
Targeting the Fibroblast Growth Factor Receptor (FGFR) Family in Lung Cancer


Abstract
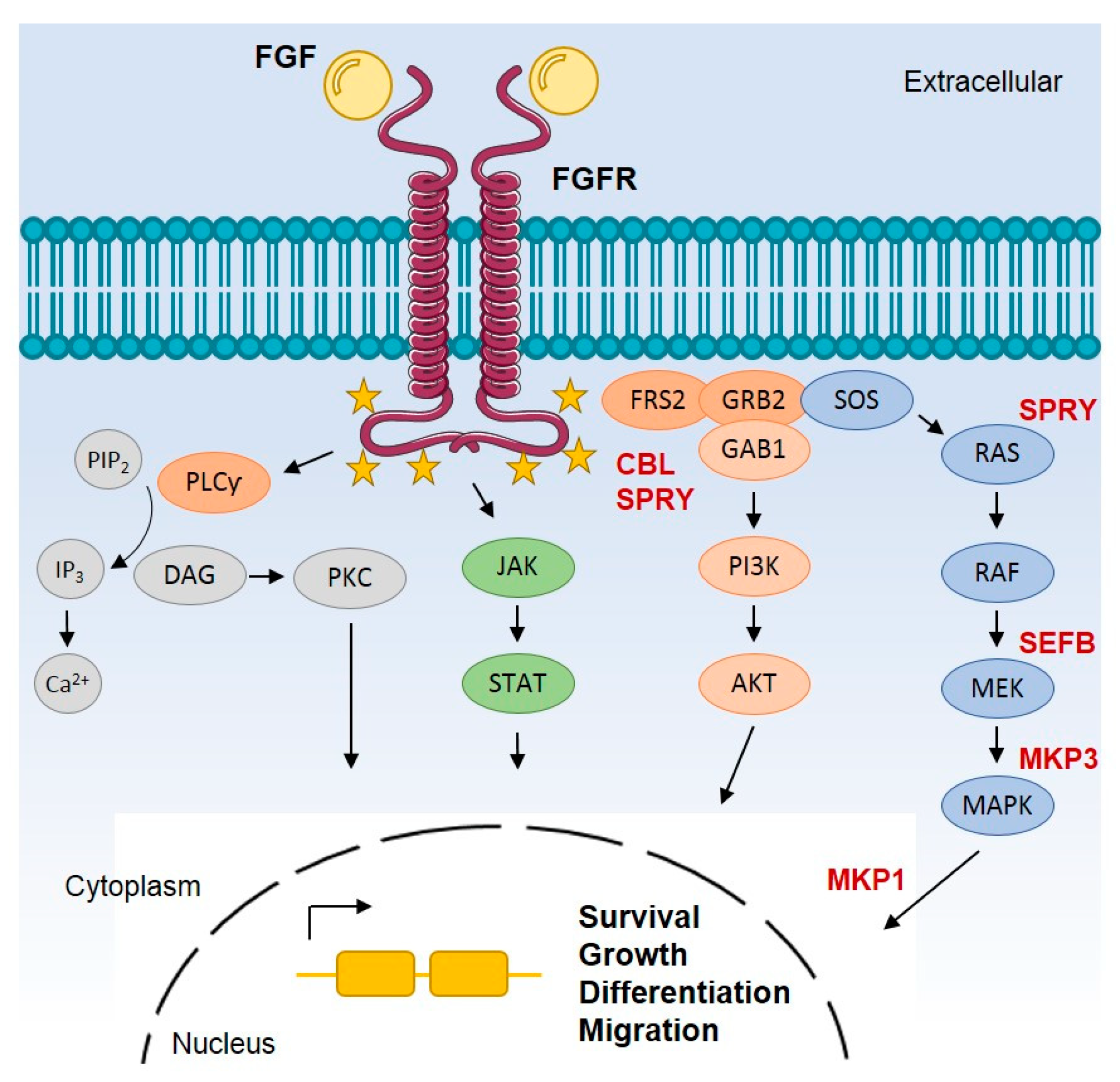
:1. Introduction
1.1. An Overview of the FGFR Family
1.2. The Landscape of FGFR Alterations in Lung Cancer
1.2.1. Gene Amplifications
1.2.2. Point Mutations
1.2.3. Chromosomal Translocations
2. FGFR as a Mechanism of Resistance to Inhibition of EGFR and KRAS Mutations
2.1. Resistance Associated with Use of EGFR Tyrosine Kinase Inhibitors
2.2. Resistance Associated to Targeting Mutant KRAS in Lung Cancer
3. Preclinical and Clinical Studies of FGFR Inhibitors in Lung Cancer
3.1. Nonselective FGFR TKIs
3.2. Selective FGFR TKIs
4. Mechanisms of Acquired Resistance to FGFR Inhibitors
5. Strategies to Overcome FGFR TKIs Resistance
5.1. Novel FGFR Therapies
5.2. Monoclonal Antibodies and FGF Traps
5.3. Combinatorial Strategies
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Moerlooze, L.; Spencer-Dene, B.; Revest, J.M.; Hajihosseini, M.; Rosewell, I.; Dickson, C. An Important Role for the IIIb Isoform of Fibroblast Growth Factor Receptor 2 in Mesenchymal-Epithelial Signalling during Mouse Organogenesis. Development 2000, 127, 483–492. [Google Scholar] [CrossRef]
- Kimelman, D.; Kirschner, M. Synergistic induction of mesoderm by FGF and TGF-β and the identification of an mRNA coding for FGF in the early xenopus embryo. Cell 1987, 51, 869–877. [Google Scholar] [CrossRef]
- Wilkie, A.O. Bad bones, absent smell, selfish testes: The pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 2005, 16, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [Green Version]
- Carter, E.P.; Fearon, A.E.; Grose, R.P. Careless talk costs lives: Fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015, 25, 221–233. [Google Scholar] [CrossRef]
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2010, 149, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Wiedemann, M.; Trueb, B. Characterization of a Novel Protein (FGFRL1) from Human Cartilage Related to FGF Receptors. Genomics 2000, 69, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornitz, D.M.; Xu, J.; Colvin, J.S.; McEwen, D.G.; MacArthur, C.A.; Coulier, F.; Gao, G.; Goldfarb, M. Receptor Specificity of the Fibroblast Growth Factor Family. J. Biol. Chem. 1996, 271, 15292–15297. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Iwata, T.; Leung, H.Y. Mechanisms of FGFR-mediated carcinogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 850–860. [Google Scholar] [CrossRef] [Green Version]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.-C. Targeting FGFR Signaling in Cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [Green Version]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Tanner, Y.; Grose, R.P. Dysregulated FGF signalling in neoplastic disorders. Semin. Cell Dev. Biol. 2016, 53, 126–135. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutt, A.; Ramos, A.H.; Hammerman, P.S.; Mermel, C.; Cho, J.; Sharifnia, T.; Chande, A.; Tanaka, K.E.; Stransky, N.; Greulich, H.; et al. Inhibitor-Sensitive FGFR1 Amplification in Human Non-Small Cell Lung Cancer. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Heist, R.S.; Mino-Kenudson, M.; Sequist, L.V.; Tammireddy, S.; Morrissey, L.; Christiani, D.C.; Engelman, J.A.; Iafrate, A.J. FGFR1 Amplification in Squamous Cell Carcinoma of The Lung. J. Thorac. Oncol. 2012, 7, 1775–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.R.; Kim, D.J.; Kang, D.R.; Lee, J.G.; Lim, S.M.; Lee, C.Y.; Rha, S.Y.; Bae, M.K.; Lee, Y.J.; Kim, S.H.; et al. Fibroblast Growth Factor Receptor 1 Gene Amplification Is Associated with Poor Survival and Cigarette Smoking Dosage in Patients with Resected Squamous Cell Lung Cancer. J. Clin. Oncol. 2013, 31, 731–737. [Google Scholar] [CrossRef]
- Schildhaus, H.-U.; Heukamp, L.C.; Merkelbach-Bruse, S.; Riesner, K.; Schmitz, K.; Binot, E.; Paggen, E.; Albus, K.; Schulte, W.; Ko, Y.-D.; et al. Definition of a fluorescence in-situ hybridization score identifies high- and low-level FGFR1 amplification types in squamous cell lung cancer. Mod. Pathol. 2012, 25, 1473–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and Focal FGFR1 Amplification Associates with Therapeutically Tractable FGFR1 Dependency in Squamous Cell Lung Cancer. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flockerzi, F.A.; Roggia, C.; Langer, F.; Holleczek, B.; Bohle, R.M. FGFR1 gene amplification in squamous cell carcinomas of the lung: A potential favorable prognostic marker for women and for patients with advanced cancer. Virchows Arch. 2018, 472, 759–769. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, A.M.; Bos, M.; Schmitz, K.; Wilsberg, L.; Binot, E.; Wolf, J.; Büttner, R.; Schildhaus, H.