [CrossRef] [Green Version]
- Posnick, L.M.; Samson, L.D. Influence of S-adenosylmethionine pool size on spontaneous mutation, dam methylation, and cell growth of Escherichia coli. J. Bacteriol. 1999, 181, 6756–6762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broderick, J.B.; Duffus, B.R.; Duschene, K.S.; Shepard, E.M. Radical S-adenosylmethionine enzymes. Chem. Rev. 2014, 114, 4229–4317. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A.; Chin, S.M.; Linn, S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 1988, 240, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Henle, E.S.; Linn, S. Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J. Biol. Chem. 1997, 272, 19095–19098. [Google Scholar] [CrossRef] [Green Version]
- Bjelland, S.; Seeberg, E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 2003, 531, 37–80. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Tanooka, H.; Nishimura, S. Formation of 8-hydroxyguanine residues in DNA by X-irradiation. Gann 1984, 75, 1037–1039. [Google Scholar]
- Dizdaroglu, M.; Bergtold, D.S. Characterization of free radical-induced base damage in DNA at biologically relevant levels. Anal. Biochem. 1986, 156, 182–188. [Google Scholar] [CrossRef]
- Schuchmann, M.N.; Steenken, S.; Wroblewski, J.; von Sonntag, C. Site of OH radical attack on dihydrouracil and some of its methyl derivatives. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1984, 46, 225–232. [Google Scholar] [CrossRef]
- Dizdaroglu, M. Free-radical-induced formation of an 8,5′-cyclo-2′-deoxyguanosine moiety in deoxyribonucleic acid. Biochem. J. 1986, 238, 247–254. [Google Scholar] [CrossRef]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef]
- Kreutzer, D.A.; Essigmann, J.M. Oxidized, deaminated cytosines are a source of C → T transitions in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 3578–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laval, J.; Boiteux, S. Repair of cyclic nucleic acid adducts and adverse effects of apurinic sites. IARC Sci. Publ. 1986, 70, 381–385. [Google Scholar]
- Basu, A.K.; Loechler, E.L.; Leadon, S.A.; Essigmann, J.M. Genetic effects of thymine glycol: Site-specific mutagenesis and molecular modeling studies. Proc. Natl. Acad. Sci. USA 1989, 86, 7677–7681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, H.; Petrullo, L.A.; Hatahet, Z.; Wallace, S.S. Processing of DNA base damage by DNA polymerases: Dihydrothymine and β-ureidoisobutyric acid as models for instructive and noninstructive lesions. J. Biol. Chem. 1991, 266, 1469–1477. [Google Scholar] [CrossRef]
- Demple, B.; DeMott, M.S. Dynamics and diversions in base excision DNA repair of oxidized abasic lesions. Oncogene 2002, 21, 8926–8934. [Google Scholar] [CrossRef] [Green Version]
- Marnett, L.J.; Burcham, P.C. Endogenous DNA adducts: Potential and paradox. Chem. Res. Toxicol. 1993, 6, 771–785. [Google Scholar] [CrossRef]
- Burcham, P.C. Genotoxic lipid peroxidation products: Their DNA damaging properties and role in formation of endogenous DNA adducts. Mutagenesis 1998, 13, 287–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, F.-L.; Nath, R.G.; Nagao, M.; Nishikawa, A.; Zhou, G.-D.; Randerath, K. Endogenous formation and significance of 1,N2-propanodeoxyguanosine adducts. Mutat. Res. 1999, 424, 71–81. [Google Scholar] [CrossRef]
- Stone, M.P.; Cho, Y.-J.; Huang, H.; Kim, H.-Y.; Kozekov, I.D.; Kozekova, A.; Wang, H.; Minko, I.G.; Lloyd, R.S.; Harris, T.M.; et al. Interstrand DNA cross-links induced by α,β-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem. Res. 2008, 41, 793–804. [Google Scholar] [CrossRef] [Green Version]
- Barker, S.; Weinfeld, M.; Murray, D. DNA–protein crosslinks: Their induction, repair, and biological consequences. Mutat. Res. 2005, 589, 111–135. [Google Scholar] [CrossRef]
- Mushegian, A.R.; Koonin, E.V. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc. Natl. Acad. Sci. USA 1996, 93, 10268–10273. [Google Scholar] [CrossRef] [Green Version]
- Hutchison, C.A., III; Peterson, S.N.; Gill, S.R.; Cline, R.T.; White, O.; Fraser, C.M.; Smith, H.O.; Venter, J.C. Global transposon mutagenesis and a minimal Mycoplasma genome. Science 1999, 286, 2165–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Ehrlich, S.D.; Albertini, A.; Amati, G.; Andersen, K.K.; Arnaud, M.; Asai, K.; Ashikaga, S.; Aymerich, S.; Bessieres, P.; et al. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. USA 2003, 100, 4678–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, R.; Silva, F.J.; Peretó, J.; Moya, A. Determination of the core of a minimal bacterial gene set. Microbiol. Mol. Biol. Rev. 2004, 68, 518–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, A.C.; Church, G.M. Towards synthesis of a minimal cell. Mol. Syst. Biol. 2006, 2, 45. [Google Scholar] [CrossRef] [Green Version]
- Glass, J.I.; Assad-Garcia, N.; Alperovich, N.; Yooseph, S.; Lewis, M.R.; Maruf, M.; Hutchison, C.A., III; Smith, H.O.; Venter, J.C. Essential genes of a minimal bacterium. Proc. Natl. Acad. Sci. USA 2006, 103, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Venetz, J.E.; Del Medico, L.; Wölfle, A.; Schächle, P.; Bucher, Y.; Appert, D.; Tschan, F.; Flores-Tinoco, C.E.; van Kooten, M.; Guennoun, R.; et al. Chemical synthesis rewriting of a bacterial genome to achieve design flexibility and biological functionality. Proc. Natl. Acad. Sci. USA 2019, 116, 8070–8079. [Google Scholar] [CrossRef] [Green Version]
- Mirkin, B.G.; Fenner, T.I.; Galperin, M.Y.; Koonin, E.V. Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol. Biol. 2003, 3, 2. [Google Scholar] [CrossRef]
