
Nanoparticle Delivered Anti-miR-141-3p for Stroke Therapy

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. PNA Synthesis
2.2. Thermal Melting Studies
2.3. Nanoparticle Preparation
2.4. Scanning Electron Microscopy (SEM)
2.5. Dynamic Light Scattering (DLS)
2.6. Nucleic Acid Release Profile
2.7. Loading Study
2.8. Safety Assessment by Cell Viability Assay
2.9. Experimental Design for In Vivo Work
2.10. Middle Cerebral Artery Occlusion (MCAO) Surgery
2.11. Treatment with miR-141 Inhibitors
2.12. Total RNA Isolation and cDNA Synthesis
2.13. Real-Time qPCR for miRNA Analysis
2.14. Immunohistochemistry
2.15. Infarct Volume Analysis
3. Results
3.1. Design and Synthesis of PNA and PS-Based Anti-miR-141-3p Probes
3.2. Formulating PLGA NPs and Its Characterizations
3.3. Nucleic Acid Release Profile
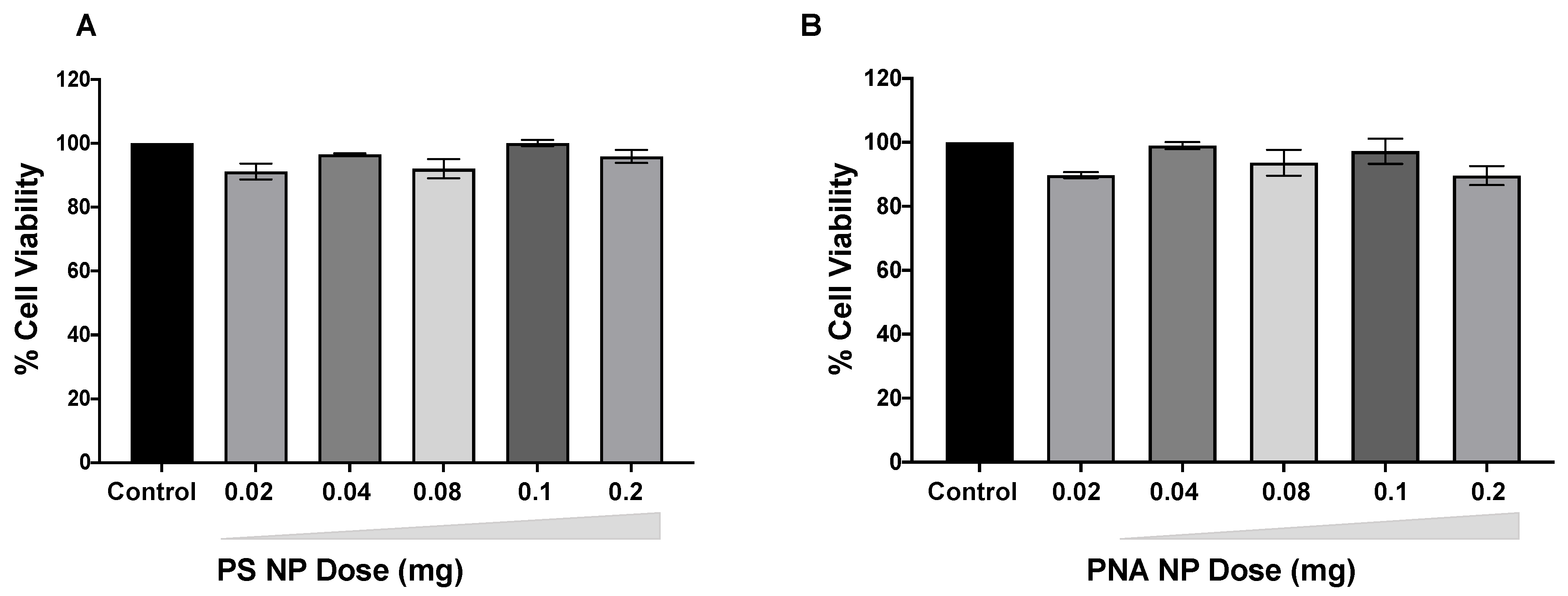
3.4. Safety of PS and PNA NPs
3.5. Visualization of TAMRA Tagged PS Inhibitor in the CNS after Systemic Delivery
3.6. Validation of In Vivo Efficacy in Stroke Mouse Model
3.7. Infarct Volume Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Van Rooij, E.; Purcell, A.L.; Levin, A.A. Developing microRNA therapeutics. Circ. Res. 2012, 110, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Khoshnam, S.E.; Winlow, W.; Farbood, Y.; Moghaddam, H.F.; Farzaneh, M. Emerging Roles of microRNAs in Ischemic Stroke: As Possible Therapeutic Agents. J. Stroke 2017, 19, 166–187. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Jeyaseelan, K.; Lim, K.Y.; Armugam, A. MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke 2008, 39, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roitbak, T. Silencing a Multifunctional microRNA Is Beneficial for Stroke Recovery. Front. Mol. Neurosci. 2018, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic. Acid. Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T. Molecular mechanisms of action of antisense drugs. Biochim. Biophys. Acta 1999, 1489, 31–44. [Google Scholar] [CrossRef]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Malik, S.; Bahal, R. Investigation of PLGA nanoparticles in conjunction with nuclear localization sequence for enhanced delivery of antimiR phosphorothioates in cancer cells in vitro. J. Nanobiotechnol. 2019, 17, 57. [Google Scholar] [CrossRef]
- Woodrow, K.A.; Cu, Y.; Booth, C.J.; Saucier-Sawyer, J.K.; Wood, M.J.; Saltzman, W.M. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat. Mater. 2009, 8, 526–533. [Google Scholar] [CrossRef]
- Lu, J.M.; Wang, X.; Marin-Muller, C.; Wang, H.; Lin, P.H.; Yao, Q.; Chen, C. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev. Mol. Diagn. 2009, 9, 325–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Bahal, R.; Gupta, M.; Glazer, P.M.; Saltzman, W.M. Nanotechnology for delivery of peptide nucleic acids (PNAs). J. Control. Release 2016, 240, 302–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosco, D.; Cilurzo, F.; Maiuolo, J.; Federico, C.; Di Martino, M.T.; Cristiano, M.C.; Tassone, P.; Fresta, M.; Paolino, D. Delivery of miR-34a by chitosan/PLGA nanoplexes for the anticancer treatment of multiple myeloma. Sci. Rep. 2015, 5, 17579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercurio, S.; Cauteruccio, S.; Manenti, R.; Candiani, S.; Scari, G.; Licandro, E.; Pennati, R. miR-7 Knockdown by Peptide Nucleic Acids in the Ascidian Ciona intestinalis. Int. J. Mol. Sci. 2019, 20, 5127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, P.E. Gene targeting and expression modulation by peptide nucleic acids (PNA). Curr. Pharm. Des. 2010, 16, 3118–3123. [Google Scholar] [CrossRef]