-U. Fibroblast growth factor receptor 1 (FGFR1) amplification is a potential therapeutic target in small-cell lung cancer. Mod. Pathol. 2014, 27, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Liao, R.G.; Jung, J.; Tchaicha, J.; Wilkerson, M.D.; Sivachenko, A.; Beauchamp, E.M.; Liu, Q.; Pugh, T.J.; Pedamallu, C.S.; Hayes, D.N.; et al. Inhibitor-Sensitive FGFR2 and FGFR3 Mutations in Lung Squamous Cell Carcinoma. Cancer Res. 2013, 73, 5195–5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Qin, A.; Johnson, A.; Ross, J.S.; Miller, V.A.; Ali, S.M.; Schrock, A.B.; Gadgeel, S.M. Detection of Known and Novel FGFR Fusions in Non–Small Cell Lung Cancer by Comprehensive Genomic Profiling. J. Thorac. Oncol. 2019, 14, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Wang, L.; Li, Y.; Hu, H.; Shen, L.; Shen, X.; Pan, Y.; Ye, T.; Zhang, Y.; Luo, X.; et al. FGFR1/3 Tyrosine Kinase Fusions Define a Unique Molecular Subtype of Non–Small Cell Lung Cancer. Clin. Cancer Res. 2014, 20, 4107–4114. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.-S.; Ju, Y.S.; Lee, W.-C.; Shin, J.-Y.; Lee, J.K.; Bleazard, T.; Jung, Y.J.; Kim, J.-O.; Yu, S.-B.; Lee, E.-R.; et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012, 22, 2109–2119. [Google Scholar] [CrossRef] [Green Version]
- Dienstmann, R.; Rodon, J.; Prat, A.; Perez-Garcia, J.; Adamo, B.; Felip, E.; Cortes, J.; Iafrate, A.J.; Nuciforo, P.; Tabernero, J. Genomic aberrations in the FGFR pathway: Opportunities for targeted therapies in solid tumors. Ann. Oncol. 2013. [Google Scholar] [CrossRef]
- Pardo, O.E.; Latigo, J.; Jeffery, R.E.; Nye, E.; Poulsom, R.; Spencer-Dene, B.; Lemoine, N.R.; Stamp, G.W.; Aboagye, E.O.; Seckl, M.J. The Fibroblast Growth Factor Receptor Inhibitor PD173074 Blocks Small Cell Lung Cancer Growth In Vitro and In Vivo. Cancer Res. 2009, 69, 8645–8651. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Ron, D.A.; Rasschaert, M.; Prenen, H.; Mehra, R.; Scilla, K.; Pauwels, P.; Rolfo, C. Is There Room for Personalized Medicine in Small-Cell Lung Cancer (SCLC)? Remarkable Activity of Pazopanib in Refractory FGFR1-Amplified ED-SCLC. JCO Precis. Oncol. 2019, 3, 1–8. [Google Scholar] [CrossRef]
- Liao, R.G.; Watanabe, H.; Meyerson, M.; Hammerman, P.S. Targeted therapy for squamous cell lung cancer. Lung Cancer Manag. 2012, 1, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, L.H.; Nelson, K.N.; Meyer, A.N.; Donoghue, D.J. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 2015, 26, 425–449. [Google Scholar] [CrossRef] [Green Version]
- Marks, J.L.; McLellan, M.D.; Zakowski, M.F.; Lash, A.E.; Kasai, Y.; Broderick, S.; Sarkaria, I.S.; Pham, D.; Singh, B.; Miner, T.L.; et al. Mutational Analysis of EGFR and Related Signaling Pathway Genes in Lung Adenocarcinomas Identifies a Novel Somatic Kinase Domain Mutation in FGFR4. PLoS ONE 2007, 2, e426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Hunter, C.A.; Bignell, G.R.; Edkins, S.; Davies, H.; Teague, J.W.; Stevens, C.; O’Meara, S.; Smith, R.; Parker, A.G.; et al. Intragenic ERBB2 kinase mutations in tumours. Nature 2004, 431, 525–526. [Google Scholar] [CrossRef]
- Schneider, L.; Essmann, F.; Kletke, A.; Rio, P.; Hanenberg, H.; Wetzel, W.; Schulze-Osthoff, K.; Nürnberg, B.; Piekorz, R.P. The Transforming Acidic Coiled Coil 3 Protein Is Essential for Spindle-dependent Chromosome Alignment and Mitotic Survival. J. Biol. Chem. 2007, 282, 29273–29283. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Ryan, E.L.; Royle, S.J. FGFR3–TACC3 cancer gene fusions cause mitotic defects by removal of endogenous TACC3 from the mitotic spindle. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming Fusions of FGFR and TACC Genes in Human Glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, B.; Ashford, P.; Moya-Garcia, A.A.; Rust, A.; Crawford, M.; Williams, S.V.; Knowles, M.A.; Katan, M.; Orengo, C.; Godovac-Zimmermann, J. Unique signalling connectivity of FGFR3-TACC3 oncoprotein revealed by quantitative phosphoproteomics and differential network analysis. Oncotarget 2017, 8, 102898–102911. [Google Scholar] [CrossRef] [Green Version]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.; Carneiro, B.A.; Taxter, T.; Tavora, F.A.; Kalyan, A.; Pai, S.A.; Chae, Y.K.; Giles, F.J. FGFR3-TACC3 fusion in solid tumors: Mini review. Oncotarget 2016, 7, 55924–55938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krook, M.A.; Lenyo, A.; Wilberding, M.; Barker, H.; Dantuono, M.; Bailey, K.M.; Chen, H.