
PD-1 and LAG-3 Checkpoint Blockade: Potential Avenues for Therapy in B-Cell Lymphoma


Abstract
:1. Introduction
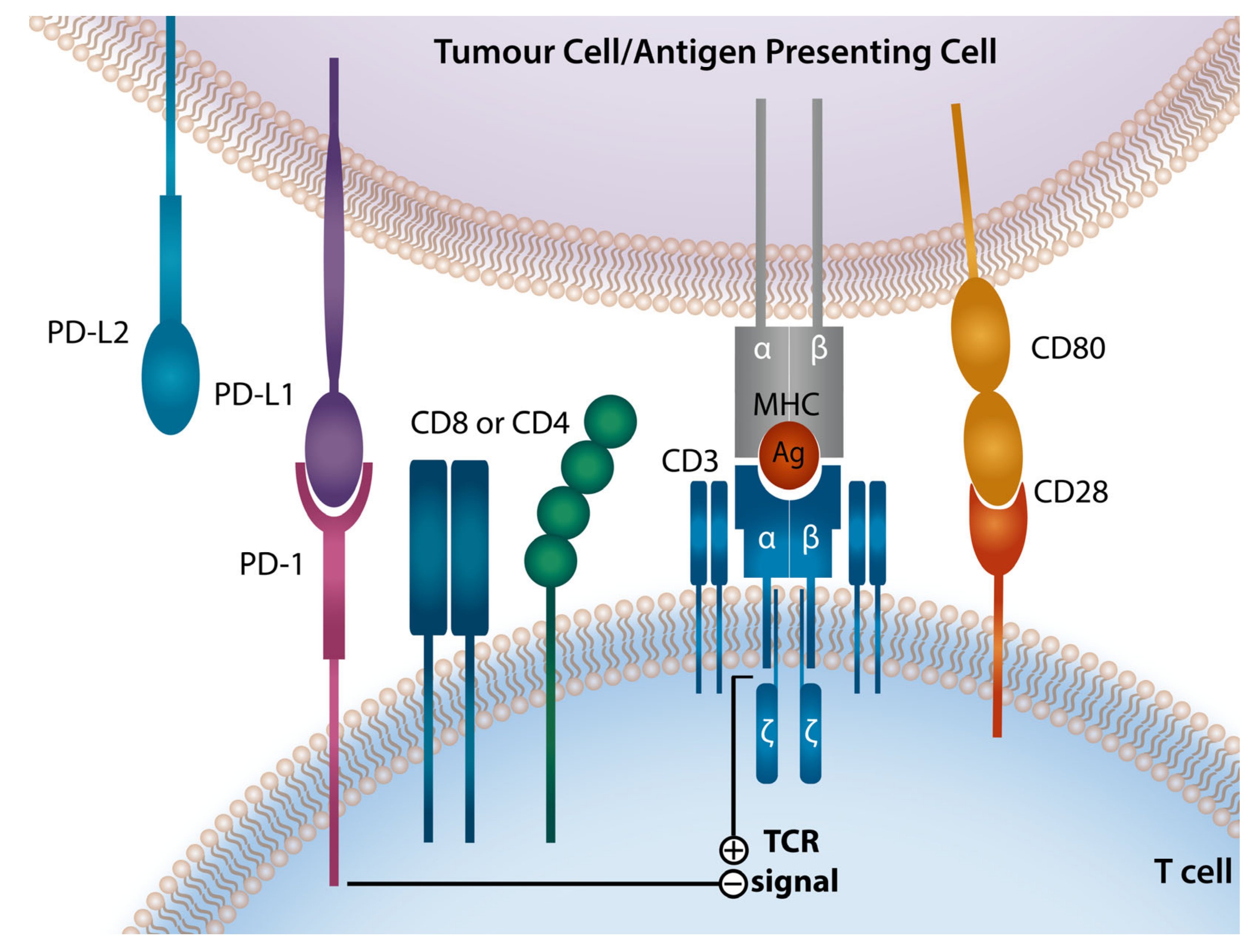
2. Programmed Death-1 Checkpoint Axis
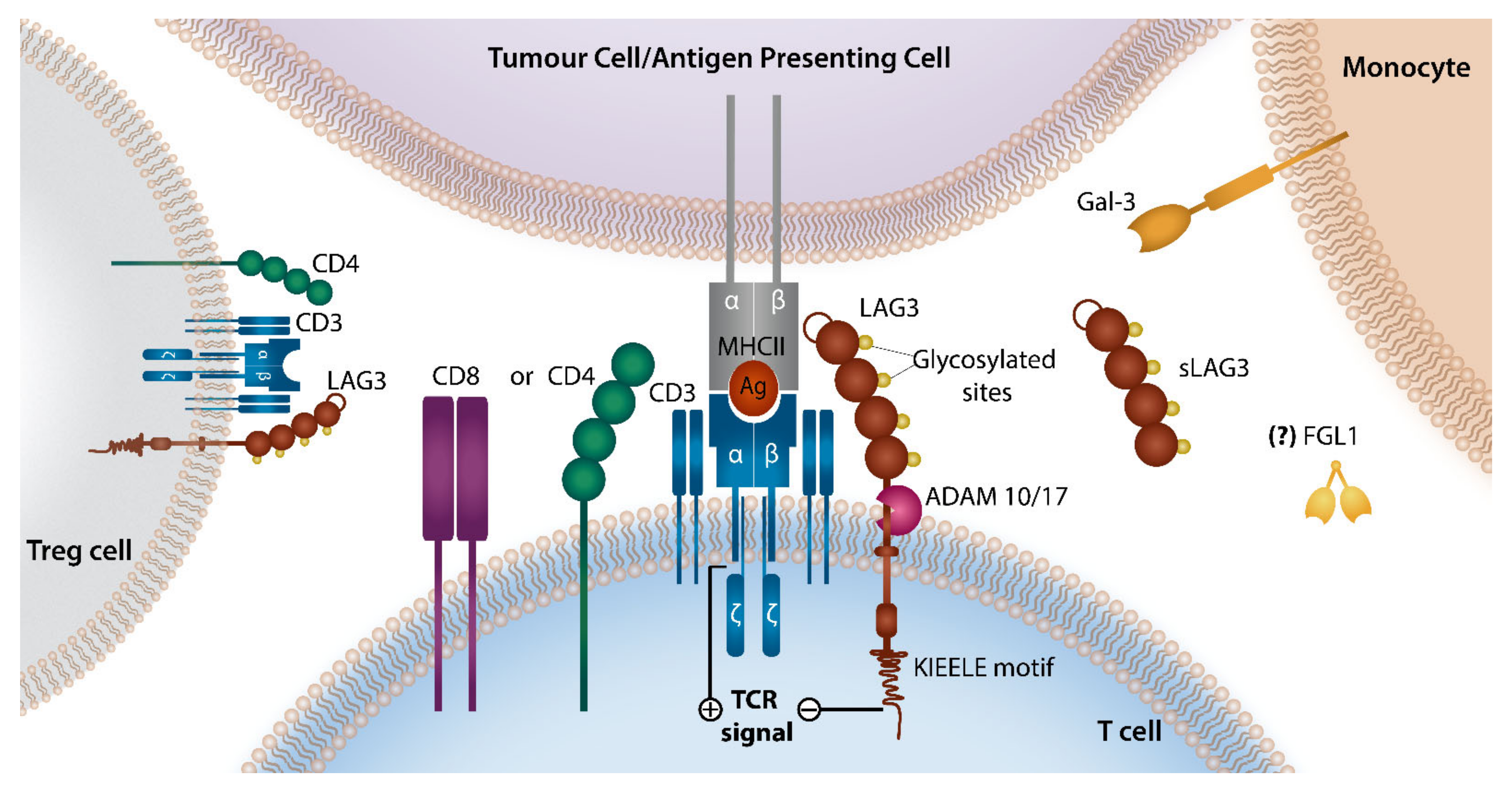
3. LAG-3 Checkpoint Axis
4. Immune Checkpoint Molecules in Classical Hodgkin Lymphoma
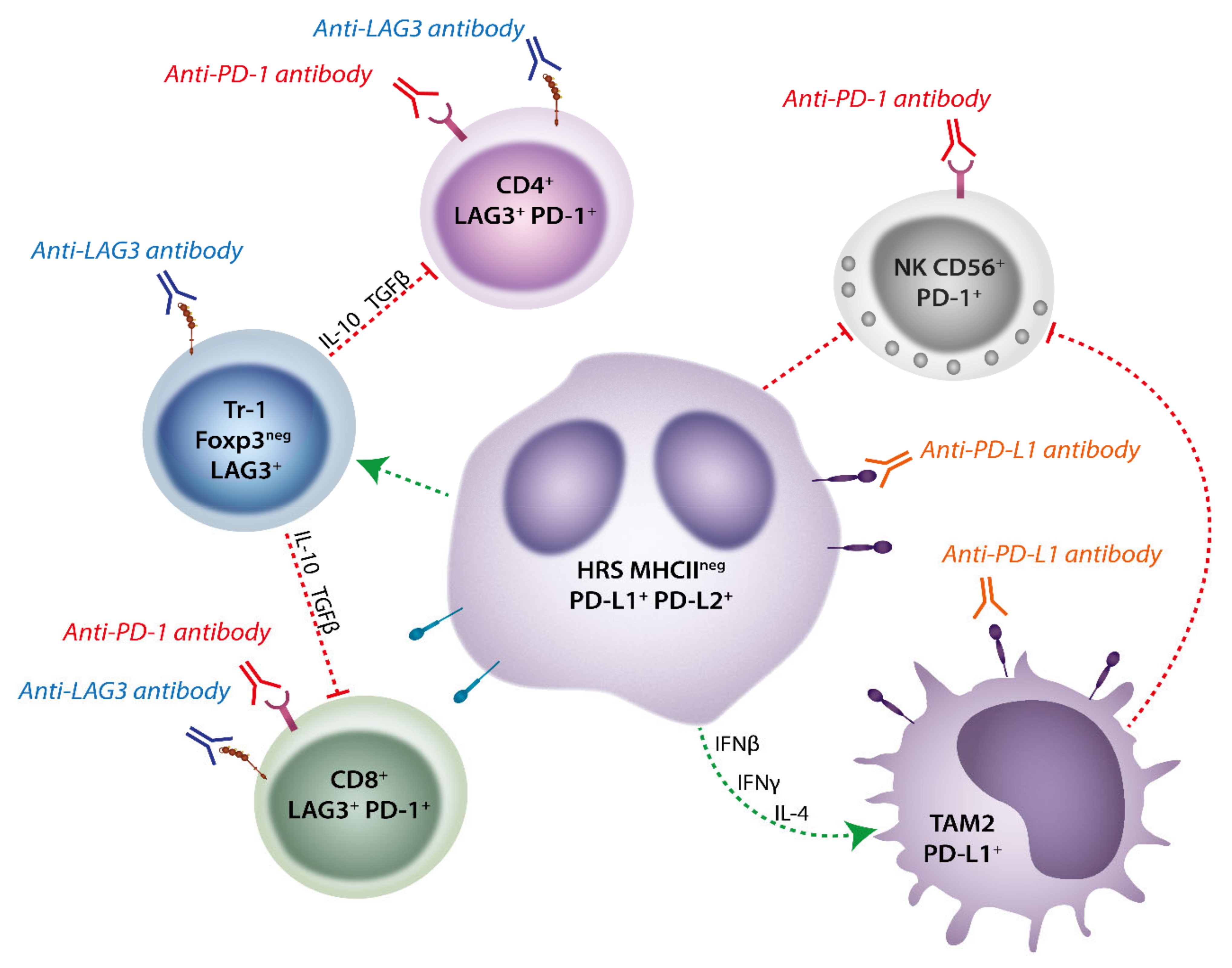
4.1. Role of PD-1 Axis in Classical Hodgkin Lymphoma
4.2. Role of LAG-3 in Classical Hodgkin Lymphoma
4.3. Immune Checkpoint Blockade Therapy in Classical Hodgkin Lymphoma
5. Checkpoint Molecules in Non-Hodgkin Lymphoma
5.1. Immune Checkpoint Molecules in Primary Mediastinal B-Cell Lymphoma
5.2. Immune Checkpoint Molecules in Primary Central Nervous System and Testicular Lymphoma
6. Diffuse Large B-Cell Lymphoma
6.1. Role of PD-1 Axis in Diffuse Large B Cell Lymphoma
6.2. Role of LAG-3 in Diffuse Large B Cell Lymphoma
6.3. Immune Checkpoint Molecules in Follicular Lymphoma
7. Immune Checkpoint Blockade Therapy in Non-Hodgkin Lymphoma
8. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Simon, S.; Labarriere, N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology 2017, 7, e1364828. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.; Blackburn, S.D.; Blattman, J.N.; Wherry, E.J. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J. Exp. Med. 2007, 204, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Olive, D.; Kuchroo, V.; Zarour, H.M. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012, 72, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 pathway in the immune response. Am. J. Transplant. 2012, 12, 2575–2587. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Ni, Z.; Liu, X.; Feng, S.; Dong, X.; Shi, X.; Zhai, J.; Mai, S.; Jiang, J.; Wang, Z.; et al. Beyond T Cells: Understanding the Role of PD-1/PD-L1 in Tumor-Associated Macrophages. J. Immunol. Res. 2019, 2019, 1919082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Jeffrey Medeiros, L.; Young, K.H. Signaling pathway and dysregulation of PD1 and its ligands in lymphoid malignancies. Biochim. Biophys. Acta 2016, 1865, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Xu-Monette, Z.Y.; Zhou, J.; Young, K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 2018, 131, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Kinter, A.L.; Godbout, E.J.; McNally, J.P.; Sereti, I.; Roby, G.A.; O’Shea, M.A.; Fauci, A.S. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J. Immunol. 2008, 181, 6738–6746. [Google Scholar] [CrossRef]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [Green Version]
- Thibult, M.L.; Mamessier, E.; Gertner-Dardenne, J.; Pastor, S.; Just-Landi, S.; Xerri, L.; Chetaille, B.; Olive, D. PD-1 is a novel regulator of human B-cell activation. Int. Immunol. 2013, 25, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Kim, J.; Myers, A.C.; Chen, L.; Pardoll, D.M.; Truong-Tran, Q.A.; Lane, A.P.; McDyer, J.F.; Fortuno, L.; Schleimer, R.P. Constitutive and inducible expression of b7 family of ligands by human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2005, 33, 280–289. [Google Scholar] [CrossRef]
- Tseng, S.Y.; Otsuji, M.; Gorski, K.; Huang, X.; Slansky, J.E.; Pai, S.I.; Shalabi, A.; Shin, T.; Pardoll, D.M.; Tsuchiya, H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 2001, 193, 839–846. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Allen, P.B.; Gordon, L.I. PD-1 blockade in Hodgkin’s lymphoma: Learning new tricks from an old teacher. Expert Rev. Hematol. 2016, 9, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Shipp, M.A.; Ribrag, V.; Michot, J.M.; Zinzani, P.L.; Kuruvilla, J.; Snyder, E.S.; Ricart, A.D.; Balakumaran, A.; Rose, S.; et al. Programmed Death-1 Blockade With Pembrolizumab in Patients With Classical Hodgkin Lymphoma After Brentuximab Vedotin Failure. J. Clin. Oncol. 2016, 34, 3733–3739. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Young, K.H. Checkpoint inhibitors in hematological malignancies. J. Hematol. Oncol. 2017, 10, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gellrich, F.F.; Schmitz, M.; Beissert, S.; Meier, F. Anti-PD-1 and Novel Combinations in the Treatment of Melanoma-An Update. J. Clin. Med. 2020, 9, 223. [Google Scholar] [CrossRef] [Green Version]
- Moser, J.C.; Chen, D.; Hu-Lieskovan, S.; Grossmann, K.F.; Patel, S.; Colonna, S.V.; Ying, J.; Hyngstrom, J.R. Real-world survival of patients with advanced BRAF V600 mutated melanoma treated with front-line BRAF/MEK inhibitors, anti-PD-1 antibodies, or nivolumab/ipilimumab. Cancer Med. 2019, 8, 7637–7643. [Google Scholar] [CrossRef] [Green Version]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef] [Green Version]
- Huard, B.; Mastrangeli, R.; Prigent, P.; Bruniquel, D.; Donini, S.; El-Tayar, N.; Maigret, B.; Dréano, M.; Triebel, F. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5744–5749. [Google Scholar] [CrossRef] [Green Version]
- Lino, A.C.; Lampropoulou, V.; Welle, A.; Joedicke, J.; Pohar, J.; Simon, Q.; Thalmensi, J.; Baures, A.; Flühler, V.; Sakwa, I. LAG-3 inhibitory receptor expression identifies immunosuppressive natural regulatory plasma cells. Immunity 2018, 49, 120–133.e129. [Google Scholar] [CrossRef]
- Gagliani, N.; Magnani, C.F.; Huber, S.; Gianolini, M.E.; Pala, M.; Licona-Limon, P.; Guo, B.; De’Broski, R.H.; Bulfone, A.; Trentini, F. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 2013, 19, 739. [Google Scholar] [CrossRef]
- Workman, C.J.; Vignali, D.A. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur. J. Immunol. 2003, 33, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Fromentin, R.; Bakeman, W.; Lawani, M.B.; Khoury, G.; Hartogensis, W.; DaFonseca, S.; Killian, M.; Epling, L.; Hoh, R.; Sinclair, E. CD4+ T cells expressing PD-1, TIGIT and LAG-3 contribute to HIV persistence during ART. PLoS Pathog. 2016, 12, e1005761. [Google Scholar] [CrossRef]