- Tuller, T.; Birin, H.; Gophna, U.; Kupiec, M.; Ruppin, E. Reconstructing ancestral gene content by coevolution. Genome Res. 2010, 20, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Lake, J.A.; Larsen, J.; Tran, D.T.; Sinsheimer, J.S. Uncovering the genomic origins of life. Genome Biol. Evol. 2018, 10, 1705–1714. [Google Scholar] [CrossRef]
- Woese, C. The universal ancestor. Proc. Natl. Acad. Sci. USA 1998, 95, 6854–6859. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat. Rev. Microbiol. 2003, 1, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Theobald, D.L. A formal test of the theory of universal common ancestry. Nature 2010, 465, 219–222. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. The neomuran origin of archaebacteria, the negibacterial root of the universal tree and bacterial megaclassification. Int. J. Syst. Evol. Microbiol. 2002, 52, 7–76. [Google Scholar] [CrossRef] [Green Version]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef] [Green Version]
- Cavalier-Smith, T.; Chao, E.E.-Y. Multidomain ribosomal protein trees and the planctobacterial origin of neomura (eukaryotes, archaebacteria). Protoplasma 2020, 257, 621–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, C.; Nelson-Sathi, S.; Roettger, M.; Sousa, F.L.; Lockhart, P.J.; Bryant, D.; Hazkani-Covo, E.; McInerney, J.O.; Landan, G.; Martin, W.F. Endosymbiotic origin and differential loss of eukaryotic genes. Nature 2015, 524, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Méheust, R.; Bhattacharya, D.; Pathmanathan, J.S.; McInerney, J.O.; Lopez, P.; Bapteste, E. Formation of chimeric genes with essential functions at the origin of eukaryotes. BMC Biol. 2018, 16, 30. [Google Scholar] [CrossRef] [Green Version]
- Fuss, J.O.; Tsai, C.-L.; Ishida, J.P.; Tainer, J.A. Emerging critical roles of Fe–S clusters in DNA replication and repair. Biochim. Biophys. Acta 2015, 1853, 1253–1271. [Google Scholar] [CrossRef] [Green Version]
- Khodour, Y.; Kaguni, L.S.; Stiban, J. Iron–sulfur clusters in nucleic acid metabolism: Varying roles of ancient cofactors. Enzymes 2019, 45, 225–256. [Google Scholar] [CrossRef]
- Copley, R.R.; Bork, P. Homology among (βα)8 barrels: Implications for the evolution of metabolic pathways. J. Mol. Biol. 2000, 303, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Sofia, H.J.; Chen, G.; Hetzler, B.G.; Reyes-Spindola, J.F.; Miller, N.E. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: Functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001, 29, 1097–1106. [Google Scholar] [CrossRef] [Green Version]
- Hitomi, K.; Iwai, S.; Tainer, J.A. The intricate structural chemistry of base excision repair machinery: Implications for DNA damage recognition, removal, and repair. DNA Repair 2007, 6, 410–428. [Google Scholar] [CrossRef]
- Mullins, E.A.; Rodriguez, A.A.; Bradley, N.P.; Eichman, B.F. Emerging roles of DNA glycosylases and the base excision repair pathway. Trends Biochem. Sci. 2019, 44, 765–781. [Google Scholar] [CrossRef]
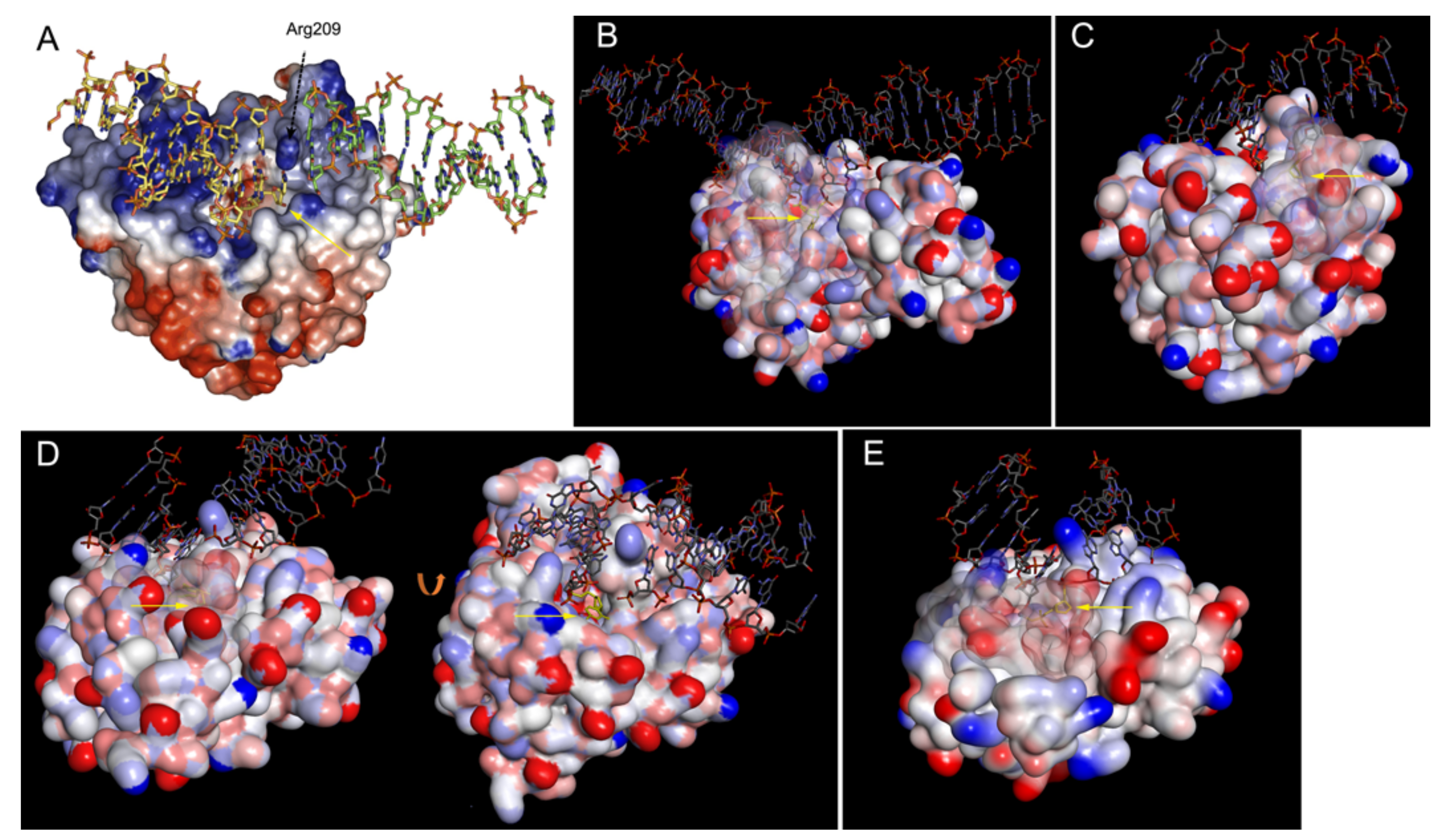
- Roberts, R.J.; Cheng, X. Base flipping. Annu. Rev. Biochem. 1998, 67, 181–198. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Cheng, X. DNA base flipping: A general mechanism for writing, reading, and erasing DNA modifications. Adv. Exp. Med. Biol. 2016, 945, 321–341. [Google Scholar] [CrossRef] [Green Version]
- Dinner, A.R.; Blackburn, G.M.; Karplus, M. Uracil-DNA glycosylase acts by substrate autocatalysis. Nature 2001, 413, 752–755. [Google Scholar] [CrossRef]
- Rios, A.C.; Tor, Y. Refining the genetic alphabet: A late-period selection pressure? Astrobiology 2012, 12, 884–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
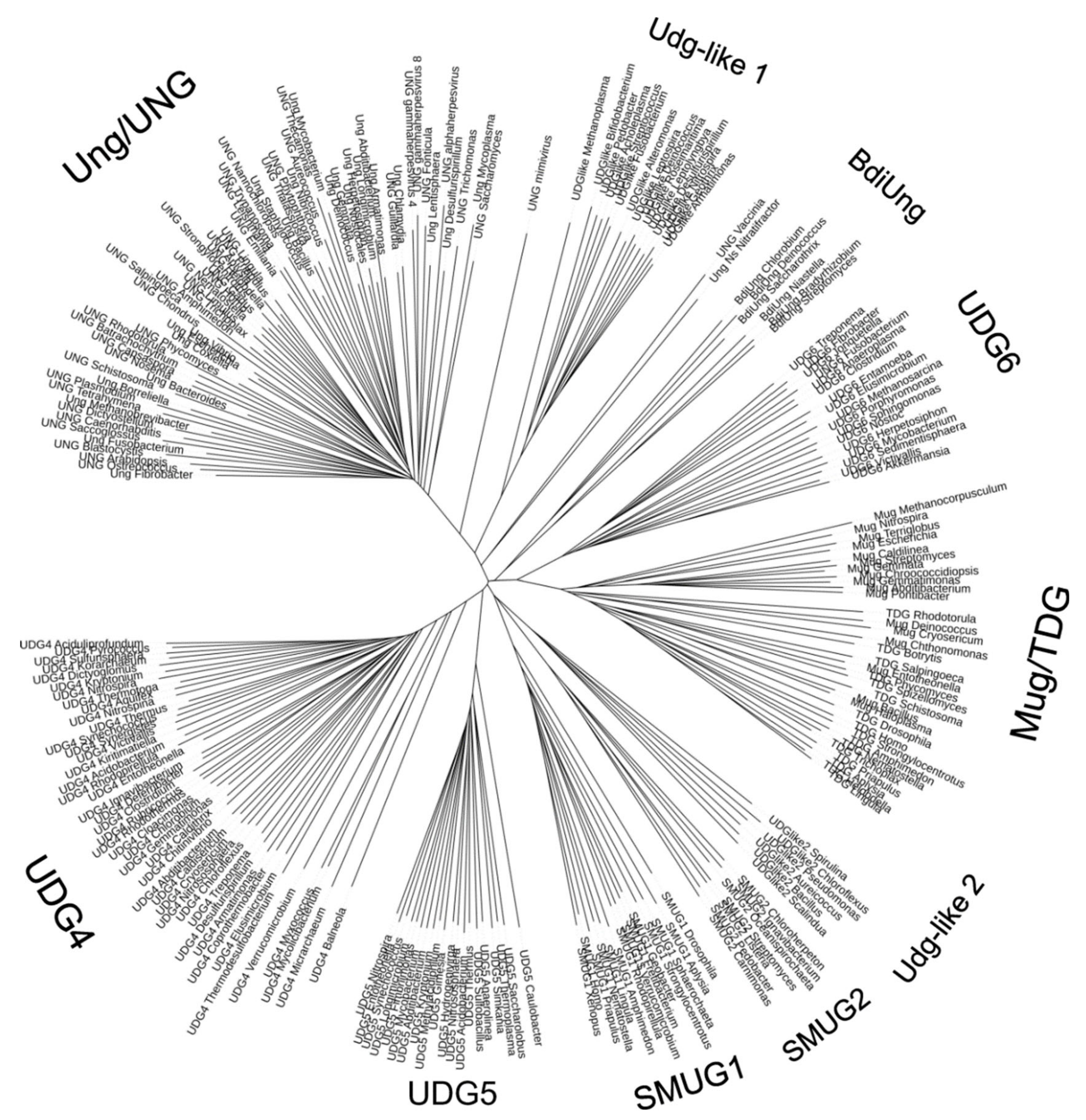
- Aravind, L.; Koonin, E.V. The α/β fold uracil DNA glycosylases: A common origin with diverse fates. Genome Biol. 2000, 1, research0007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas-Lledó, J.I.; Maddamsetti, R.; Lynch, M. Phylogenomic analysis of the uracil-DNA glycosylase superfamily. Mol. Biol. Evol. 2011, 28, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Hoseki, J.; Okamoto, A.; Masui, R.; Shibata, T.; Inoue, Y.; Yokoyama, S.; Kuramitsu, S. Crystal structure of a family 4 uracil-DNA glycosylase from Thermus thermophilus HB8. J. Mol. Biol. 2003, 333, 515–526. [Google Scholar] [CrossRef]
- Kosaka, H.; Hoseki, J.; Nakagawa, N.; Kuramitsu, S.; Masui, R. Crystal structure of family 5 uracil-DNA glycosylase bound to DNA. J. Mol. Biol. 2007, 373, 839–850. [Google Scholar] [CrossRef]
- Lee, H.-W.; Dominy, B.N.; Cao, W. New family of deamination repair enzymes in uracil-DNA glycosylase superfamily. J. Biol. Chem. 2011, 286, 31282–31287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engstrom, L.M.; Partington, O.A.; David, S.S. An iron–sulfur cluster loop motif in the Archaeoglobus fulgidus uracil–DNA glycosylase mediates efficient uracil recognition and removal. Biochemistry 2012, 51, 5187–5197. [Google Scholar] [CrossRef] [Green Version]
- Wibley, J.E.A.; Waters, T.R.; Haushalter, K.; Verdine, G.L.; Pearl, L.H. Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol. Cell 2003, 11, 1647–1659. [Google Scholar] [CrossRef]
- Xia, B.; Liu, Y.; Li, W.; Brice, A.R.; Dominy, B.N.; Cao, W. Specificity and catalytic mechanism in family 5 uracil DNA glycosylase. J. Biol. Chem. 2014, 289, 18413–18426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, P.; Yang, Y.; Li, J.; Wang, Z.; Cao, W.; Xie, W. SMUG2 DNA glycosylase from Pedobacter heparinus as a new subfamily of the UDG superfamily. Biochem. J. 2017, 474, 923–938. [Google Scholar] [CrossRef] [Green Version]