-Z.; Reeser, J.W.; Wing, M.R.; Miya, J.; et al. Efficacy of FGFR Inhibitors and Combination Therapies for Acquired Resistance in FGFR2-Fusion Cholangiocarcinoma. Mol. Cancer Ther. 2020, 19, 847–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churi, C.R.; Shroff, R.; Wang, Y.; Rashid, A.; Kang, H.C.; Weatherly, J.; Zuo, M.; Zinner, R.; Hong, D.; Meric-Bernstam, F.; et al. Mutation Profiling in Cholangiocarcinoma: Prognostic and Therapeutic Implications. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive Molecular Profiling of Lung Adenocarcinoma: The Cancer Genome Atlas Research Network. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Pakkala, S.; Ramalingam, S.S. Personalized therapy for lung cancer: Striking a moving target. JCI Insight 2018. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.P.; Pietanza, M.C.; Johnson, M.L.; Riely, G.J.; Miller, V.A.; Sima, C.S.; Zakowski, M.F.; Rusch, V.W.; Ladanyi, M.; Kris, M.G. Incidence of EGFR Exon 19 Deletions and L858R in Tumor Specimens from Men and Cigarette Smokers with Lung Adenocarcinomas. J. Clin. Oncol. 2011, 29, 2066–2070. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Yoshida, K.; Hida, T.; Tsuboi, M.; Tada, H.; Kuwano, H.; Mitsudomi, T. Analysis of Epidermal Growth Factor Receptor Gene Mutation in Patients with Non–Small Cell Lung Cancer and Acquired Resistance to Gefitinib. Clin. Cancer Res. 2006, 12, 5764–5769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, A.M.; Huang, P.H. Receptor Tyrosine Kinase Coactivation Networks in Cancer. Cancer Res. 2010, 70, 3857–3860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.-C.; Vyse, S.; Huang, P.H. Exploiting receptor tyrosine kinase co-activation for cancer therapy. Drug Discov. Today 2017, 22, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Azuma, K.; Kawahara, A.; Sonoda, K.; Nakashima, K.; Tashiro, K.; Watari, K.; Izumi, H.; Kage, M.; Kuwano, M.; Ono, M.; et al. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget 2014, 5, 5908–5919. [Google Scholar] [CrossRef] [Green Version]
- Terai, H.; Soejima, K.; Yasuda, H.; Nakayama, S.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Ikemura, S.; Sato, T.; et al. Activation of the FGF2-FGFR1 Autocrine Pathway: A Novel Mechanism of Acquired Resistance to Gefitinib in NSCLC. Mol. Cancer Res. 2013, 11, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ware, K.E.; Marshall, M.E.; Heasley, L.R.; Marek, L.; Hinz, T.K.; Hercule, P.; Helfrich, B.A.; Doebele, R.C.; Heasley, L.E. Rapidly Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in NSCLC Cell Lines through De-Repression of FGFR2 and FGFR3 Expression. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Ware, K.E.; Hinz, T.K.; Kleczko, E.K.; Singleton, K.R.; Marek, L.A.; Helfrich, B.; Cummings, C.T.; Graham, D.K.; Astling, D.P.; Tan, A.-C.; et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis 2013, 2, e39. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.T.; Tsunematsu, T.; Yanagisawa, S.; Kudo, Y.; Miyauchi, M.; Kamata, N.; Takata, T. The FGFR1 inhibitor PD173074 induces mesenchymal–epithelial transition through the transcription factor AP-1. Br. J. Cancer 2013, 109, 2248–2258. [Google Scholar] [CrossRef] [Green Version]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Shien, K.; Toyooka, S.; Yamamoto, H.; Soh, J.; Jida, M.; Thu, K.L.; Hashida, S.; Maki, Y.; Ichihara, E.; Asano, H.; et al. Acquired Resistance to EGFR Inhibitors Is Associated with a Manifestation of Stem Cell–like Properties in Cancer Cells. Cancer Res. 2015, 73, 3051–3061. [Google Scholar] [CrossRef] [Green Version]
- Weng, C.-H.; Chen, L.-Y.; Lin, Y.-C.; Shih, J.-Y.; Lin, Y.-C.; Tseng, R.-Y.; Chiu, A.-C.; Yeh, Y.-H.; Liu, C.; Lin, Y.-T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, P.T.; Huang, P.H. Exploiting vulnerabilities in cancer signalling networks to combat targeted therapy resistance. Essays Biochem. 2018, 62, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, K.; Hensing, T.; Malik, R.; Salgia, R. Prognostic and Predictive Value inKRASin Non–Small-Cell Lung Cancer. JAMA Oncol. 2016, 2, 805–812. [Google Scholar] [CrossRef]
- Blasco, R.B.; Francoz, S.; Santamaría, D.; Cañamero, M.; Dubus, P.; Charron, J.; Baccarini, M.; Barbacid, M. C-Raf, but Not B-Raf, Is Essential for Development of K-Ras Oncogene-Driven Non-Small Cell Lung Carcinoma. Cancer Cell 2011, 19, 652–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puyol, M.; Martín, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaría, D.; Barbacid, M. A Synthetic Lethal Interaction between K-Ras Oncogenes and Cdk4 Unveils a Therapeutic Strategy for Non-small Cell Lung Carcinoma. Cancer Cell 2010, 18, 63–73. [Google Scholar] [CrossRef]
- Haines, E.; Chen, T.; Kommajosyula, N.; Chen, Z.; Herter-Sprie, G.S.; Cornell, L.; Wong, K.-K.; Shapiro, G.I. Palbociclib resistance confers dependence on an FGFR-MAP kinase-mTOR-driven pathway in KRAS-mutant non-small cell lung cancer. Oncotarget 2018, 9, 31572–31589. [Google Scholar] [CrossRef] [Green Version]