- Hurst, J.; Hoffmann, M.; Pace, M.; Williams, J.P.; Thornhill, J.; Hamlyn, E.; Meyerowitz, J.; Willberg, C.; Koelsch, K.K.; Robinson, N. Immunological biomarkers predict HIV-1 viral rebound after treatment interruption. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 2014, 74, 3418–3428. [Google Scholar] [CrossRef] [Green Version]
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol. Res. 2015, 3, 412–423. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 2019, 176, 334–347. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wang, Y.; Forbes, K.; Vignali, K.M.; Heale, B.S.; Saftig, P.; Hartmann, D.; Black, R.A.; Rossi, J.J.; Blobel, C.P.; et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. Embo J. 2007, 26, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG 3 (CD 223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brignone, C.; Gutierrez, M.; Mefti, F.; Brain, E.; Jarcau, R.; Cvitkovic, F.; Bousetta, N.; Medioni, J.; Gligorov, J.; Grygar, C. First-line chemoimmunotherapy in metastatic breast carcinoma: Combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J. Transl. Med. 2010, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Romano, E.; Michielin, O.; Voelter, V.; Laurent, J.; Bichat, H.; Stravodimou, A.; Romero, P.; Speiser, D.E.; Triebel, F.; Leyvraz, S. MART-1 peptide vaccination plus IMP321 (LAG-3Ig fusion protein) in patients receiving autologous PBMCs after lymphodepletion: Results of a Phase I trial. J. Transl. Med. 2014, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Melero, I.; Bhatia, S.; Bono, P.; Sanborn, R.E.; Lipson, E.J.; Callahan, M.K.; Gajewski, T.; Gomez-Roca, C.A.; Hodi, F.S. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J. Clin. Oncol. 2017, 35, 9520. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Kraman, M.; Faroudi, M.; Allen, N.L.; Kmiecik, K.; Gliddon, D.; Seal, C.; Koers, A.; Wydro, M.M.; Batey, S.; Winnewisser, J.; et al. FS118, a Bispecific Antibody Targeting LAG-3 and PD-L1, Enhances T-Cell Activation Resulting in Potent Antitumor Activity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3333–3344. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [Green Version]
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 942–950. [Google Scholar] [CrossRef]
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, R.; Stanelle, J.; Hansmann, M.L.; Küppers, R. Pathogenesis of classical and lymphocyte-predominant Hodgkin lymphoma. Annu. Rev. Pathol. 2009, 4, 151–174. [Google Scholar] [CrossRef]
- Fang, W.; Zhang, J.; Hong, S.; Zhan, J.; Chen, N.; Qin, T.; Tang, Y.; Zhang, Y.; Kang, S.; Zhou, T.; et al. EBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget 2014, 5, 12189–12202. [Google Scholar] [CrossRef] [PubMed]
- Panjwani, P.K.; Charu, V.; DeLisser, M.; Molina-Kirsch, H.; Natkunam, Y.; Zhao, S. Programmed death-1 ligands PD-L1 and PD-L2 show distinctive and restricted patterns of expression in lymphoma subtypes. Hum. Pathol. 2018, 71, 91–99. [Google Scholar] [CrossRef]
- De Goycoechea, D.; Stalder, G.; Martins, F.; Duchosal, M.A. Immune Checkpoint Inhibition in Classical Hodgkin Lymphoma: From Early Achievements towards New Perspectives. J. Oncol. 2019, 2019, 9513701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, C.D.; Gusenleitner, D.; Lipschitz, M.; Roemer, M.G.M.; Stack, E.C.; Gjini, E.; Hu, X.; Redd, R.; Freeman, G.J.; Neuberg, D.; et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood 2017, 130, 2420–2430. [Google Scholar] [CrossRef]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Zaki, M.A.; Wada, N.; Ikeda, J.; Shibayama, H.; Hashimoto, K.; Yamagami, T.; Tatsumi, Y.; Tsukaguchi, M.; Take, H.; Tsudo, M.; et al. Prognostic implication of types of tumor-associated macrophages in Hodgkin lymphoma. Virchows Arch. Int. J. Pathol. 2011, 459, 361–366. [Google Scholar] [CrossRef]
- Tan, K.L.; Scott, D.W.; Hong, F.; Kahl, B.S.; Fisher, R.I.; Bartlett, N.L.; Advani, R.H.; Buckstein, R.; Rimsza, L.M.; Connors, J.M.; et al. Tumor-associated macrophages predict inferior outcomes in classic Hodgkin lymphoma: A correlative study from the E2496 Intergroup trial. Blood 2012, 120, 3280–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Espiridión, B.; Martin-Moreno, A.M.; Montalbán, C.; Medeiros, L.J.; Vega, F.; Younes, A.; Piris, M.A.; Garcia, J.F. Immunohistochemical markers for tumor associated macrophages and survival in advanced classical Hodgkin’s lymphoma. Haematologica 2012, 97, 1080–1084. [Google Scholar] [CrossRef] [Green Version]
- Jakovic, L.R.; Mihaljevic, B.S.; Perunicic Jovanovic, M.D.; Bogdanovic, A.D.; Andjelic, B.M.; Bumbasirevic, V.Z. The prognostic relevance of tumor associated macrophages in advanced stage classical Hodgkin lymphoma. Leuk. Lymphoma 2011, 52, 1913–1919. [Google Scholar] [CrossRef] [PubMed]
- Kamper, P.; Bendix, K.; Hamilton-Dutoit, S.; Honoré, B.; Nyengaard, J.R.; d’Amore, F. Tumor-infiltrating macrophages correlate with adverse prognosis and Epstein-Barr virus status in classical Hodgkin’s lymphoma. Haematologica 2011, 96, 269–276. [Google Scholar] [CrossRef]
- Casulo, C.; Arcila, M.; Bohn, O.L.; Teruya-Feldstein, J.; Maragulia, J.; Moskowitz, C.H. Tumor associated macrophages in relapsed and refractory Hodgkin lymphoma. Leuk. Res. 2013, 37, 1178–1183. [Google Scholar] [CrossRef]
- Greaves, P.; Clear, A.; Coutinho, R.; Wilson, A.; Matthews, J.; Owen, A.; Shanyinde, M.; Lister, T.A.; Calaminici, M.; Gribben, J.G. Expression of FOXP3, CD68, and CD20 at diagnosis in the microenvironment of classical Hodgkin lymphoma is predictive of outcome. J. Clin. Oncol. 2013, 31, 256–262. [Google Scholar] [CrossRef] [Green Version]