- Chembazhi, U.V.; Patil, V.V.; Sah, S.; Reeve, W.; Tiwari, R.P.; Woo, E.; Varshney, U. Uracil DNA glycosylase (UDG) activities in Bradyrhizobium diazoefficiens: Characterization of a new class of UDG with broad substrate specificity. Nucleic Acids Res. 2017, 45, 5863–5876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortázar, D.; Kunz, C.; Selfridge, J.; Lettieri, T.; Saito, Y.; MacDougall, E.; Wirz, A.; Schuermann, D.; Jacobs, A.L.; Siegrist, F.; et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 2011, 470, 419–423. [Google Scholar] [CrossRef]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Saparbaev, M.; Laval, J. 3,N4-ethenocytosine, a highly mutagenic adduct, is a primary substrate for Escherichia coli double-stranded uracil-DNA glycosylase and human mismatch-specific thymine-DNA glycosylase. Proc. Natl. Acad. Sci. USA 1998, 95, 8508–8513. [Google Scholar] [CrossRef] [Green Version]
- Talhaoui, I.; Couvé, S.; Ishchenko, A.A.; Kunz, C.; Schär, P.; Saparbaev, M. 7,8-dihydro-8-oxoadenine, a highly mutagenic adduct, is repaired by Escherichia coli and human mismatch-specific uracil/thymine-DNA glycosylases. Nucleic Acids Res. 2013, 41, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Doherty, A.J.; Serpell, L.C.; Ponting, C.P. The helix-hairpin-helix DNA-binding motif: A structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res. 1996, 24, 2488–2497. [Google Scholar] [CrossRef]
- Aravind, L.; Walker, D.R.; Koonin, E.V. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999, 27, 1223–1242. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Grishin, N.V. Common fold in helix–hairpin–helix proteins. Nucleic Acids Res. 2000, 28, 2643–2650. [Google Scholar] [CrossRef]
- Deterding, L.J.; Prasad, R.; Mullen, G.P.; Wilson, S.H.; Tomer, K.B. Mapping of the 5′-2-deoxyribose-5-phosphate lyase active site in DNA polymerase β by mass spectrometry. J. Biol. Chem. 2000, 275, 10463–10471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Díaz, M.; Bebenek, K.; Kunkel, T.A.; Blanco, L. Identification of an intrinsic 5′-deoxyribose-5-phosphate lyase activity in human DNA polymerase λ: A possible role in base excision repair. J. Biol. Chem. 2001, 276, 34659–34663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thayer, M.M.; Ahern, H.; Xing, D.; Cunningham, R.P.; Tainer, J.A. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J. 1995, 14, 4108–4120. [Google Scholar] [CrossRef] [PubMed]
- Nash, H.M.; Bruner, S.D.; Schärer, O.D.; Kawate, T.; Addona, T.A.; Spooner, E.; Lane, W.S.; Verdine, G.L. Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol. 1996, 6, 968–980. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Golan, G.; Gilboa, R.; Fernandes, A.S.; Gerchman, S.E.; Kycia, J.H.; Rieger, R.A.; Grollman, A.P.; Shoham, G. Structural analysis of an Escherichia coli endonuclease VIII covalent reaction intermediate. EMBO J. 2002, 21, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Prakash, A.; Doublié, S.; Wallace, S.S. The Fpg/Nei family of DNA glycosylases: Substrates, structures, and search for damage. Prog. Mol. Biol. Transl. Sci. 2012, 110, 71–91. [Google Scholar] [CrossRef] [Green Version]
- Verly, W.G.; Paquette, Y. An endonuclease for depurinated DNA in Escherichia coli B. Can. J. Biochem. 1972, 50, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Barzilay, G.; Hickson, I.D. Structure and function of apurinic/apyrimidinic endonucleases. Bioessays 1995, 17, 713–719. [Google Scholar] [CrossRef]
- Mol, C.D.; Hosfield, D.J.; Tainer, J.A. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: The 3′ ends justify the means. Mutat. Res. 2000, 460, 211–229. [Google Scholar] [CrossRef]
- Wilson, D.M., III; Barsky, D. The major human abasic endonuclease: Formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 2001, 485, 283–307. [Google Scholar] [CrossRef]
- Suck, D.; Oefner, C. Structure of DNase I at 2.0 Å resolution suggests a mechanism for binding to and cutting DNA. Nature 1986, 321, 620–625. [Google Scholar] [CrossRef]
- Mol, C.D.; Kuo, C.-F.; Thayer, M.M.; Cunningham, R.P.; Tainer, J.A. Structure and function of the multifunctional DNA-repair enzyme exonuclease III. Nature 1995, 374, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Mol, C.D.; Izumi, T.; Mitra, S.; Tainer, J.A. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature 2000, 403, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Whisstock, J.C.; Romero, S.; Gurung, R.; Nandurkar, H.; Ooms, L.M.; Bottomley, S.P.; Mitchell, C.A. The inositol polyphosphate 5-phosphatases and the apurinic/apyrimidinic base excision repair endonucleases share a common mechanism for catalysis. J. Biol. Chem. 2000, 275, 37055–37061. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Kurahashi, K.; Gao, R.; Tsutakawa, S.E.; Tainer, J.A.; Pommier, Y.; Aihara, H. Structural basis for recognition of 5′-phosphotyrosine adducts by Tdp2. Nat. Struct. Mol. Biol. 2012, 19, 1372–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosfield, D.J.; Guan, Y.; Haas, B.J.; Cunningham, R.P.; Tainer, J.A. Structure of the DNA repair enzyme endonuclease IV and its DNA complex: Double-nucleotide flipping at abasic sites and three-metal-ion catalysis. Cell 1999, 98, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Tsutakawa, S.E.; Shin, D.S.; Mol, C.D.; Izumi, T.; Arvai, A.S.; Mantha, A.K.; Szczesny, B.; Ivanov, I.N.; Hosfield, D.J.; Maiti, B.; et al. Conserved structural chemistry for incision activity in structurally non-homologous apurinic/apyrimidinic endonuclease APE1 and endonuclease IV DNA repair enzymes. J. Biol. Chem. 2013, 288, 8445–8455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demple, B.; Halbrook, J.; Linn, S. Echerichia coli xth mutants are hypersensitive to hydrogen peroxide. J. Bacteriol. 1983, 153, 1079–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, R.P.; Saporito, S.M.; Spitzer, S.G.; Weiss, B. Endonuclease IV (nfo) mutant of Escherichia coli. J. Bacteriol. 1986, 168, 1120–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillet, M.; Boiteux, S. Endogenous DNA abasic sites cause cell death in the absence of Apn1, Apn2 and Rad1/Rad10 in Saccharomyces cerevisiae. EMBO J. 2002, 21, 2833–2841. [Google Scholar] [CrossRef] [Green Version]