- Kitai, H.; Ebi, H.; Tomida, S.; Floros, K.V.; Kotani, H.; Adachi, Y.; Oizumi, S.; Nishimura, M.; Faber, A.C.; Yano, S. Epithelial-to-Mesenchymal Transition Defines Feedback Activation of Receptor Tyrosine Kinase Signaling Induced by MEK Inhibition in KRAS-Mutant Lung Cancer. Cancer Discov. 2016, 6, 754–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manchado, E.; Weissmueller, S.; Morris, J.P.; Chen, C.-C.; Wullenkord, R.; Lujambio, A.; De Stanchina, E.; Poirier, J.T.; Gainor, J.F.; Corcoran, R.B.; et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016, 534, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, W.; Franchini, F.; Alexander, M.; Officer, A.; Wong, H.-L.; Ijzerman, M.; Desai, J.; Solomon, B.J. Real world outcomes in KRAS G12C mutation positive non-small cell lung cancer. Lung Cancer 2020, 146, 310–317. [Google Scholar] [CrossRef]
- O’Bryan, J.P. Pharmacological targeting of RAS: Recent success with direct inhibitors. Pharmacol. Res. 2019, 139, 503–511. [Google Scholar] [CrossRef]
- Jiao, D.; Yang, S. Overcoming Resistance to Drugs Targeting KRAS Mutation. Innovation 2020, 1, 100035. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Solomon, M.; Li, L.-S.; Hansen, R.; Rosen, N. Cancer therapeutics: Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.B.; De La Cruz, F.F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRASG12C Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [Green Version]
- Misale, S.; Fatherree, J.P.; Cortez, E.; Li, C.; Bilton, S.J.; Timonina, D.; Myers, D.T.; Lee, D.; Gomez-Caraballo, M.; Greenberg, M.; et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin. Cancer Res. 2019, 25, 796–807. [Google Scholar] [CrossRef] [Green Version]
- Hallinan, N.; Finn, S.; Cuffe, S.; Rafee, S.; O’Byrne, K.; Gately, K. Targeting the fibroblast growth factor receptor family in cancer. Cancer Treat. Rev. 2016, 46, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craddock, K.J.; Ludkovski, O.; Sykes, J.; Shepherd, F.A.; Tsao, M.-S. Prognostic Value of Fibroblast Growth Factor Receptor 1 Gene Locus Amplification in Resected Lung Squamous Cell Carcinoma. J. Thorac. Oncol. 2013, 8, 1371–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cihoric, N.; Savic, S.; Schneider, S.; Ackermann, I.; Bichsel-Naef, M.; A Schmid, R.; Lardinois, D.; Gugger, M.; Bubendorf, L.; Zlobec, I.; et al. Prognostic role of FGFR1 amplification in early-stage non-small cell lung cancer. Br. J. Cancer 2014, 110, 2914–2922. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.L.; Yu, H.; Smith, D.E.; Boyle, T.A.; York, E.R.; Leedy, S.; Gao, D.; Aisner, D.L.; Van Bokhoven, A.; Heasley, L.E.; et al. Preselection of Lung Cancer Cases Using FGFR1 mRNA and Gene Copy Number for Treatment with Ponatinib. Clin. Lung Cancer 2019, 20, e39–e51. [Google Scholar] [CrossRef]
- Lim, S.H.; Sun, J.-M.; Choi, Y.; Kim, H.R.; Ahn, S.; Lee, J.Y.; Lee, S.-H.; Ahn, J.S.; Park, K.; Kim, J.H.; et al. Efficacy and safety of dovitinib in pretreated patients with advanced squamous non-small cell lung cancer withFGFR1amplification: A single-arm, phase 2 study. Cancer 2016, 122, 3024–3031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, J.-C.; De Braud, F.G.; Bahleda, R.; Adamo, B.; Cereda, R.; Camboni, M.G.; Robert, R.; Isaacson, J.D.; Litten, J.B.; Allen, A.R.; et al. A phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. J. Clin. Oncol. 2014, 32, 2500. [Google Scholar] [CrossRef]
- Paik, P.K.; Shen, R.; Berger, M.F.; Ferry, D.; Soria, J.-C.; Mathewson, A.; Rooney, C.; Smith, N.R.; Cullberg, M.; Kilgour, E.; et al. A Phase Ib Open-Label Multicenter Study of AZD4547 in Patients with Advanced Squamous Cell Lung Cancers. Clin. Cancer Res. 2017, 23, 5366–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, C.; Redman, M.W.; Lara, P.N.; Borghaei, H.; Hoffman, P.; Bradley, J.D.; Newman, A.J.; Feldman, M.J.; Minichiello, K.; Miao, J.; et al. SWOG S1400D (NCT02965378), a Phase II Study of the Fibroblast Growth Factor Receptor Inhibitor AZD4547 in Previously Treated Patients with Fibroblast Growth Factor Pathway–Activated Stage IV Squamous Cell Lung Cancer (Lung-MAP Substudy). J. Thorac. Oncol. 2019, 14, 1847–1852. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Zhang, T.; Cheng, P.; Li, D.; Ogorodniitchouk, O.; Lahmamssi, C.; Wang, G.; Lan, M. Therapeutic implications of fibroblast growth factor receptor inhibitors in a combination regimen for solid tumors (Review). Oncol. Lett. 2020, 20, 2525–2536. [Google Scholar] [CrossRef]