- Touati, M.; Delage-Corre, M.; Monteil, J.; Abraham, J.; Moreau, S.; Remenieras, L.; Gourin, M.P.; Dmytruk, N.; Olivrie, A.; Turlure, P.; et al. CD68-positive tumor-associated macrophages predict unfavorable treatment outcomes in classical Hodgkin lymphoma in correlation with interim fluorodeoxyglucose-positron emission tomography assessment. Leuk. Lymphoma 2015, 56, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Medeiros, L.J.; Li, S.; Yin, C.C.; Khoury, J.D.; Xu, J. PD-1/PD-L1 Pathway and Its Blockade in Patients with Classic Hodgkin Lymphoma and Non-Hodgkin Large-Cell Lymphomas. Curr. Hematol. Malig. Rep. 2020, 15, 372–381. [Google Scholar] [CrossRef]
- Arlt, A.; von Bonin, F.; Rehberg, T.; Perez-Rubio, P.; Engelmann, J.C.; Limm, K.; Reinke, S.; Dullin, C.; Sun, X.; Specht, R.; et al. High CD206 levels in Hodgkin lymphoma-educated macrophages are linked to matrix-remodeling and lymphoma dissemination. Mol. Oncol. 2020, 14, 571–589. [Google Scholar] [CrossRef] [Green Version]
- Merryman, R.W.; Armand, P.; Wright, K.T.; Rodig, S.J. Checkpoint blockade in Hodgkin and non-Hodgkin lymphoma. Blood Adv. 2017, 1, 2643–2654. [Google Scholar] [CrossRef] [Green Version]
- Meti, N.; Esfahani, K.; Johnson, N.A. The Role of Immune Checkpoint Inhibitors in Classical Hodgkin Lymphoma. Cancers 2018, 10, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Halabi, L.; Adam, J.; Gravelle, P.; Marty, V.; Danu, A.; Lazarovici, J.; Ribrag, V.; Bosq, J.; Camara-Clayette, V.; Laurent, C. Expression of the Immune Checkpoint Regulators LAG-3 and TIM-3 in Classical Hodgkin Lymphoma. Clin. Lymphoma Myeloma Leuk. 2020. [Google Scholar] [CrossRef]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T. Single-cell transcriptome analysis reveals disease-defining T-cell subsets in the tumor microenvironment of classic Hodgkin lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Nagasaki, J.; Togashi, Y.; Sugawara, T.; Itami, M.; Yamauchi, N.; Yuda, J.; Sugano, M.; Ohara, Y.; Minami, Y.; Nakamae, H. The critical role of CD4+ T cells in PD-1 blockade against MHC-II–expressing tumors such as classic Hodgkin lymphoma. Blood Adv. 2020, 4, 4069–4082. [Google Scholar] [CrossRef]
- Keane, C.; Law, S.C.; Gould, C.; Francis, S.; Abro, E.U.; Tobin, J.W.D.; Birch, S.; Gifford, G.; Gabreilli, S.; Stevenson, W.S.; et al. Elevated LAG-3 Expression in the Tumor Microenvironement of Patients with DLBCL Is Associated with a Non-GCB Phenotype and Poor Prognosis. Blood 2018, 132, 1576. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Armand, P.; Engert, A.; Younes, A.; Fanale, M.; Santoro, A.; Zinzani, P.L.; Timmerman, J.M.; Collins, G.P.; Ramchandren, R.; Cohen, J.B.; et al. Nivolumab for Relapsed/Refractory Classic Hodgkin Lymphoma After Failure of Autologous Hematopoietic Cell Transplantation: Extended Follow-Up of the Multicohort Single-Arm Phase II CheckMate 205 Trial. J. Clin. Oncol. 2018, 36, 1428–1439. [Google Scholar] [CrossRef]
- Chen, R.; Zinzani, P.L.; Lee, H.J.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Pembrolizumab in relapsed or refractory Hodgkin lymphoma: 2-year follow-up of KEYNOTE-087. Blood 2019, 134, 1144–1153. [Google Scholar] [CrossRef] [Green Version]
- Kuruvilla, J.; Ramchandren, R.; Santoro, A.; Paszkiewicz-Kozik, E.; Gasiorowski, R.; Johnson, N.A.; Fogliatto, L.M.; Goncalves, I.; de Oliveira, J.S.R.; Buccheri, V.; et al. Pembrolizumab versus brentuximab vedotin in relapsed or refractory classical Hodgkin lymphoma (KEYNOTE-204): An interim analysis of a multicentre, randomised, open-label, phase 3 study. Lancet Oncol. 2021. [Google Scholar] [CrossRef]
- Younes, A.; Santoro, A.; Shipp, M.; Zinzani, P.L.; Timmerman, J.M.; Ansell, S.; Armand, P.; Fanale, M.; Ratanatharathorn, V.; Kuruvilla, J.; et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: A multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol. 2016, 17, 1283–1294. [Google Scholar] [CrossRef] [Green Version]
- Herrera, A.F.; Moskowitz, A.J.; Bartlett, N.L.; Vose, J.M.; Ramchandren, R.; Feldman, T.A.; LaCasce, A.S.; Ansell, S.M.; Moskowitz, C.H.; Fenton, K.; et al. Interim results of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2018, 131, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, A.J.; Advani, R.H.; Bartlett, N.L.; Vose, J.M.; Ramchandren, R.; Feldman, T.A.; LaCasce, A.S.; Christian, B.A.; Ansell, S.M.; Moskowitz, C.H.; et al. Brentuximab Vedotin and Nivolumab for Relapsed or Refractory Classic Hodgkin Lymphoma: Long-Term Follow-up Results from the Single-Arm Phase 1/2 Study. Blood 2019, 134, 238. [Google Scholar] [CrossRef]
- Diefenbach, C.S.; Hong, F.; Ambinder, R.F.; Cohen, J.B.; Robertson, M.J.; David, K.A.; Advani, R.H.; Fenske, T.S.; Barta, S.K.; Palmisiano, N.D.; et al. Ipilimumab, nivolumab, and brentuximab vedotin combination therapies in patients with relapsed or refractory Hodgkin lymphoma: Phase 1 results of an open-label, multicentre, phase 1/2 trial. Lancet Haematol. 2020, 7, e660–e670. [Google Scholar] [CrossRef]
- Kao, S.C.; Cheng, Y.Y.; Williams, M.; Kirschner, M.B.; Madore, J.; Lum, T.; Sarun, K.H.; Linton, A.; McCaughan, B.; Klebe, S.; et al. Tumor Suppressor microRNAs Contribute to the Regulation of PD-L1 Expression in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2017, 12, 1421–1433. [Google Scholar] [CrossRef] [Green Version]
- Zinzani, P.L.; Lee, H.J.; Armand, P.; Johnson, N.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; Tomita, A.; et al. Three-Year Follow-up of Keynote-087: Pembrolizumab Monotherapy in Relapsed/Refractory Classic Hodgkin Lymphoma. Blood 2019, 134, 240. [Google Scholar] [CrossRef]