- Karumbati, A.S.; Deshpande, R.A.; Jilani, A.; Vance, J.R.; Ramotar, D.; Wilson, T.E. The role of yeast DNA 3′-phosphatase Tpp1 and Rad1/Rad10 endonuclease in processing spontaneous and induced base lesions. J. Biol. Chem. 2003, 278, 31434–31443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fryxell, K.J.; Zuckerkandl, E. Cytosine deamination plays a primary role in the evolution of mammalian isochores. Mol. Biol. Evol. 2000, 17, 1371–1383. [Google Scholar] [CrossRef] [Green Version]
- Hershberg, R.; Petrov, D.A. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet. 2010, 6, e1001115. [Google Scholar] [CrossRef] [Green Version]
- Ognjanovic, S.; Olivier, M.; Bergemann, T.L.; Hainaut, P. Sarcomas in TP53 germline mutation carriers: A review of the IARC TP53 database. Cancer 2012, 118, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Warner, H.R.; Duncan, B.K.; Garrett, C.; Neuhard, J. Synthesis and metabolism of uracil-containing deoxyribonucleic acid in Escherichia coli. J. Bacteriol. 1981, 145, 687–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillet, M.; Auffret Van Der Kemp, P.; Boiteux, S. dUTPase activity is critical to maintain genetic stability in Saccharomyces cerevisiae. Nucleic Acids Res. 2006, 34, 2056–2066. [Google Scholar] [CrossRef] [PubMed]
- Slesarev, A.I.; Mezhevaya, K.V.; Makarova, K.S.; Polushin, N.N.; Shcherbinina, O.V.; Shakhova, V.V.; Belova, G.I.; Aravind, L.; Natale, D.A.; Rogozin, I.B.; et al. The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens. Proc. Natl. Acad. Sci. USA 2002, 99, 4644–4649. [Google Scholar] [CrossRef] [Green Version]
- Bult, C.J.; White, O.; Olsen, G.J.; Zhou, L.; Fleischmann, R.D.; Sutton, G.G.; Blake, J.A.; FitzGerald, L.M.; Clayton, R.A.; Gocayne, J.D.; et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science 1996, 273, 1058–1073. [Google Scholar] [CrossRef]
- Hendrickson, E.L.; Kaul, R.; Zhou, Y.; Bovee, D.; Chapman, P.; Chung, J.; Conway de Macario, E.; Dodsworth, J.A.; Gillett, W.; Graham, D.E.; et al. Complete genome sequence of the genetically tractable hydrogenotrophic methanogen Methanococcus maripaludis. J. Bacteriol. 2004, 186, 6956–6969. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.R.; Doucette-Stamm, L.A.; Deloughery, C.; Lee, H.; Dubois, J.; Aldredge, T.; Bashirzadeh, R.; Blakely, D.; Cook, R.; Gilbert, K.; et al. Complete genome sequence of Methanobacterium thermoautotrophicum ΔH: Functional analysis and comparative genomics. J. Bacteriol. 1997, 179, 7135–7155. [Google Scholar] [CrossRef] [Green Version]
- Fricke, W.F.; Seedorf, H.; Henne, A.; Krüer, M.; Liesegang, H.; Hedderich, R.; Gottschalk, G.; Thauer, R.K. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J. Bacteriol. 2006, 188, 642–658. [Google Scholar] [CrossRef] [Green Version]
- Schomacher, L.; Chong, J.P.J.; McDermott, P.; Kramer, W.; Fritz, H.-J. DNA uracil repair initiated by the archaeal ExoIII homologue Mth212 via direct strand incision. Nucleic Acids Res. 2009, 37, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Schomacher, L.; Schürer, K.A.; Ciirdaeva, E.; McDermott, P.; Chong, J.P.J.; Kramera, W.; Fritz, H.-J. Archaeal DNA uracil repair via direct strand incision: A minimal system reconstituted from purified components. DNA Repair 2010, 9, 438–447. [Google Scholar] [CrossRef]
- Lakomek, K.; Dickmanns, A.; Ciirdaeva, E.; Schomacher, L.; Ficner, R. Crystal structure analysis of DNA uridine endonuclease Mth212 bound to DNA. J. Mol. Biol. 2010, 399, 604–617. [Google Scholar] [CrossRef]
- Prorok, P.; Alili, D.; Saint-Pierre, C.; Gasparutto, D.; Zharkov, D.O.; Ishchenko, A.A.; Tudek, B.; Saparbaev, M. Uracil in duplex DNA is a substrate for the human nucleotide incision repair pathway. Proc. Natl. Acad. Sci. USA 2013, 110, E3695–E3703. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, M.; Ishino, S.; Yamagami, T.; Egashira, Y.; Kiyonari, S.; Ishino, Y. A novel endonuclease that may be responsible for damaged DNA base repair in Pyrococcus furiosus. Nucleic Acids Res. 2015, 43, 2853–2863. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, M.; Iwai, S. Molecular basis of substrate recognition of endonuclease Q from the euryarchaeon Pyrococcus furiosus. J. Bacteriol. 2020, 202, e00542. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, M.; Ishino, S.; Heffernan, M.; Cann, I.; Ishino, Y. The mesophilic archaeon Methanosarcina acetivorans counteracts uracil in DNA with multiple enzymes: EndoQ, ExoIII, and UDG. Sci. Rep. 2018, 8, 15791. [Google Scholar] [CrossRef]
- Miyazono, K.-i.; Ishino, S.; Makita, N.; Ito, T.; Ishino, Y.; Tanokura, M. Crystal structure of the novel lesion-specific endonuclease PfuEndoQ from Pyrococcus furiosus. Nucleic Acids Res. 2018, 46, 4807–4818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazouzi, A.; Vigouroux, A.; Aikeshev, B.; Brooks, P.J.; Saparbaev, M.K.; Morera, S.; Ishchenko, A.A. Insight into mechanisms of 3′-5′ exonuclease activity and removal of bulky 8,5′-cyclopurine adducts by apurinic/apyrimidinic endonucleases. Proc. Natl. Acad. Sci. USA 2013, 110, E3071–E3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcin, E.D.; Hosfield, D.J.; Desai, S.A.; Haas, B.J.; Björas, M.; Cunningham, R.P.; Tainer, J.A. DNA apurinic-apyrimidinic site binding and excision by endonuclease IV. Nat. Struct. Mol. Biol. 2008, 15, 515–522. [Google Scholar] [CrossRef]