- Smyth, E.C.; Turner, N.C.; Pearson, A.; Peckitt, C.; Chau, I.; Watkins, D.J.; Starling, N.; Rao, S.; Gillbanks, A.; Kilgour, E.; et al. Phase II study of AZD4547 in FGFR amplified tumours: Gastroesophageal cancer (GC) cohort pharmacodynamic and biomarker results. J. Clin. Oncol. 2016, 34, 154. [Google Scholar] [CrossRef]
- Nogova, L.; Sequist, L.V.; Garcia, J.M.P.; Andre, F.; Delord, J.-P.; Hidalgo, M.; Schellens, J.H.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients with Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [CrossRef]
- Schuler, M.; Cho, B.C.; Sayehli, C.M.; Navarro, A.; A Soo, R.; Richly, H.; Cassier, P.A.; Tai, D.; Penel, N.; Nogova, L.; et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: A phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2019, 20, 1454–1466. [Google Scholar] [CrossRef]
- Yamania, A.; Bieleckab, D.Z.-; Lipnera, J.; Stańczakbc, A.; Piórkowskaa, N.; Seweryna Stańczakb, P.; Olejkowskaa, P.; Hucz-Kalitowska, J.; Magdycza, M.; Dzwonekb, K.; et al. Discovery and optimization of novel pyrazole-benzimidazole CPL304110, as a potent and selective inhibitor of fibroblast growth factor receptors FGFR (1–3). Eur. J. Med. Chem. 2021, 210. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, S.; Petrelli, A. From Single- to Multi-Target Drugs in Cancer Therapy: When Aspecificity Becomes an Advantage. Curr. Med. Chem. 2008, 15, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Levitzki, A.; Mishani, E. Tyrphostins and Other Tyrosine Kinase Inhibitors. Annu. Rev. Biochem. 2006, 75, 93–109. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.-F.; et al. Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [Green Version]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: A novel multi-tyrosine kinase inhibitor against human malignancies. OncoTargets Ther. 2019, 12, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.; Hong, M.; Liu, G.; Wang, H.; Patel, V.; Biddinger, P.; Silva, J.; Cowell, J.; Hao, Z. Novel FGFR inhibitor ponatinib suppresses the growth of non-small cell lung cancer cells overexpressing FGFR1. Oncol. Rep. 2013, 29, 2181–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, K.R.; Hinz, T.K.; Kleczko, E.K.; Marek, L.A.; Kwak, J.; Harp, T.; Kim, J.; Tan, A.C.; Heasley, L.E. Kinome RNAi Screens Reveal Synergistic Targeting of MTOR and FGFR1 Pathways for Treatment of Lung Cancer and HNSCC. Cancer Res. 2015, 75, 4398–4406. [Google Scholar] [CrossRef] [Green Version]
- Wynes, M.W.; Hinz, T.K.; Gao, D.; Martini, M.; Marek, L.A.; Ware, K.E.; Edwards, M.G.; Böhm, D.; Perner, S.; Helfrich, B.A.; et al. FGFR1 mRNA and Protein Expression, not Gene Copy Number, Predict FGFR TKI Sensitivity across All Lung Cancer Histologies. Clin. Cancer Res. 2014, 20, 3299–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.E.; Williams, L.T. Structural and Functional Diversity in the FGf Receptor Multigene Family. Adv. Cancer Res. 1992, 60, 1–41. [Google Scholar] [CrossRef]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Senthilkumar, G.; Fisher, M.M.; Skiba, J.H.; Miller, M.C.; Brennan, S.R.; Kaushik, S.; Bradley, S.T.; Longhurst, C.A.; Buehler, D.; Nickel, K.P.; et al. FGFR Inhibition Enhances Sensitivity to Radiation in Non–Small Cell Lung Cancer. Mol. Cancer Ther. 2020, 19, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Quintanal-Villalonga, Á.; Ferrer, I.; Guruceaga, E.; Cirauqui, C.; Marrugal, Á.; Ojeda, L.; García, S.; Zugazagoitia, J.; Muñoz-Galván, S.; Lopez-Rios, F.; et al. FGFR1 and FGFR4 oncogenicity depends on n-cadherin and their co-expression may predict FGFR-targeted therapy efficacy. EBioMedicine 2020, 53. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, L.; Su, X.; Li, M.; Xie, L.; Malchers, F.; Fan, S.; Yin, X.; Xu, Y.; Liu, K.; et al. Translating the Therapeutic Potential of AZD4547 in FGFR1-Amplified Non–Small Cell Lung Cancer through the Use of Patient-Derived Tumor Xenograft Models. Clin. Cancer Res. 2012, 18, 6658–6667. [Google Scholar] [CrossRef] [Green Version]
- Markham, A. Erdafitinib: First Global Approval. Drugs 2019, 79, 1017–1021. [Google Scholar] [CrossRef]