- Armand, P.; Chen, Y.-B.; Redd, R.A.; Joyce, R.M.; Bsat, J.; Jeter, E.; Merryman, R.W.; Coleman, K.C.; Dahi, P.B.; Nieto, Y.; et al. PD-1 blockade with pembrolizumab for classical Hodgkin lymphoma after autologous stem cell transplantation. Blood 2019, 134, 22–29. [Google Scholar] [CrossRef]
- Armand, P.; Kuruvilla, J.; Michot, J.-M.; Ribrag, V.; Zinzani, P.L.; Zhu, Y.; Marinello, P.; Nahar, A.; Moskowitz, C.H. KEYNOTE-013 4-year follow-up of pembrolizumab in classical Hodgkin lymphoma after brentuximab vedotin failure. Blood Adv. 2020, 4, 2617–2622. [Google Scholar] [CrossRef]
- Ramchandren, R.; Domingo-Domènech, E.; Rueda, A.; Trněný, M.; Feldman, T.A.; Lee, H.J.; Provencio, M.; Sillaber, C.; Cohen, J.B.; Savage, K.J.; et al. Nivolumab for Newly Diagnosed Advanced-Stage Classic Hodgkin Lymphoma: Safety and Efficacy in the Phase II CheckMate 205 Study. J. Clin. Oncol. 2019, 37, 1997–2007. [Google Scholar] [CrossRef]
- Ansell, S.; Ramchandren, R.; Domingo-Domènech, E.; Rueda, A.; Trněný, M.; Feldman, T.; Lee, H.; Provencio, M.; Sillaber, C.; Cohen, J.; et al. NIVOLUMAB PLUS DOXORUBICIN, VINBLASTINE AND DACARBAZINE FOR NEWLY DIAGNOSED ADVANCED-STAGE CLASSICAL HODGKIN LYMPHOMA: CHECKMATE 205 COHORT D 2-YEAR FOLLOW-UP. Hematol. Oncol. 2019, 37, 146–147. [Google Scholar] [CrossRef] [Green Version]
- Bröckelmann, P.J.; Goergen, H.; Keller, U.; Meissner, J.; Ordemann, R.; Halbsguth, T.V.; Sasse, S.; Sökler, M.; Kerkhoff, A.; Mathas, S.; et al. Efficacy of Nivolumab and AVD in Early-Stage Unfavorable Classic Hodgkin Lymphoma: The Randomized Phase 2 German Hodgkin Study Group NIVAHL Trial. JAMA Oncol. 2020, 6, 872–880. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Paz-Ares, L.; Horn, L.; Borghaei, H.; Spigel, D.R.; Steins, M.; Ready, N.; Chow, L.Q.M.; Vokes, E.E.; Felip, E.; Holgado, E. Phase III, randomized trial (CheckMate 057) of nivolumab (NIVO) versus docetaxel (DOC) in advanced non-squamous cell (non-SQ) non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2015, 33, 109. [Google Scholar] [CrossRef]
- Roemer, M.G.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin Lymphoma with Reduced β2M/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol. Res. 2016, 4, 910–916. [Google Scholar] [CrossRef] [Green Version]
- Cader, F.Z.; Hu, X.; Goh, W.L.; Wienand, K.; Ouyang, J.; Mandato, E.; Redd, R.; Lawton, L.N.; Chen, P.H.; Weirather, J.L.; et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat. Med. 2020, 26, 1468–1479. [Google Scholar] [CrossRef]
- Wienand, K.; Chapuy, B.; Stewart, C.; Dunford, A.J.; Wu, D.; Kim, J.; Kamburov, A.; Wood, T.R.; Cader, F.Z.; Ducar, M.D.; et al. Genomic analyses of flow-sorted Hodgkin Reed-Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv 2019, 3, 4065–4080. [Google Scholar] [CrossRef] [Green Version]
- Cader, F.Z.; Schackmann, R.C.J.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; et al. Mass cytometry of Hodgkin lymphoma reveals a CD4+ regulatory T-cell–rich and exhausted T-effector microenvironment. Blood 2018, 132, 825–836. [Google Scholar] [CrossRef]
- Reinke, S.; Bröckelmann, P.J.; Iaccarino, I.; Garcia-Marquez, M.; Borchmann, S.; Jochims, F.; Kotrova, M.; Pal, K.; Brüggemann, M.; Hartmann, E.; et al. Tumor and microenvironment response but no cytotoxic T-cell activation in classic Hodgkin lymphoma treated with anti-PD1. Blood 2020, 136, 2851–2863. [Google Scholar] [CrossRef] [PubMed]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef]
- Broccoli, A.; Zinzani, P.L. The unique biology and treatment of primary mediastinal B-cell lymphoma. Best Pract. Res. Clin. Haematol. 2018, 31, 241–250. [Google Scholar] [CrossRef]
- Twa, D.D.; Chan, F.C.; Ben-Neriah, S.; Woolcock, B.W.; Mottok, A.; Tan, K.L.; Slack, G.W.; Gunawardana, J.; Lim, R.S.; McPherson, A.W.; et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood 2014, 123, 2062–2065. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Xu-Monette, Z.Y.; Xiao, L.; Strati, P.; Hagemeister, F.B.; He, Y.; Chen, H.; Li, Y.; Manyam, G.C.; Li, Y.; et al. Prognostic factors, therapeutic approaches, and distinct immunobiologic features in patients with primary mediastinal large B-cell lymphoma on long-term follow-up. Blood Cancer J. 2020, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Shah, S.P.; Woolcock, B.W.; Rui, L.; Kawahara, M.; Farinha, P.; Johnson, N.A.; Zhao, Y.; Telenius, A.; Neriah, S.B.; et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011, 471, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Roemer, M.G.; Chapuy, B.; Liao, X.; Sun, H.; Pinkus, G.S.; Shipp, M.A.; Freeman, G.J.; Rodig, S.J. Expression of programmed cell death 1 ligand 2 (PD-L2) is a distinguishing feature of primary mediastinal (thymic) large B-cell lymphoma and associated with PDCD1LG2 copy gain. Am. J. Surg. Pathol. 2014, 38, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Aoki, T.; Chong, L.C.; Milne, K.; Miyata-Takata, T.; Singh, K.; Goodyear, T.; Farinha, P.; Slack, G.W.; Sehn, L.H. Identification of LAG3+ T Cell Populations in the Tumor Microenvironment of Classical Hodgkin Lymphoma and B-Cell Non-Hodgkin Lymphoma. Blood 2020, 136. [Google Scholar] [CrossRef]
- Chapuy, B.; Roemer, M.G.; Stewart, C.; Tan, Y.; Abo, R.P.; Zhang, L.; Dunford, A.J.; Meredith, D.M.; Thorner, A.R.; Jordanova, E.S.; et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood 2016, 127, 869–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, T.K.; Kovach, A.E.; Grover, N.S.; Huang, L.C.; Lee, L.A.; Rubinstein, S.M.; Wang, Y.; Morgan, D.S.; Greer, J.P.; Park, S.I.; et al. Clinicopathologic correlates of MYD88 L265P mutation and programmed cell death (PD-1) pathway in primary central nervous system lymphoma. Leuk. Lymphoma 2019, 60, 2880–2889. [Google Scholar] [CrossRef]