- Shiraishi, M.; Ishino, S.; Yoshida, K.; Yamagami, T.; Cann, I.; Ishino, Y. PCNA is involved in the EndoQ-mediated DNA repair process in Thermococcales. Sci. Rep. 2016, 6, 25532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, M.; Ishino, S.; Cann, I.; Ishino, Y. A functional endonuclease Q exists in the bacterial domain: Identification and characterization of endonuclease Q from Bacillus pumilus. Biosci. Biotechnol. Biochem. 2017, 81, 931–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, K.; Moeller, N.H.; Banerjee, S.; McCann, J.L.; Carpenter, M.A.; Yin, L.; Moorthy, R.; Orellana, K.; Harki, D.A.; Harris, R.S.; et al. Structural basis for recognition of distinct deaminated DNA lesions by endonuclease Q. Proc. Natl. Acad. Sci. USA 2021, 118, e2021120118. [Google Scholar] [CrossRef]
- Lahm, A.; Suck, D. DNase I-induced DNA conformation. 2 Å structure of a DNase I-octamer complex. J. Mol. Biol. 1991, 222, 645–667. [Google Scholar] [CrossRef]
- Weston, S.A.; Lahm, A.; Suck, D. X-ray structure of the DNase I-d(GGTATACC)2 complex at 2.3 Å resolution. J. Mol. Biol. 1992, 226, 1237–1256. [Google Scholar] [CrossRef]
- Redrejo-Rodríguez, M.; Vigouroux, A.; Mursalimov, A.; Grin, I.; Alili, D.; Koshenov, Z.; Akishev, Z.; Maksimenko, A.; Bissenbaev, A.K.; Matkarimov, B.T.; et al. Structural comparison of AP endonucleases from the exonuclease III family reveals new amino acid residues in human AP endonuclease 1 that are involved in incision of damaged DNA. Biochimie 2016, 128–129, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Van Houten, B.; Snowden, A. Mechanism of action of the Escherichia coli UvrABC nuclease: Clues to the damage recognition problem. Bioessays 1993, 15, 51–59. [Google Scholar] [CrossRef]
- Yang, W. Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair 2006, 5, 654–666. [Google Scholar] [CrossRef]
- Min, J.-H.; Pavletich, N.P. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 2007, 449, 570–575. [Google Scholar] [CrossRef]
- Scrima, A.; Koníčková, R.; Czyzewski, B.K.; Kawasaki, Y.; Jeffrey, P.D.; Groisman, R.; Nakatani, Y.; Iwai, S.; Pavletich, N.P.; Thomä, N.H. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell 2008, 135, 1213–1223. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.E.; Chudaev, M.; Mustaev, A. Key features of magnesium that underpin its role as the major ion for electrophilic biocatalysis. FEBS J. 2020, 287, 5439–5463. [Google Scholar] [CrossRef] [PubMed]
- Ide, H.; Tedzuka, K.; Shimzu, H.; Kimura, Y.; Purmal, A.A.; Wallace, S.S.; Kow, Y.W. α-Deoxyadenosine, a major anoxic radiolysis product of adenine in DNA, is a substrate for Escherichia coli endonuclease IV. Biochemistry 1994, 33, 7842–7847. [Google Scholar] [CrossRef]
- Gros, L.; Ishchenko, A.A.; Ide, H.; Elder, R.H.; Saparbaev, M.K. The major human AP endonuclease (Ape1) is involved in the nucleotide incision repair pathway. Nucleic Acids Res. 2004, 32, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Bowman, K.K.; Sidik, K.; Smith, C.A.; Taylor, J.-S.; Doetsch, P.W.; Freyer, G.A. A new ATP-independent DNA endonuclease from Schizosaccharomyces pombe that recognizes cyclobutane pyrimidine dimers and 6–4 photoproducts. Nucleic Acids Res. 1994, 22, 3026–3032. [Google Scholar] [CrossRef] [Green Version]
- Yajima, H.; Takao, M.; Yasuhira, S.; Zhao, J.H.; Ishii, C.; Inoue, H.; Yasui, A. A eukaryotic gene encoding an endonuclease that specifically repairs DNA damaged by ultraviolet light. EMBO J. 1995, 14, 2393–2399. [Google Scholar] [CrossRef]
- Avery, A.M.; Kaur, B.; Taylor, J.-S.; Mello, J.A.; Essigmann, J.M.; Doetsch, P.W. Substrate specificity of ultraviolet DNA endonuclease (UVDE/Uve1p) from Schizosaccharomyces pombe. Nucleic Acids Res. 1999, 27, 2256–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanno, S.-i.; Shigenori, I.; Takao, M.; Yasui, A. Repair of apurinic/apyrimidinic sites by UV damage endonuclease; a repair protein for UV and oxidative damage. Nucleic Acids Res. 1999, 27, 3096–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paspaleva, K.; Thomassen, E.; Pannu, N.S.; Iwai, S.; Moolenaar, G.F.; Goosen, N.; Abrahams, J.P. Crystal structure of the DNA repair enzyme ultraviolet damage endonuclease. Structure 2007, 15, 1316–1324. [Google Scholar] [CrossRef] [Green Version]
- Meulenbroek, E.M.; Peron Cane, C.; Jala, I.; Iwai, S.; Moolenaar, G.F.; Goosen, N.; Pannu, N.S. UV damage endonuclease employs a novel dual-dinucleotide flipping mechanism to recognize different DNA lesions. Nucleic Acids Res. 2013, 41, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Nick McElhinny, S.A.; Kumar, D.; Clark, A.B.; Watt, D.L.; Watts, B.E.; Lundström, E.-B.; Johansson, E.; Chabes, A.; Kunkel, T.A. Genome instability due to ribonucleotide incorporation into DNA. Nat. Chem. Biol. 2010, 6, 774–781. [Google Scholar] [CrossRef] [Green Version]
- Reijns, M.A.M.; Rabe, B.; Rigby, R.E.; Mill, P.; Astell, K.R.; Lettice, L.A.; Boyle, S.; Leitch, A.; Keighren, M.; Kilanowski, F.; et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 2012, 149, 1008–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eder, P.S.; Walder, R.Y.; Walder, J.A. Substrate specificity of human RNase H1 and its role in excision repair of ribose residues misincorporated in DNA. Biochimie 1993, 75, 123–126. [Google Scholar] [CrossRef]
- Rydberg, B.; Game, J. Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN-1 in cell-free extracts. Proc. Natl. Acad. Sci. USA 2002, 99, 16654–16659. [Google Scholar] [CrossRef] [Green Version]