- Perera, T.P.; Jovcheva, E.; Mevellec, L.; Vialard, J.; De Lange, D.; Verhulst, T.; Paulussen, C.; Van De Ven, K.; King, P.; Freyne, E.; et al. Discovery and Pharmacological Characterization of JNJ-42756493 (Erdafitinib), a Functionally Selective Small-Molecule FGFR Family Inhibitor. Mol. Cancer Ther. 2017, 16, 1010–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palakurthi, S.; Kuraguchi, M.; Zacharek, S.J.; Zudaire, E.; Huang, W.; Bonal, D.M.; Liu, J.; Dhaneshwar, A.; Depeaux, K.; Gowaski, M.R.; et al. The Combined Effect of FGFR Inhibition and PD-1 Blockade Promotes Tumor-Intrinsic Induction of Antitumor Immunity. Cancer Immunol. Res. 2019, 7, 1457–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grünewald, S.; Politz, O.; Bender, S.; Héroult, M.; Lustig, K.; Thuss, U.; Kneip, C.; Kopitz, C.; Zopf, D.; Collin, M.; et al. Rogaratinib: A potent and selective pan-FGFR inhibitor with broad antitumor activity in FGFR-overexpressing preclinical cancer models. Int. J. Cancer 2019, 145, 1346–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitowska, K.; Gorska-Arcisz, M.; Antoun, D.; Zarczynska, I.; Czaplinska, D.; Szczepaniak, A.; Skladanowski, A.C.; Wieczorek, M.; Stanczak, A.; Skupinska, M.; et al. MET-Pyk2 Axis Mediates Acquired Resistance to FGFR Inhibition in Cancer Cells. Front. Oncol. 2021, 11, 1. [Google Scholar] [CrossRef]
- Chell, V.; Balmanno, K.; Little, A.S.; Wilson, M.L.; Andrews, S.; Blockley, L.Y.; Hampson, M.; Gavine, P.R.; Cook, S.J. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene 2013, 32, 3059–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byron, S.A.; Chen, H.; Wortmann, A.; Loch, D.; Gartside, M.G.; Dehkhoda, F.; Blais, S.P.; Neubert, T.A.; Mohammadi, M.; Pollock, P.M. The N550K/H Mutations in FGFR2 Confer Differential Resistance to PD173074, Dovitinib, and Ponatinib ATP-Competitive Inhibitors. Neoplasia 2013, 15, 975–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohl, C.D.; Ryan, M.R.; Luo, B.; Frey, K.M.; Anderson, K.S. Illuminating the Molecular Mechanisms of Tyrosine Kinase Inhibitor Resistance for the FGFR1 Gatekeeper Mutation: The Achilles’ Heel of Targeted Therapy. ACS Chem. Biol. 2015, 10, 1319–1329. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.R.; Sohl, C.D.; Luo, B.; Anderson, K.S. The FGFR1 V561M Gatekeeper Mutation Drives AZD4547 Resistance through STAT3 Activation and EMT. Mol. Cancer Res. 2019, 17, 532–543. [Google Scholar] [CrossRef] [Green Version]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion–Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.G., 6th; Cheuk, A.T.; Tsang, P.S.; Chung, J.-Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.-R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 119, 3395–3407. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.; Chen, Q.; Guo, Y.; Zhang, T.; Guo, W. Insight into resistance mechanisms of AZD4547 and E3810 to FGFR1 gatekeeper mutation via theoretical study. Drug Des. Dev. Ther. 2017, 11, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Harbinski, F.; Craig, V.J.; Sanghavi, S.; Jeffery, D.; Liu, L.; Sheppard, K.A.; Wagner, S.; Stamm, C.; Buness, A.; Chatenay-Rivauday, C.; et al. Rescue Screens with Secreted Proteins Reveal Compensatory Potential of Receptor Tyrosine Kinases in Driving Cancer Growth. Cancer Discov. 2012, 2, 948–959. [Google Scholar] [CrossRef] [Green Version]
- Jänne, P.A.; Gray, N.; Settleman, J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat. Rev. Drug Discov. 2009, 8, 709–723. [Google Scholar] [CrossRef]
- Chandarlapaty, S. Negative Feedback and Adaptive Resistance to the Targeted Therapy of Cancer. Cancer Discov. 2012, 2, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Datta, J.; Damodaran, S.; Parks, H.; Ocrainiciuc, C.; Miya, J.; Yu, L.; Gardner, E.P.; Samorodnitsky, E.; Wing, M.R.; Bhatt, D.; et al. Akt Activation Mediates Acquired Resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol. Cancer Ther. 2017, 16, 614–624. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ai, J.; Liu, H.; Peng, X.; Chen, H.; Chen, Y.; Su, Y.; Shen, A.; Huang, X.; Ding, J.; et al. The Secretome Engages STAT3 to Favor a Cytokine-rich Microenvironment in Mediating Acquired Resistance to FGFR Inhibitors. Mol. Cancer Ther. 2019, 18, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotani, H.; Ebi, H.; Kitai, H.; Nanjo, S.; Kita, K.; Huynh, T.G.; Ooi, A.; Faber, A.C.; Minokenudson, M.; Yano, S. Co-active receptor tyrosine kinases mitigate the effect of FGFR inhibitors in FGFR1-amplified lung cancers with low FGFR1 protein expression. Oncogene 2016, 35, 3587–3597. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Watanabe, K.; Kita, K.; Kitai, H.; Kotani, H.; Sato, Y.; Inase, N.; Yano, S.; Ebi, H. Resistance mediated by alternative receptor tyrosine kinases in FGFR1-amplified lung cancer. Carcinogenesis 2017, 38, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Malchers, F.; Ercanoglu, M.; Schütte, D.; Castiglione, R.; Tischler, V.; Michels, S.; Dahmen, I.; Brägelmann, J.; Menon, R.; Heuckmann, J.M.; et al. Mechanisms of Primary Drug Resistance in FGFR1-Amplified Lung Cancer. Clin. Cancer Res. 2017, 23, 5527–5536. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-M.; Kim, H.R.; Yun, M.R.; Kang, H.N.; Pyo, K.-H.; Park, H.J.; Lee, J.M.; Choi, H.M.; Ellinghaus, P.; Ocker, M.; et al. Activation of the Met kinase confers acquired drug resistance in FGFR-targeted lung cancer therapy. Oncogene 2016, 5, e241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockorny, B.; Rusan, M.; Chen, W.; Liao, R.G.; Li, Y.; Piccioni, F.; Wang, J.; Tan, L.; Thorner, A.R.; Li, T.; et al. RAS–MAPK Reactivation Facilitates Acquired Resistance in FGFR1-Amplified Lung Cancer and Underlies a Rationale for Upfront FGFR–MEK Blockade. Mol. Cancer Ther. 2018, 17, 1526–1539. [Google Scholar] [CrossRef] [Green Version]