- Pellestor, F.; Paulasova, P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur. J. Hum. Genet. 2004, 12, 694–700. [Google Scholar] [CrossRef]
- Swenson, C.S.; Heemstra, J.M. Peptide nucleic acids harness dual information codes in a single molecule. Chem. Commun. 2020, 56, 1926–1935. [Google Scholar] [CrossRef]
- Christensen, L.; Fitzpatrick, R.; Gildea, B.; Petersen, K.H.; Hansen, H.F.; Koch, T.; Egholm, M.; Buchardt, O.; Nielsen, P.E.; Coull, J.; et al. Solid-phase synthesis of peptide nucleic acids. J. Pept. Sci. 1995, 1, 175–183. [Google Scholar] [CrossRef]
- Malik, S.; Slack, F.J.; Bahal, R. Formulation of PLGA nanoparticles containing short cationic peptide nucleic acids. MethodsX 2020, 7, 101115. [Google Scholar] [CrossRef]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Verma, R.; Ritzel, R.M.; Harris, N.M.; Lee, J.; Kim, T.; Pandi, G.; Vemuganti, R.; McCullough, L.D. Inhibition of miR-141-3p Ameliorates the Negative Effects of Poststroke Social Isolation in Aged Mice. Stroke 2018, 49, 1701–1707. [Google Scholar] [CrossRef]
- Swanson, R.A.; Morton, M.T.; Tsao-Wu, G.; Savalos, R.A.; Davidson, C.; Sharp, F.R. A semiautomated method for measuring brain infarct volume. J. Cereb. Blood Flow. Metab. 1990, 10, 290–293. [Google Scholar] [CrossRef]
- Manna, A.; Rapireddy, S.; Bahal, R.; Ly, D.H. MiniPEG-gammaPNA. Methods Mol. Biol. 2014, 1050, 1–12. [Google Scholar] [CrossRef]
- Oyaghire, S.N.; Quijano, E.; Piotrowski-Daspit, A.S.; Saltzman, W.M.; Glazer, P.M. Poly(Lactic-co-Glycolic Acid) Nanoparticle Delivery of Peptide Nucleic Acids In Vivo. Methods Mol. Biol. 2020, 2105, 261–281. [Google Scholar] [CrossRef]
- Ji, W.; Sun, B.; Su, C. Targeting MicroRNAs in Cancer Gene Therapy. Genes 2017, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Chitkara, D.; Kumar, V.; Behrman, S.W.; Mahato, R.I. miRNA profiling in pancreatic cancer and restoration of chemosensitivity. Cancer Lett. 2013, 334, 211–220. [Google Scholar] [CrossRef]
- Sliwinska, A.; Kasinska, M.A.; Drzewoski, J. MicroRNAs and metabolic disorders—Where are we heading? Arch. Med. Sci. 2017, 13, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Seto, A.G.; Beatty, X.; Lynch, J.M.; Hermreck, M.; Tetzlaff, M.; Duvic, M.; Jackson, A.L. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 2018, 183, 428–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiadou, E.; Seto, A.; Beatty, X.; Hermreck, M.; Gilles, M.E.; Stroopinsky, D.; Pinter-Brown, L.C.; Pestano, L.; Marchese, C.; Avigan, D.; et al. Cobomarsen, an oligonucleotide inhibitor of miR-155, slows DLBCL tumor cell growth in vitro and in vivo. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Furdon, P.J.; Dominski, Z.; Kole, R. RNase H cleavage of RNA hybridized to oligonucleotides containing methylphosphonate, phosphorothioate and phosphodiester bonds. Nucleic Acids Res. 1989, 17, 9193–9204. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Quijano, E.; Liu, Y.; Bahal, R.; Scanlon, S.E.; Song, E.; Hsieh, W.C.; Braddock, D.E.; Ly, D.H.; Saltzman, W.M.; et al. Anti-tumor Activity of miniPEG-gamma-Modified PNAs to Inhibit MicroRNA-210 for Cancer Therapy. Mol. Ther. Nucleic Acids 2017, 9, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Philippen, L.E.; Dirkx, E.; Wit, J.B.; Burggraaf, K.; de Windt, L.J.; da Costa Martins, P.A. Antisense MicroRNA Therapeutics in Cardiovascular Disease: Quo Vadis? Mol. Ther. 2015, 23, 1810–1818. [Google Scholar] [CrossRef]