- Sparks, J.L.; Chon, H.; Cerritelli, S.M.; Kunkel, T.A.; Johansson, E.; Crouch, R.J.; Burgers, P.M. RNase H2-initiated ribonucleotide excision repair. Mol. Cell 2012, 47, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Craigie, R.; Davies, D.R. Retroviral integrases and their cousins. Curr. Opin. Struct. Biol. 1996, 6, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and mechanism. DNA Repair 2019, 84, 102672. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Kanaya, S. Ribonuclease H: Molecular diversities, substrate binding domains, and catalytic mechanism of the prokaryotic enzymes. FEBS J. 2009, 276, 1482–1493. [Google Scholar] [CrossRef]
- Gates, F.T., III; Linn, S. Endonuclease V of Escherichia coli. J. Biol. Chem. 1977, 252, 1647–1653. [Google Scholar] [CrossRef]
- Yao, M.; Hatahet, Z.; Melamede, R.J.; Kow, Y.W. Purification and characterization of a novel deoxyinosine-specific enzyme, deoxyinosine 3′ endonuclease, from Escherichia coli. J. Biol. Chem. 1994, 269, 16260–16268. [Google Scholar] [CrossRef]
- Yao, M.; Kow, Y.W. Cleavage of insertion/deletion mismatches, flap and pseudo-Y DNA structures by deoxyinosine 3′-endonuclease from Escherichia coli. J. Biol. Chem. 1996, 271, 30672–30676. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Kow, Y.W. Further characterization of Escherichia coli endonuclease V: Mechanism of recognition for deoxyinosine, deoxyuridine, and base mismatches in DNA. J. Biol. Chem. 1997, 272, 30774–30779. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Lu, J.; Barany, F.; Cao, W. Multiple cleavage activities of endonuclease V from Thermotoga maritima: Recognition and strand nicking mechanism. Biochemistry 2001, 40, 8738–8748. [Google Scholar] [CrossRef]
- Kiyonari, S.; Egashira, Y.; Ishino, S.; Ishino, Y. Biochemical characterization of endonuclease V from the hyperthermophilic archaeon, Pyrococcus furiosus. J. Biochem. 2014, 155, 325–333. [Google Scholar] [CrossRef]
- Ishino, S.; Makita, N.; Shiraishi, M.; Yamagami, T.; Ishino, Y. EndoQ and EndoV work individually for damaged DNA base repair in Pyrococcus furiosus. Biochimie 2015, 118, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Schouten, K.A.; Weiss, B. Endonuclease V protects Escherichia coli against specific mutations caused by nitrous acid. Mutat. Res. 1999, 435, 245–254. [Google Scholar] [CrossRef]
- Weiss, B. Endonuclease V of Escherichia coli prevents mutations from nitrosative deamination during nitrate/nitrite respiration. Mutat. Res. 2001, 461, 301–309. [Google Scholar] [CrossRef]
- Pang, B.; McFaline, J.L.; Burgis, N.E.; Dong, M.; Taghizadeh, K.; Sullivan, M.R.; Elmquist, C.E.; Cunningham, R.P.; Dedon, P.C. Defects in purine nucleotide metabolism lead to substantial incorporation of xanthine and hypoxanthine into DNA and RNA. Proc. Natl. Acad. Sci. USA 2012, 109, 2319–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saparbaev, M.; Laval, J. Excision of hypoxanthine from DNA containing dIMP residues by the Escherichia coli, yeast, rat, and human alkylpurine DNA glycosylases. Proc. Natl. Acad. Sci. USA 1994, 91, 5873–5877. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, P.J.; Ellenberger, T. Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J. Biol. Chem. 2004, 279, 9750–9757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moe, A.; Ringvoll, J.; Nordstrand, L.M.; Eide, L.; Bjørås, M.; Seeberg, E.; Rognes, T.; Klungland, A. Incision at hypoxanthine residues in DNA by a mammalian homologue of the Escherichia coli antimutator enzyme endonuclease V. Nucleic Acids Res. 2003, 31, 3893–3900. [Google Scholar] [CrossRef] [Green Version]
- Fladeby, C.; Vik, E.S.; Laerdahl, J.K.; Neurauter, C.G.; Heggelund, J.E.; Thorgaard, E.; Strøm-Andersen, P.; Bjørås, M.; Dalhus, B.; Alseth, I. The human homolog of Escherichia coli endonuclease V is a nucleolar protein with affinity for branched DNA structures. PLoS ONE 2012, 7, e47466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, R.; Alford-Zappala, M.; Kow, Y.W.; Cunningham, R.P.; Cao, W. Human endonuclease V as a repair enzyme for DNA deamination. Mutat. Res. 2012, 735, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Morita, Y.; Shibutani, T.; Nakanishi, N.; Nishikura, K.; Iwai, S.; Kuraoka, I. Human endonuclease V is a ribonuclease specific for inosine-containing RNA. Nat. Commun. 2013, 4, 2273. [Google Scholar] [CrossRef] [Green Version]
- Vik, E.S.; Nawaz, M.S.; Strøm Andersen, P.; Fladeby, C.; Bjørås, M.; Dalhus, B.; Alseth, I. Endonuclease V cleaves at inosines in RNA. Nat. Commun. 2013, 4, 2271. [Google Scholar] [CrossRef] [Green Version]
- Dalhus, B.; Alseth, I.; Bjørås, M. Structural basis for incision at deaminated adenines in DNA and RNA by endonuclease V. Prog. Biophys. Mol. Biol. 2015, 117, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Laval, J.; Boiteux, S. Substrate specificity of the Escherichia coli endonuclease III: Excision of thymine- and cytosine-derived lesions in DNA produced by radiation-generated free radicals. Biochemistry 1993, 32, 12105–12111. [Google Scholar] [CrossRef]
- Jurado, J.; Saparbaev, M.; Matray, T.J.; Greenberg, M.M.; Laval, J. The ring fragmentation product of thymidine C5-hydrate when present in DNA is repaired by the Escherichia coli Fpg and Nth proteins. Biochemistry 1998, 37, 7757–7763. [Google Scholar] [CrossRef]
- Daniels, D.S.; Woo, T.T.; Luu, K.X.; Noll, D.M.; Clarke, N.D.; Pegg, A.E.; Tainer, J.A. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat. Struct. Mol. Biol. 2004, 11, 714–720. [Google Scholar] [CrossRef]