- Bemanian, V.; Sauer, T.; Touma, J.; Lindstedt, B.A.; Chen, Y.; Ødegård, H.P.; Vetvik, K.M.; Bukholm, I.R.; Geißler, J. The Epidermal Growth Factor Receptor (EGFR / HER-1) Gatekeeper Mutation T790M Is Present in European Patients with Early Breast Cancer. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.-E.C.; Radhakrishna, V.K.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Tan, L.; Wang, J.; Tanizaki, J.; Huang, Z.; Aref, A.R.; Rusan, M.; Zhu, S.-J.; Zhang, Y.; Ercan, D.; Liao, R.G.; et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4869–E4877. [Google Scholar] [CrossRef] [Green Version]
- Fumarola, C.; Bozza, N.; Castelli, R.; Ferlenghi, F.; Marseglia, G.; Lodola, A.; Bonelli, M.; La Monica, S.; Cretella, D.; Alfieri, R.; et al. Expanding the Arsenal of FGFR Inhibitors: A Novel Chloroacetamide Derivative as a New Irreversible Agent with Anti-proliferative Activity Against FGFR1-Amplified Lung Cancer Cell Lines. Front. Oncol. 2019, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.S.; Tan, L.; Smith, A.; Gray, N.S.; Wendt, M.K. Covalent Targeting of Fibroblast Growth Factor Receptor Inhibits Metastatic Breast Cancer. Mol. Cancer Ther. 2016, 15, 2096–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiseo, M.; Gelsomino, F.; Alfieri, R.; Cavazzoni, A.; Bozzetti, C.; De Giorgi, A.M.; Petronini, P.G.; Ardizzoni, A. FGFR as potential target in the treatment of squamous non small cell lung cancer. Cancer Treat. Rev. 2015, 41, 527–539. [Google Scholar] [CrossRef]
- Qing, J.; Du, X.; Chen, Y.; Chan, P.; Li, H.; Wu, P.; Marsters, S.; Stawicki, S.; Tien, J.; Totpal, K.; et al. Antibody-based targeting of FGFR3 in bladder carcinoma and t(4;14)-positive multiple myeloma in mice. J. Clin. Investig. 2009, 119, 1216–1229. [Google Scholar] [CrossRef] [Green Version]
- Sommer, A.; Kopitz, C.; Schatz, C.A.; Nising, C.F.; Mahlert, C.; Lerchen, H.-G.; Stelte-Ludwig, B.; Hammer, S.; Greven, S.; Schuhmacher, J.; et al. Preclinical Efficacy of the Auristatin-Based Antibody–Drug Conjugate BAY 1187982 for the Treatment of FGFR2-Positive Solid Tumors. Cancer Res. 2016, 76, 6331–6339. [Google Scholar] [CrossRef] [Green Version]
- Sievers, E.L.; Senter, P.D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Chiodelli, P.; Giacomini, A.; Rusnati, M.; Ronca, R. Fibroblast growth factors (FGFs) in cancer: FGF traps as a new therapeutic approach. Pharmacol. Ther. 2017, 179, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Harding, T.C.; Long, L.; Palencia, S.; Zhang, H.; Sadra, A.; Hestir, K.; Patil, N.; Levin, A.; Hsu, A.W.; Charych, D.; et al. Blockade of Nonhormonal Fibroblast Growth Factors by FP-1039 Inhibits Growth of Multiple Types of Cancer. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017, 14, 57–66. [Google Scholar] [CrossRef]
- Englinger, B.; Lötsch, D.; Pirker, C.; Mohr, T.; Van Schoonhoven, S.; Boidol, B.; Lardeau, C.-H.; Spitzwieser, M.; Szabó, P.; Heffeter, P.; et al. Acquired nintedanib resistance in FGFR1-driven small cell lung cancer: Role of endothelin-A receptor-activated ABCB1 expression. Oncotarget 2016, 7, 50161–50179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, V.D.; Blakely, C.M.; Lin, L.; Asthana, S.; Matni, N.; Olivas, V.; Pazarentzos, E.; Gubens, M.A.; Bastian, B.C.; Taylor, B.S.; et al. Novel computational method for predicting polytherapy switching strategies to overcome tumor heterogeneity and evolution. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Hemann, M.T.; Lauffenburger, D.A. Modeling Tumor Clonal Evolution for Drug Combinations Design. Trends Cancer 2016, 2, 144–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Sedlak, J.C.; Srinivas, R.; Creixell, P.; Pritchard, J.R.; Tidor, B.; Lauffenburger, D.A.; Hemann, M.T. Exploiting Temporal Collateral Sensitivity in Tumor Clonal Evolution. Cell 2016, 165, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Acar, A.; Nichol, D.; Fernandez-Mateos, J.; Cresswell, G.D.; Barozzi, I.; Hong, S.P.; Trahearn, N.; Spiteri, I.; Stubbs, M.; Burke, R.; et al. Exploiting evolutionary steering to induce collateral drug sensitivity in cancer. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Malchers, F.; Dietlein, F.; Schöttle, J.; Lu, X.; Nogova, L.; Albus, K.; Fernandez-Cuesta, L.; Heuckmann, J.M.; Gautschi, O.; Diebold, J.; et al. Cell-Autonomous and Non–Cell-Autonomous Mechanisms of Transformation by Amplified FGFR1 in Lung Cancer. Cancer Discov. 2014, 4, 246–257. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Rivard, C.J.; Yu, H.; Genova, C.; Rozenboom, L.; Gao, D.; Hinz, T.K.; Rikke, B.A.; Wynes, M.W.; Caldwell, C.; et al. A miRNA Panel Predicts Sensitivity of FGFR Inhibitor in Lung Cancer Cell Lines. Clin. Lung Cancer 2018, 19, 450–456. [Google Scholar] [CrossRef]