- Furuse, M.; Kuwabara, H.; Ikeda, N.; Hattori, Y.; Ichikawa, T.; Kagawa, N.; Kikuta, K.; Tamai, S.; Nakada, M.; Wakabayashi, T.; et al. PD-L1 and PD-L2 expression in the tumor microenvironment including peritumoral tissue in primary central nervous system lymphoma. Bmc Cancer 2020, 20, 277. [Google Scholar] [CrossRef] [PubMed]
- Pollari, M.; Brück, O.; Pellinen, T.; Vähämurto, P.; Karjalainen-Lindsberg, M.L.; Mannisto, S.; Kallioniemi, O.; Kellokumpu-Lehtinen, P.L.; Mustjoki, S.; Leivonen, S.K.; et al. PD-L1(+) tumor-associated macrophages and PD-1(+) tumor-infiltrating lymphocytes predict survival in primary testicular lymphoma. Haematologica 2018, 103, 1908–1914. [Google Scholar] [CrossRef] [Green Version]
- Pollari, M.; Pellinen, T.; Karjalainen-Lindsberg, M.L.; Kellokumpu-Lehtinen, P.L.; Leivonen, S.K.; Leppä, S. Adverse prognostic impact of regulatory T-cells in testicular diffuse large B-cell lymphoma. Eur. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Cho, H.; Kim, S.H.; Kim, S.J.; Chang, J.H.; Yang, W.I.; Suh, C.O.; Kim, Y.R.; Jang, J.E.; Cheong, J.W.; Min, Y.H.; et al. Programmed cell death 1 expression is associated with inferior survival in patients with primary central nervous system lymphoma. Oncotarget 2017, 8, 87317–87328. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.; Lee, H.; Yoon, S.E.; Ryu, K.J.; Ko, Y.H.; Kim, W.S.; Kim, S.J. Serum levels of soluble programmed death-ligand 1 (sPD-L1) in patients with primary central nervous system diffuse large B-cell lymphoma. BMC Cancer 2020, 20, 120. [Google Scholar] [CrossRef] [Green Version]
- Four, M.; Cacheux, V.; Tempier, A.; Platero, D.; Fabbro, M.; Marin, G.; Leventoux, N.; Rigau, V.; Costes-Martineau, V.; Szablewski, V. PD1 and PDL1 expression in primary central nervous system diffuse large B-cell lymphoma are frequent and expression of PD1 predicts poor survival. Hematol. Oncol. 2017, 35, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Hayano, A.; Komohara, Y.; Takashima, Y.; Takeya, H.; Homma, J.; Fukai, J.; Iwadate, Y.; Kajiwara, K.; Ishizawa, S.; Hondoh, H.; et al. Programmed Cell Death Ligand 1 Expression in Primary Central Nervous System Lymphomas: A Clinicopathological Study. Anticancer Res. 2017, 37, 5655–5666. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Kawaguchi, A.; Sato, R.; Yoshida, K.; Hayano, A.; Homma, J.; Fukai, J.; Iwadate, Y.; Kajiwara, K.; Ishizawa, S.; et al. Differential expression of individual transcript variants of PD-1 and PD-L2 genes on Th-1/Th-2 status is guaranteed for prognosis prediction in PCNSL. Sci. Rep. 2019, 9, 10004. [Google Scholar] [CrossRef] [Green Version]
- Ok, C.Y.; Li, L.; Xu-Monette, Z.Y.; Visco, C.; Tzankov, A.; Manyam, G.C.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; Orazi, A.; et al. Prevalence and clinical implications of epstein-barr virus infection in de novo diffuse large B-cell lymphoma in Western countries. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2338–2349. [Google Scholar] [CrossRef] [Green Version]
- Marcelis, L.; Antoranz, A.; Delsupehe, A.M.; Biesemans, P.; Ferreiro, J.F.; Debackere, K.; Vandenberghe, P.; Verhoef, G.; Gheysens, O.; Cattoretti, G.; et al. In-depth characterization of the tumor microenvironment in central nervous system lymphoma reveals implications for immune-checkpoint therapy. Cancer Immunol. Immunother. 2020, 69, 1751–1766. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.K.; Keane, C.; Tobin, J.W.D.; Talaulikar, D.; Jain, S.; Vari, F.; Kruze, L.; Murigneux, V.; Fink, L.; Gunawardana, J.; et al. The Impact of EBV upon the Tumor Microenvironment and Mutational Profile of Primary CNS Lymphoma in PTLD. Blood 2017, 130, 2731. [Google Scholar] [CrossRef]
- Jones, K.; Vari, F.; Keane, C.; Crooks, P.; Nourse, J.P.; Seymour, L.A.; Gottlieb, D.; Ritchie, D.; Gill, D.; Gandhi, M.K. Serum CD163 and TARC as disease response biomarkers in classical Hodgkin lymphoma. Clin. Cancer Res. 2013, 19, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.; Xu, M.L.; Yu, H.; Fletcher, C.D.; Freeman, G.J.; Shipp, M.A.; et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, M.K.; Lambley, E.; Duraiswamy, J.; Dua, U.; Smith, C.; Elliott, S.; Gill, D.; Marlton, P.; Seymour, J.; Khanna, R. Expression of LAG-3 by tumor-infiltrating lymphocytes is coincident with the suppression of latent membrane antigen-specific CD8+ T-cell function in Hodgkin lymphoma patients. Blood 2006, 108, 2280–2289. [Google Scholar] [CrossRef]
- Miyasato, Y.; Takashima, Y.; Takeya, H.; Yano, H.; Hayano, A.; Nakagawa, T.; Makino, K.; Takeya, M.; Yamanaka, R.; Komohara, Y. The expression of PD-1 ligands and IDO1 by macrophage/microglia in primary central nervous system lymphoma. J. Clin. Exp. Hematop. 2018, 58, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Georgiou, K.; Chen, L.; Berglund, M.; Ren, W.; de Miranda, N.F.; Lisboa, S.; Fangazio, M.; Zhu, S.; Hou, Y.; Wu, K.; et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood 2016, 127, 3026–3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, D.; Kim, S.; Kim, P.J.; Go, H.; Nam, S.J.; Paik, J.H.; Kim, Y.A.; Kim, T.M.; Heo, D.S.; Kim, C.W.; et al. Clinicopathological analysis of programmed cell death 1 and programmed cell death ligand 1 expression in the tumour microenvironments of diffuse large B cell lymphomas. Histopathology 2016, 68, 1079–1089. [Google Scholar] [CrossRef]
- Laurent, C.; Charmpi, K.; Gravelle, P.; Tosolini, M.; Franchet, C.; Ysebaert, L.; Brousset, P.; Bidaut, A.; Ycart, B.; Fournié, J.J. Several immune escape patterns in non-Hodgkin’s lymphomas. Oncoimmunology 2015, 4, e1026530. [Google Scholar] [CrossRef] [Green Version]