- Aas, P.A.; Otterlei, M.; Falnes, P.Ø.; Vågbø, C.B.; Skorpen, F.; Akbari, M.; Sundheim, O.; Bjørås, M.; Slupphaug, G.; Seeberg, E.; et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 2003, 421, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Falnes, P.Ø.; Rognes, T. DNA repair by bacterial AlkB proteins. Res. Microbiol. 2003, 154, 531–538. [Google Scholar] [CrossRef]
- Sedgwick, B. Repairing DNA-methylation damage. Nat. Rev. Mol. Cell Biol. 2004, 5, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Trewick, S.C.; Henshaw, T.F.; Hausinger, R.P.; Lindahl, T.; Sedgwick, B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 2002, 419, 174–178. [Google Scholar] [CrossRef]
- Falnes, P.Ø.; Johansen, R.F.; Seeberg, E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature 2002, 419, 178–182. [Google Scholar] [CrossRef]
- Leiros, I.; Nabong, M.P.; Grøsvik, K.; Ringvoll, J.; Haugland, G.T.; Uldal, L.; Reite, K.; Olsbu, I.K.; Knævelsrud, I.; Moe, E.; et al. Structural basis for enzymatic excision of N1-methyladenine and N3-methylcytosine from DNA. EMBO J. 2007, 26, 2206–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schul, W.; Jans, J.; Rijksen, Y.M.A.; Klemann, K.H.M.; Eker, A.P.M.; de Wit, J.; Nikaido, O.; Nakajima, S.; Yasui, A.; Hoeijmakers, J.H.J.; et al. Enhanced repair of cyclobutane pyrimidine dimers and improved UV resistance in photolyase transgenic mice. EMBO J. 2002, 21, 4719–4729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancar, A. Structure and function of photolyase and in vivo enzymology: 50th anniversary. J. Biol. Chem. 2008, 283, 32153–32157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, M.; Jarillo, J.A.; Smirnova, O.; Cashmore, A.R. Cryptochrome blue-light photoreceptors of Arabidopsis implicated in phototropism. Nature 1998, 392, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Hogenesch, J.B.; Kay, S.A. Circadian rhythms from flies to human. Nature 2002, 417, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-W.; Kim, S.-T.; Sancar, A.; Deisenhofer, J. Crystal structure of DNA photolyase from Escherichia coli. Science 1995, 268, 1866–1872. [Google Scholar] [CrossRef] [PubMed]
- Tamada, T.; Kitadokoro, K.; Higuchi, Y.; Inaka, K.; Yasui, A.; de Ruiter, P.E.; Eker, A.P.M.; Miki, K. Crystal structure of DNA photolyase from Anacystis nidulans. Nat. Struct. Biol. 1997, 4, 887–891. [Google Scholar] [CrossRef]
- Eker, A.P.M.; Dekker, R.H.; Berends, W. Photoreactivating enzyme from Streptomyces griseus—IV. On the nature of the chromophoric cofactor in Streptomyces griseus photoreactivating enzyme. Photochem. Photobiol. 1981, 33, 65–72. [Google Scholar] [CrossRef]
- Eker, A.P.M.; Kooiman, P.; Hessels, J.K.C.; Yasui, A. DNA photoreactivating enzyme from the cyanobacterium Anacystis nidulans. J. Biol. Chem. 1990, 265, 8009–8015. [Google Scholar] [CrossRef]
- Johnson, J.L.; Hamm-Alvarez, S.; Payne, G.; Sancar, G.B.; Rajagopalan, K.V.; Sancar, A. Identification of the second chromophore of Escherichia coli and yeast DNA photolyases as 5,10-methenyltetrahydrofolate. Proc. Natl. Acad. Sci. USA 1988, 85, 2046–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.F.; Sancar, A. Cloning, sequencing, expression and characterization of DNA photolyase from Salmonella typhimurium. Nucleic Acids Res. 1991, 19, 4885–4890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravind, L.; Anantharaman, V.; Koonin, E.V. Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: Implications for protein evolution in the RNA world. Proteins 2002, 48, 1–14. [Google Scholar] [CrossRef]
- Oberpichler, I.; Pierik, A.J.; Wesslowski, J.; Pokorny, R.; Rosen, R.; Vugman, M.; Zhang, F.; Neubauer, O.; Ron, E.Z.; Batschauer, A.; et al. A photolyase-like protein from Agrobacterium tumefaciens with an iron-sulfur cluster. PLoS ONE 2011, 6, e26775. [Google Scholar] [CrossRef]
- Setlow, P.; Li, L. Photochemistry and photobiology of the spore photoproduct: A 50-year journey. Photochem. Photobiol. 2015, 91, 1263–1290. [Google Scholar] [CrossRef] [Green Version]
- Berteau, O.; Benjdia, A. DNA repair by the radical SAM enzyme spore photoproduct lyase: From biochemistry to structural investigations. Photochem. Photobiol. 2017, 93, 67–77. [Google Scholar] [CrossRef]
- Krishnan, A.; Burroughs, A.M.; Iyer, L.M.; Aravind, L. Unexpected evolution of lesion-recognition modules in eukaryotic NER and kinetoplast DNA dynamics proteins from bacterial mobile elements. iScience 2018, 9, 192–208. [Google Scholar] [CrossRef]
- Geourjon, C.; Orelle, C.; Steinfels, E.; Blanchet, C.; Deléage, G.; Di Pietro, A.; Jault, J.-M. A common mechanism for ATP hydrolysis in ABC transporter and helicase superfamilies. Trends Biochem. Sci. 2001, 26, 539–544. [Google Scholar] [CrossRef]
- White, M.F.; Allers, T. DNA repair in the archaea—an emerging picture. FEMS Microbiol. Rev. 2018, 42, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Matelska, D.; Steczkiewicz, K.; Ginalski, K. Comprehensive classification of the PIN domain-like superfamily. Nucleic Acids Res. 2017, 45, 6995–7020. [Google Scholar] [CrossRef] [PubMed]
- Senissar, M.; Manav, M.C.; Brodersen, D.E. Structural conservation of the PIN domain active site across all domains of life. Protein Sci. 2017, 26, 1474–1492. [Google Scholar] [CrossRef] [Green Version]
- Bujnicki, J.M. Phylogeny of the restriction endonuclease-like superfamily inferred from comparison of protein structures. J. Mol. Evol. 2000, 50, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Kinch, L.N.; Ginalski, K.; Rychlewski, L.; Grishin, N.V. Identification of novel restriction endonuclease-like fold families among hypothetical proteins. Nucleic Acids Res. 2005, 33, 3598–3605. [Google Scholar] [CrossRef] [PubMed]