- Lau, D.K.; Jenkins, L.; Weickhardt, A. Mechanisms of acquired resistance to fibroblast growth factor receptor targeted therapy. Cancer Drug Resist. 2019, 2, 568–579. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Gene | Alteration | Histology | Incidence (%) | Ref. |
|---|---|---|---|---|
| FGFR1 | Amplification | NSCLC SqCC | 6–22 | [15,19,20,21,22,23,24] |
| FGFR1 | Amplification | SCLC | 7 | [25,26,27] |
| FGFR2 | Somatic mutations; W290C, S320C, K660E/N | NSCLC SqCC | 3 | [15,28] |
| FGFR3 | Somatic mutations; R248C, S249C | NSCLC SqCC | 3 | [15,28] |
| FGFR4 | Somatic mutations; G2041A | NSCLC adenocarcinoma | 2 | [29] |
| FGFR3 | Translocations; FGFR3-TACC3 | NSCLC, prevalently SqCC | 0.1–1.1% | [30,31] |
| FGFR2 | Translocations; FGFR2-SHTN1, FGFR2-CIT | NSCLC adenocarcinoma | rare | [30,32] |
| FGFR1 | Translocations BAG4-FGFR1 | NSCLC SqCC | rare | [30,31] |
| Inhibitor (Manufacturer) | Target | Clinical Trial Identifier | Patient Characteristics | Regimen | Phase Study | Status/Ref. |
|---|---|---|---|---|---|---|
| Nonselective inhibitors | ||||||
| Ponatinib (ARIAD, Pharmaceuticals) | FGFR, PDGFR, VEGFR, ABL, SRC, KIT | NCT01761747 | Advanced NSCLC; FGFR1 alterations | Ponatinib monotherapy | II | Terminated [85] |
| NCT01935336 | Advanced lung cancer, all histologies; FGFR SISH/ISH 1 | Ponatinib monotherapy | II | Active, not recruiting [85] | ||
| Dovitinib (Allarity Therapeutics) | FGFR1-3, VEGFR1-3, PDGFRβ, FLT3, KIT, RET, TRKA, CSF1 | NCT01861197 | Advanced SqCC; FGFR1 amplification | Dovitinib monotherapy | II | Unknown [86] |
| Pazopanib (Novartis | FGFR1-3, VEGFR1-3, PDGFR, KIT | Case report study | Advanced SCLC; FGFR1 amplification | Pazopanib monotherapy | [35] | |
| Nintedanib (Boehringer-ingelheim) | FGFR1-4, VEGFR1-3, PDGFRα-β | NCT01948141 | Advanced SqCC; FGFR1 amplification | Nintedanib monotherapy | II | Completed |
| Lucitanib (HaiHe Biopharma) | FGFR1, VEGFR1-3 | NCT01283945 | Advanced NSCLC; FGFR1 amplification | E3810 monotherapy | I/II | Completed [87] |
| Selective inhibitors | ||||||
| AZD4547 (AstraZeneca) | FGFR1-3 | NCT00979134 | Advanced SqCC; FGFR1 amplification | AZD4547 monotherapy | I | Terminated [88] |
| NCT02965378 | Advanced SqCC; FGFR alterations | AZD4547, docetaxel | II/III | Active, not recruiting [89] | ||
| NCT01824901 | Advanced SqCC; FGFR1 amplification | Docetaxel with or without AZD4547 | I/II | Completed [90] | ||
| NCT01795768 | Advanced SqCC; FGFR1-or FGFR2-amplified tumours | AZD4547 monotherapy | II | Unknown [91] | ||
| NCT02154490 | Advanced SqCC; FGFR1-3 positive tumours | AZD4547, docetaxel | II/III | Active, not recruiting | ||
| Infigratinib (QED Therapeutics) | FGFR1-3 | NCT01004224 | Advanced SqCC; FGFR1 amplification | Infigratinib monotherapy | I/II | Completed [92] |
| Erdafitinib (Janssen Pharmaceuticals) | FGFR1-4 | NCT03827850 | Advanced NSCLC; FGFR alterations | Erdafitinib monotherapy | II | Recruiting |
| NCT04083976 | Advanced NSCLC; FGFR alterations | Erdafitinib monotherapy | II | Recruiting | ||
| Rogaratinib (Bayer) | FGFR1-4 | NCT01976741 | Advanced NSCLC; FGFR alterations | Rogaratinib monotherapy | I | Completed [93] |
| NCT03762122 | Advanced SqCC; FGFR mRNA overexpression | Rogaratinib monotherapy | II | Active, not recruiting | ||
| CPL304110 (Celon Pharma) | FGFR1-3 | NCT04149691 | Advanced SqCC; FGFR1-3 alterations | CPL304110 monotherapy | I | Recruiting [94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacini, L.; Jenks, A.D.; Lima, N.C.; Huang, P.H. Targeting the Fibroblast Growth Factor Receptor (FGFR) Family in Lung Cancer. Cells 2021, 10, 1154. https://doi.org/10.3390/cells10051154
Pacini L, Jenks AD, Lima NC, Huang PH. Targeting the Fibroblast Growth Factor Receptor (FGFR) Family in Lung Cancer. Cells. 2021; 10(5):1154. https://doi.org/10.3390/cells10051154
Chicago/Turabian StylePacini, Laura, Andrew D. Jenks, Nadia Carvalho Lima, and Paul H. Huang. 2021. "Targeting the Fibroblast Growth Factor Receptor (FGFR) Family in Lung Cancer" Cells 10, no. 5: 1154. https://doi.org/10.3390/cells10051154