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar] [PubMed]
- Gupta, A.; Mishra, A.; Puri, N. Peptide nucleic acids: Advanced tools for biomedical applications. J. Biotechnol. 2017, 259, 148–159. [Google Scholar] [CrossRef] [PubMed]
- White, P.J.; Anastasopoulos, F.; Pouton, C.W.; Boyd, B.J. Overcoming biological barriers to in vivo efficacy of antisense oligonucleotides. Expert Rev. Mol. Med. 2009, 11, e10. [Google Scholar] [CrossRef]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef]
- Soni, S.; Ruhela, R.K.; Medhi, B. Nanomedicine in Central Nervous System (CNS) Disorders: A Present and Future Prospective. Adv. Pharm. Bull. 2016, 6, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Harris, N.M.; Ritzel, R.; Mancini, N.S.; Jiang, Y.; Yi, X.; Manickam, D.S.; Banks, W.A.; Kabanov, A.V.; McCullough, L.D.; Verma, R. Nano-particle delivery of brain derived neurotrophic factor after focal cerebral ischemia reduces tissue injury and enhances behavioral recovery. Pharmacol. Biochem. Behav. 2016, 150–151, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Perni, S.; Prokopovich, P. Poly-beta-amino-esters nano-vehicles based drug delivery system for cartilage. Nanomedicine 2017, 13, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Wahane, A.; Waghmode, A.; Kapphahn, A.; Dhuri, K.; Gupta, A.; Bahal, R. Role of Lipid-Based and Polymer-Based Non-Viral Vectors in Nucleic Acid Delivery for Next-Generation Gene Therapy. Molecules 2020, 25, 2866. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, M.J.; Rapireddy, S.; Bahal, R.; Sacui, I.; Ly, D.H. Effect of Steric Constraint at the gamma-Backbone Position on the Conformations and Hybridization Properties of PNAs. J. Nucleic Acids 2011, 2011, 652702. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoformulation | DLS (nm ± SEM) | PDI (± SEM) | Zeta Potential (mV ± SEM) |
|---|---|---|---|
| Blank-1 | 376.3 ± 09.1 | 0.22 ± 0.03 | −21.03 ± 2.55 |
| PS-141 | 349.5 ± 15.8 | 0.21 ± 0.02 | −19.97 ± 2.71 |
| Scr-PS-141 | 350.6 ± 15.8 | 0.20 ± 0.01 | −18.83 ± 3.45 |
| Blank-2 | 336.3 ± 28.3 | 0.21 ± 0.04 | −24.17 ± 0.64 |
| PNA-141 | 330.9 ± 41.4 | 0.18 ± 0.07 | −18.03 ± 1.07 |
| Scr-PNA-141 | 315.9 ± 28.1 | 0.18 ± 0.07 | −22.03 ± 1.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhuri, K.; Vyas, R.N.; Blumenfeld, L.; Verma, R.; Bahal, R. Nanoparticle Delivered Anti-miR-141-3p for Stroke Therapy. Cells 2021, 10, 1011. https://doi.org/10.3390/cells10051011
Dhuri K, Vyas RN, Blumenfeld L, Verma R, Bahal R. Nanoparticle Delivered Anti-miR-141-3p for Stroke Therapy. Cells. 2021; 10(5):1011. https://doi.org/10.3390/cells10051011
Chicago/Turabian StyleDhuri, Karishma, Rutesh N. Vyas, Leslie Blumenfeld, Rajkumar Verma, and Raman Bahal. 2021. "Nanoparticle Delivered Anti-miR-141-3p for Stroke Therapy" Cells 10, no. 5: 1011. https://doi.org/10.3390/cells10051011