- Karakas, E.; Truglio, J.J.; Croteau, D.; Rhau, B.; Wang, L.; Van Houten, B.; Kisker, C. Structure of the C-terminal half of UvrC reveals an RNase H endonuclease domain with an Argonaute-like catalytic triad. EMBO J. 2007, 26, 613–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunin-Horkawicz, S.; Feder, M.; Bujnicki, J.M. Phylogenomic analysis of the GIY-YIG nuclease superfamily. BMC Genom. 2006, 7, 98. [Google Scholar] [CrossRef] [Green Version]
- Furuya, K.; Hutchinson, C.R. The DrrC protein of Streptomyces peucetius, a UvrA-like protein, is a DNA-binding protein whose gene is induced by daunorubicin. FEMS Microbiol. Lett. 1998, 168, 243–249. [Google Scholar] [CrossRef]
- Moolenaar, G.F.; van Rossum-Fikkert, S.; van Kesteren, M.; Goosen, N. Cho, a second endonuclease involved in Escherichia coli nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2002, 99, 1467–1472. [Google Scholar] [CrossRef] [Green Version]
- Perera, A.V.; Mendenhall, J.B.; Courcelle, C.T.; Courcelle, J. Cho endonuclease functions during DNA interstrand cross-link repair in Escherichia coli. J. Bacteriol. 2016, 198, 3099–3108. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Superfamily | Process | E. coli | Human | Function a |
|---|---|---|---|---|
| LUCA genome reconstruction: Mirkin et al. [89] b | ||||
| O6-methylguanine-DNA–protein-cysteine methyltransferase | DR | Ada, Ogt, Atl1 | MGMT | exocyclic O-alkyltransferases |
| NUDIX hydrolases | nucleotide pool sanitization | MutT | MTH1 | oxidized NTP/dNTP hydrolysis |
| Uracil–DNA glycosylase | BER | Ung, Mug | UNG, TDG, SMUG1 | uracil–DNA glycosylase |
| Helix–hairpin–helix | BER | Nth, MutY, AlkA | NTHL1, OGG1, MUTYH, MBD4 | DNA glycosylases for various lesions |
| TIM barrel | BER | Nfo | – | AP endonuclease |
| 5′ → 3′ exonucleases | various | PolA, Exo | FEN1 | normal and damaged DNA degradation; flap endonucleases |
| Family A DNA polymerases; DnaQ-like 3′ → 5′ exonucleases | repair DNA synthesis, proofreading | PolA | POLN, POLQ | DNA polymerases; proofreading activity may or may not be present |
| DEAD-box helicases | NER, recombination repair | UvrD, RecB | PARPBP1 | monomeric DNA helicases involved in recombination, replication, and repair |
| ATP-binding cassette transporters | MMR | MutS | MSH2–MSH6 | mismatch detection |
| S5-like fold;histidine kinase-, DNA gyrase B-, and HSP90-like ATPase | MMR | MutL | MLH1, MLH3, PMS1, PMS2 | mismatch signal transduction |
| P-loop NTPases | recombination repair; non-homologous end joining | SbcC | SMC3, RAD50 | DSB recognition and/or processing |
| Calcineurin-like phosphoesterases | recombination repair; non-homologous end joining | SbcD | MRE11 | DSB recognition and/or processing |
| Haloacid dehalogenases | recombination repair | RecJ | – | 5′ → 3′ exonucleases |
| P-loop NTPases | recombination repair | RecA | RAD51, DMC1 | strand exchange |
| LUCA genome reconstruction: Weiss et al. [12] | ||||
| PDDEXK nucleases | specialized mismatch repair | Vsr | – | T:G mismatch cleavage |
| PDDEXK nucleases | specialized mismatch repair | – | – | archaeal mismatch endonucleases (NucS/ EndoMS) |
| RNase H-like | AER | Nfi | ENDOV | deoxyinosine 3′-endonuclease |
| P-loop NTPases | recombination repair | RuvB | WRNIP1 | hexameric DNA helicases driving branch migration during recombination |
| Family Y DNA polymerases | translesion synthesis | DinB, UmuC | POLH, POLI, POLK, REV1 | DNA polymerases capable of translesion synthesis |
| DEAD-box helicases | various | RecQ | BLM, WRN | monomeric DNA helicases involved in recombination, replication, and repair |
| DEAD-box helicases | unknown, DNA damage-induced | Lhr | ASCC3 | DNA damage-inducible DNA:RNA helicase |
| DEAD-box helicases | NER | RadD (YejH) | ERCC3 | DNA helicase that functions in transcription and NER |
| DNA topoisomerases, subtype IA | possibly break repair and chromatin remodeling/maintenance | – | – | archaeal reverse gyrase; introduces positive supercoils into DNA |
| Radical_SAM | ? | – | – | putative DNA modification/repair FeS-containing radical SAM protein |
| Minimal genome: Glass et al. [87] c | ||||
| Uracil–DNA glycosylase | BER | Ung | UNG | uracil–DNA glycosylase |
| TIM barrel | BER | Nfo | – | AP endonuclease |
| ATP-binding cassette transporters | NER | UvrA | – | NER lesion search |
| ATP-binding cassette transporters | NER | UvrB | – | NER lesion search |
| RNase H-like | NER | UvrC | – | NER nuclease |
| Minimal genome: Venetz et al. [88] d | ||||
| dUTPases | nucleotide pool sanitization | Dut | DUT | trimeric dUTP diphosphatase |
| Uracil–DNA glycosylase | BER | Ung | UNG | uracil–DNA glycosylase |
| Haloacid dehalogenases | recombination repair | RecJ | – | 5′ → 3′ exonucleases |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prorok, P.; Grin, I.R.; Matkarimov, B.T.; Ishchenko, A.A.; Laval, J.; Zharkov, D.O.; Saparbaev, M. Evolutionary Origins of DNA Repair Pathways: Role of Oxygen Catastrophe in the Emergence of DNA Glycosylases. Cells 2021, 10, 1591. https://doi.org/10.3390/cells10071591
Prorok P, Grin IR, Matkarimov BT, Ishchenko AA, Laval J, Zharkov DO, Saparbaev M. Evolutionary Origins of DNA Repair Pathways: Role of Oxygen Catastrophe in the Emergence of DNA Glycosylases. Cells. 2021; 10(7):1591. https://doi.org/10.3390/cells10071591
Chicago/Turabian StyleProrok, Paulina, Inga R. Grin, Bakhyt T. Matkarimov, Alexander A. Ishchenko, Jacques Laval, Dmitry O. Zharkov, and Murat Saparbaev. 2021. "Evolutionary Origins of DNA Repair Pathways: Role of Oxygen Catastrophe in the Emergence of DNA Glycosylases" Cells 10, no. 7: 1591. https://doi.org/10.3390/cells10071591