- Boussiotis, V.A. Cell-specific PD-L1 expression in DLBCL. Blood 2015, 126, 2171–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, K.; Shiraishi, Y.; Takeda, Y.; Sakata, S.; Matsumoto, M.; Nagano, S.; Maeda, T.; Nagata, Y.; Kitanaka, A.; Mizuno, S.; et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature 2016, 534, 402–406. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell. Signal. 2015, 27, 443–452. [Google Scholar] [CrossRef]
- Zhao, L.; Yu, H.; Yi, S.; Peng, X.; Su, P.; Xiao, Z.; Liu, R.; Tang, A.; Li, X.; Liu, F.; et al. The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget 2016, 7, 45370–45384. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.; Vistarop, A.G.; Huaman, F.; Narbaitz, M.; Metrebian, F.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Cytotoxic response against Epstein Barr virus coexists with diffuse large B-cell lymphoma tolerogenic microenvironment: Clinical features and survival impact. Sci. Rep. 2017, 7, 10813. [Google Scholar] [CrossRef]
- Cristino, A.S.; Nourse, J.; West, R.A.; Sabdia, M.B.; Law, S.C.; Gunawardana, J.; Vari, F.; Mujaj, S.; Thillaiyampalam, G.; Snell, C.; et al. EBV microRNA-BHRF1-2-5p targets the 3′UTR of immune checkpoint ligands PD-L1 and PD-L2. Blood 2019, 134, 2261–2270. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, D.; Xie, G.; Yin, Y.; Zhao, E.; Tao, K.; Li, R. MicroRNA-152 regulates immune response via targeting B7-H1 in gastric carcinoma. Oncotarget 2017, 8, 28125–28134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Yan, F.; Wu, C. Overexpressed miR-195 attenuated immune escape of diffuse large B-cell lymphoma by targeting PD-L1. Biomed. Pharmacother. 2018, 98, 95–101. [Google Scholar] [CrossRef]
- Sun, J.R.; Zhang, X.; Zhang, Y. MiR-214 prevents the progression of diffuse large B-cell lymphoma by targeting PD-L1. Cell. Mol. Biol. Lett. 2019, 24, 68. [Google Scholar] [CrossRef]
- Panda, A.; Rosenfeld, J.A.; Singer, E.A.; Bhanot, G.; Ganesan, S. Genomic and immunologic correlates of LAG-3 expression in cancer. Oncoimmunology 2020, 9, 1756116. [Google Scholar] [CrossRef]
- Chen, B.J.; Dashnamoorthy, R.; Galera, P.; Makarenko, V.; Chang, H.; Ghosh, S.; Evens, A.M. The immune checkpoint molecules PD-1, PD-L1, TIM-3 and LAG-3 in diffuse large B-cell lymphoma. Oncotarget 2019, 10, 2030. [Google Scholar] [CrossRef] [Green Version]
- Keane, C.; Law, S.C.; Gould, C.; Birch, S.; Sabdia, M.B.; Merida de Long, L.; Thillaiyampalam, G.; Abro, E.; Tobin, J.W.; Tan, X. LAG3: A novel immune checkpoint expressed by multiple lymphocyte subsets in diffuse large B-cell lymphoma. Blood Adv. 2020, 4, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Myklebust, J.H.; Irish, J.M.; Brody, J.; Czerwinski, D.K.; Houot, R.; Kohrt, H.E.; Timmerman, J.; Said, J.; Green, M.R.; Delabie, J.; et al. High PD-1 expression and suppressed cytokine signaling distinguish T cells infiltrating follicular lymphoma tumors from peripheral T cells. Blood 2013, 121, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.Z.; Grote, D.M.; Ziesmer, S.C.; Xiu, B.; Novak, A.J.; Ansell, S.M. PD-1 expression defines two distinct T-cell sub-populations in follicular lymphoma that differentially impact patient survival. Blood Cancer J. 2015, 5, e281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreras, J.; Lopez-Guillermo, A.; Roncador, G.; Villamor, N.; Colomo, L.; Martinez, A.; Hamoudi, R.; Howat, W.J.; Montserrat, E.; Campo, E. High numbers of tumor-infiltrating programmed cell death 1-positive regulatory lymphocytes are associated with improved overall survival in follicular lymphoma. J. Clin. Oncol. 2009, 27, 1470–1476. [Google Scholar] [CrossRef]
- Richendollar, B.G.; Pohlman, B.; Elson, P.; Hsi, E.D. Follicular programmed death 1-positive lymphocytes in the tumor microenvironment are an independent prognostic factor in follicular lymphoma. Hum. Pathol. 2011, 42, 552–557. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Kim, H.J.; Villasboas, J.C.; Chen, Y.P.; Price-Troska, T.; Jalali, S.; Wilson, M.; Novak, A.J.; Ansell, S.M. Expression of LAG-3 defines exhaustion of intratumoral PD-1(+) T cells and correlates with poor outcome in follicular lymphoma. Oncotarget 2017, 8, 61425–61439. [Google Scholar] [CrossRef] [Green Version]
- Blaker, Y.N.; Spetalen, S.; Brodtkorb, M.; Lingjaerde, O.C.; Beiske, K.; Østenstad, B.; Sander, B.; Wahlin, B.E.; Melen, C.M.; Myklebust, J.H.; et al. The tumour microenvironment influences survival and time to transformation in follicular lymphoma in the rituximab era. Br. J. Haematol. 2016, 175, 102–114. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M.; Minnema, M.C.; Johnson, P.; Timmerman, J.M.; Armand, P.; Shipp, M.A.; Rodig, S.J.; Ligon, A.H.; Roemer, M.G.M.; Reddy, N.; et al. Nivolumab for Relapsed/Refractory Diffuse Large B-Cell Lymphoma in Patients Ineligible for or Having Failed Autologous Transplantation: A Single-Arm, Phase II Study. J. Clin. Oncol. 2019, 37, 481–489. [Google Scholar] [CrossRef]
- Tobin, J.W.D.; Keane, C.; Gunawardana, J.; Mollee, P.; Birch, S.; Hoang, T.; Lee, J.; Li, L.; Huang, L.; Murigneux, V.; et al. Progression of Disease Within 24 Months in Follicular Lymphoma Is Associated With Reduced Intratumoral Immune Infiltration. J. Clin. Oncol. 2019, 37, 3300–3309. [Google Scholar] [CrossRef]
- Wahlin, B.E.; Aggarwal, M.; Montes-Moreno, S.; Gonzalez, L.F.; Roncador, G.; Sanchez-Verde, L.; Christensson, B.; Sander, B.; Kimby, E. A unifying microenvironment model in follicular lymphoma: Outcome is predicted by programmed death-1--positive, regulatory, cytotoxic, and helper T cells and macrophages. Clin. Cancer Res. 2010, 16, 637–650. [Google Scholar] [CrossRef] [Green Version]
- De Jong, D.; Koster, A.; Hagenbeek, A.; Raemaekers, J.; Veldhuizen, D.; Heisterkamp, S.; de Boer, J.P.; van Glabbeke, M. Impact of the tumor microenvironment on prognosis in follicular lymphoma is dependent on specific treatment protocols. Haematologica 2009, 94, 70–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, W.B.C.; Mendeville, M.; Redd, R.; Clear, A.J.; Bladergroen, R.; Calaminici, M.; Rosenwald, A.; Hoster, E.; Hiddemann, W.; Gaulard, P.; et al. Prognostic relevance of CD163 and CD8 combined with EZH2 and gain of chromosome 18 in follicular lymphoma: A study by the Lunenburg Lymphoma Biomarker Consortium. Haematologica 2017, 102, 1413–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Tomita, N.; Sakata, S.; Tsuyama, N.; Hashimoto, C.; Ohshima, R.; Matsuura, S.; Ogawa, K.; Yamamoto, W.; Kameda, Y.; et al. Prognostic significance of programmed cell death-1-positive cells in follicular lymphoma patients may alter in the rituximab era. Eur. J. Haematol. 2013, 90, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.J.; Armand, P.; Redd, R.A.; Jeter, E.; Merryman, R.W.; Coleman, K.C.; Herrera, A.F.; Dahi, P.; Nieto, Y.; LaCasce, A.S.; et al. PD-1 blockade for diffuse large B-cell lymphoma after autologous stem cell transplantation. Blood Adv. 2020, 4, 122–126. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Ribrag, V.; Moskowitz, C.H.; Michot, J.M.; Kuruvilla, J.; Balakumaran, A.; Zhang, Y.; Chlosta, S.; Shipp, M.A.; Armand, P. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017, 130, 267–270. [Google Scholar] [CrossRef] [Green Version]
- Zinzani, P.L.; Santoro, A.; Gritti, G.; Brice, P.; Barr, P.M.; Kuruvilla, J.; Cunningham, D.; Kline, J.; Johnson, N.A.; Mehta-Shah, N.; et al. Nivolumab Combined With Brentuximab Vedotin for Relapsed/Refractory Primary Mediastinal Large B-Cell Lymphoma: Efficacy and Safety From the Phase II CheckMate 436 Study. J. Clin. Oncol. 2019, 37, 3081–3089. [Google Scholar] [CrossRef]
- Armand, P.; Rodig, S.; Melnichenko, V.; Thieblemont, C.; Bouabdallah, K.; Tumyan, G.; Özcan, M.; Portino, S.; Fogliatto, L.; Caballero, M.D.; et al. Pembrolizumab in Relapsed or Refractory Primary Mediastinal Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 3291–3299. [Google Scholar] [CrossRef]
- Hoang-Xuan, K.; Houot, R.; Soussain, C.; Blonski, M.; Schmitt, A.; Delwail, V.; Damaj, G.L.; Ghesquieres, H.; Peyrade, F.; Tempescul, A.; et al. First Results of the Acsé Pembrolizumab Phase II in the Primary CNS Lymphoma (PCNSL) Cohort. Blood 2020, 136, 15–16. [Google Scholar] [CrossRef]
- Nayak, L.; Iwamoto, F.M.; LaCasce, A.; Mukundan, S.; Roemer, M.G.M.; Chapuy, B.; Armand, P.; Rodig, S.J.; Shipp, M.A. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood 2017, 129, 3071–3073. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Westin, J.R.; Chu, F.; Zhang, M.; Fayad, L.E.; Kwak, L.W.; Fowler, N.; Romaguera, J.; Hagemeister, F.; Fanale, M.; Samaniego, F.; et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: A single group, open-label, phase 2 trial. Lancet Oncol. 2014, 15, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Armand, P.; Janssens, A.; Gritti, G.; Radford, J.; Timmerman, J.; Pinto, A.; Mercadal Vilchez, S.; Johnson, P.; Cunningham, D.; Leonard, J.P. Efficacy and safety results from CheckMate 140, a phase 2 study of nivolumab for relapsed/refractory follicular lymphoma. BloodJ. Am. Soc. Hematol. 2021, 137, 637–645. [Google Scholar]
- Barraclough, A.; Chong, G.; Gilbertson, M.; Grigg, A.; Churilov, L.; Fancourt, T.; Ritchie, D.; Koldej, R.; Agarwal, R.; Manos, K.; et al. Immune Priming with Single-Agent Nivolumab Followed By Combined Nivolumab & Rituximab Is Safe and Efficacious for First-Line Treatment of Follicular Lymphoma; Interim Analysis of the ‘1st FLOR’ Study. Blood 2019, 134, 1523. [Google Scholar] [CrossRef]
- Godfrey, J.; Tumuluru, S.; Bao, R.; Leukam, M.; Venkataraman, G.; Phillip, J.; Fitzpatrick, C.; McElherne, J.; MacNabb, B.W.; Orlowski, R.; et al. PD-L1 gene alterations identify a subset of diffuse large B-cell lymphoma harboring a T-cell-inflamed phenotype. Blood 2019, 133, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Hyeon, J.; Cho, I.; Ko, Y.H.; Kim, W.S. Comparison of Efficacy of Pembrolizumab between Epstein-Barr Virus‒Positive and ‒Negative Relapsed or Refractory Non-Hodgkin Lymphomas. Cancer Res. Treat. 2019, 51, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Lipson, E.; Gopal, A.; Neelapu, S.; Armand, P.; Spurgeon, S.; Leonard, J.; Hodi, F.; Sanborn, R.; Melero, I.; Gajewski, T. Initial experience administering BMS-986016, a monoclonal antibody that targets lymphocyte activation gene (LAG)-3, alone and in combination with nivolumab to patients with hematologic and solid malignancies. J. Immunother. Cancer 2016, 4, P232. [Google Scholar]
- Zhang, Y.; Zhang, X.; Cheng, C.; Mu, W.; Liu, X.; Li, N.; Wei, X.; Liu, X.; Xia, C.; Wang, H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017, 11, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Zou, Y.; Zhang, L.; Tang, J.; Niedermann, G.; Firat, E.; Huang, X.; Zhu, X. Nucleofection with Plasmid DNA for CRISPR/Cas9-Mediated Inactivation of Programmed Cell Death Protein 1 in CD133-Specific CAR T Cells. Hum. Gene Ther. 2019, 30, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Clinical Trial (Year) | Pt Characteristics | Experimental Arm | Study | ORR | CR Rate (%) | PFS, OS, DOR | Grade 3–5 AEs | Ref. |
|---|---|---|---|---|---|---|---|---|
| R/R cHL Trials | ||||||||
| CheckMate 039 (2015) | R/R cHL | Nivolumab 3 mg/kg q2 weekly CR, PD or excessive toxicity | Phase 1 23 pts | 87% | 17% | PFS at 24 weeks = 86% | 22% | [76] |
| CheckMate 205 (2016–2018) | R/R cHL, Previous ASCT Pt Subsets: A: BV Naïve B: Prev BV after ASCT C: Prev BV before +/− after ASCT | Nivolumab 3 mg/kg q2 weekly until PD excessive toxicity | Phase 2 243 pts A = 63 B = 80 C = 100 | Overall 71% A = 65% B = 71% C = 75% | Overall 21% A = 32% B = 14% C = 20% | Median PFS = 15 months Median OS = Not reached Median DOR = 18 months | 40% 1 | [77,80] |
| NCT 03004833 (2017–2019) | R/R cHL, No prior ASCT | BV 1.8 mg/kg + Nivolumab 3 mg/kg q3 weekly × 4 cycles +/− ASCT | Phase 1/2 91 pts | 85% 74% proceed to ASCT | 67% | PFS at 24 months = 78% 2 OS at 24 months = 93% | 31% | [81,82] |
| ECOG-ACRIN Research Group NCT01896999 (2020) | R/R cHL Included previous allogeneic (n = 8) and autologous (n = 21) transplant patients | 3 Experimental Arms: A = BV (1.8 mg/kg) + Ipilimumab (1 mg/kg or 3 mg/kg) B = BV (1.2 mg/kg or 1.8 mg/kg) + Nivolumab (3 mg/kg) C = BV (1.2 mg/kg) + Ipilimumab (1 mg/kg) + Nivolumab (3 mg/kg) | Phase 1/2 61 pts A = 21 B = 18 C = 22 | A = 76% B = 89% C = 82% | A = 57% B = 61% C = 73% | A: Median PFS = 14.4 months B: Median PFS = Not reached C: Median PFS = Not reached | A = 43% B = 16% C = 50% | [83] |
| KEYNOTE-087 (2017–2019) | R/R cHL Pt Subsets: A: BV Naïve, Previous ASCT B: Previous BV after ASCT C: Previous BV, no ASCT | Pembrolizumab (200 mg) q3 weekly for 2 years | Phase 2 210 pts A = 60 B = 69 C = 81 | Overall 71% A = 71.7% B = 78.3% B = 64.2% | Overall 27.6% A = 31.7% B = 26.1% B = 25.9% | Overall: Median PFS 13.7 months Median DOR 16.6 months A: Median PFS 16.4 months B: Median PFS 11.1 months C: Median PFS 19.4 months A: Median DOR 22.1 months B: Median DOR 11.1 months C: Median DOR 24.2 months | 11.9% | [84,85] |
| NCT02362997 (2019) | R/R cHL Consolidation post-ASCT | Pembrolizumab 200 mg 3 weekly × 8 cycles 3 | Phase 2 30 pts | N/a | N/a | PFS at 18 months = 82% OS at 18 months = 100% | 30% | [86] |
| KEYNOTE-013 (2020) | R/R cHL, Previous BV Previously failed or ineligible for ASCT | Pembrolizumab 10 mg/kg 2 weekly for 2 years | Phase 2 31 pts | 58% | 19% | Median PFS 11.4 months OS at 36 months = 81% DOR at 36 months = 50% | 6% | [87] |
| Frontline Hodgkin Lymphoma Trials | ||||||||
| CheckMate 205 (2019) | Advanced-Stage cHL Treatment Naïve | Nivolumab (240 mg) q2 weekly × 4 cycles Followed by Nivolumab + AVD q2 weekly × 12 cycles | Phase 2 51 pts | 75% | 84% | PFS at 21 month 80% | 59% | [88,89] |
| GHSG NIVAHL 2020 | Early-Stage Unfavorable cHL Treatment Naïve | 2 Experimental Arms 4: A = Nivolumab (240 mg) + AVD × 4 cycles B = Nivolumab q2 weekly for 2 cycles followed by Nivolumab-AVD × 2 cycles followed by AVD × 2 cycles. | Phase 2 109 pts A = 55 B = 54 | A = 100 B = 98 | A = 83 B = 84 | PFS at 12 months = 99% OS at 12 months = 100% | A = 28% B = 38% | [90] |
| Clinical Trial (Year) | Pt Characteristics | Experimental Arm | Study | ORR | CR Rate (%) | PFS, OS, DOR | Grade 3–5 AEs | Ref. |
|---|---|---|---|---|---|---|---|---|
| DLBCL Trials | ||||||||
| NCT02038933 | R/R DLBCL Pt Subsets: A: Previously Failed ASCT B: Ineligible for ASCT | Nivolumab (3 mg/kg) q2 weekly | Phase 2 121 pts | A = 10% B = 3% | N/a | A: Median PFS = 1.9 months B: Median PFS 1.4 months | 20% | [143] |
| NCT02362997 (2019) | R/R DLBCL Following ASCT 1 | Pembrolizumab 200 mg q3 weekly × 8 cycles. | Phase 2 29 pts | N/a | N/a | PFS at 18 months = 59% 2 | 79% | [149] |
| PMBCL Trials | ||||||||
| KEYNOTE-013 (2017) | R/R PMBCL | Pembrolizumab 200 mg q3 weekly for 2 years or until toxicity or PD | Phase 1 21 pts | 48% | 33% | Median PFS = 10.4 months Median OS = 31.4 months Median DOR not reached | 24% | [87,150] |
| CheckMate 436 (2019) | R/R PMBCL | Nivolumab (240 mg) + BV q3 weekly | Phase 2 30 pts | 70% | 43% | Median PFS = not reached Median OS = not reached | 53% | [151] |
| KEYNOTE 170 (2019) | R/R PMBCL | Pembrolizumab 200 mg q3 weekly for 2 years (or until PD toxicity or patient withdrawal | Phase 2 53 pts | 45% | 21% | Median PFS = 5.5 months Median OS = not reached Median DOR = not reached | 23% | [152] |
| Other NHL Trials | ||||||||
| Acsé Trial (2020) | R/R PCNSL | Pembrolizumab 200 mg q3 weekly for 2 years | Phase 2 50 pts | 26% | 16% | Median PFS = 2.6 months Median DOR = 10.0 months | 10% | [153] |
| DFCI Case Series (2017) | R/R PCNSL and PTL | Nivolumab 3 mg/kg q2 weekly | Case Series 5 pts | 10% | 80% | PFS at 17 months = 60% OS at 17 months= 100% | 20% | [154] |
| NCT01592370 (2016) | R/R NHL and Myeloma Pt Subsets: A: FL B: DLBCL C: Other B-NHL | Nivolumab 1 mg/kg then 3 mg/kg | Phase 1 B-NHL 31 pts A = 10 pts B = 11 pts C = 10 pts | A = 40% B = 36% C = 0% | N/a | N/a | 21% | [155] |
| NCT00904722 (2014) | R/R FL | Rituximab 375 mg/m2 weekly × 4 cycles + Pidilizumab 3 mg/kg q4 weekly × 12 cycles 3 | Phase 2 32 pts | 66% | 52% | Median PFS = 18.8 months Median DOR = 20.2 months | 0% | [156] |
| CheckMate 140 (2020) | R/R FL | Nivolumab 3 mg/kg q2 weekly | Phase 2 92 pts | 1% | 4% | Median PFS 2.2 months Median DOR 11 months | 49% | [157] |
| 1st FLOR (2019) | Frontline FL Treatment Naïve | Nivolumab (240 mg) q2 weekly × 8 cycles + Rituximab 375 mg/m2 2 weekly × 4 cycles Followed by Nivolumab maintenance q4 weekly × 12 cycles + Rituximab maintenance q12 weekly × 8 cycles | Phase 2 39 pts | 84% | 37% | N/A | 15% | [158] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tobin, J.W.D.; Bednarska, K.; Campbell, A.; Keane, C. PD-1 and LAG-3 Checkpoint Blockade: Potential Avenues for Therapy in B-Cell Lymphoma. Cells 2021, 10, 1152. https://doi.org/10.3390/cells10051152
Tobin JWD, Bednarska K, Campbell A, Keane C. PD-1 and LAG-3 Checkpoint Blockade: Potential Avenues for Therapy in B-Cell Lymphoma. Cells. 2021; 10(5):1152. https://doi.org/10.3390/cells10051152
Chicago/Turabian StyleTobin, Joshua W. D., Karolina Bednarska, Ashlea Campbell, and Colm Keane. 2021. "PD-1 and LAG-3 Checkpoint Blockade: Potential Avenues for Therapy in B-Cell Lymphoma" Cells 10, no. 5: 1152. https://doi.org/10.3390/cells10051152