
Broad-Spectrum HDAC Inhibitors Promote Autophagy through FOXO Transcription Factors in Neuroblastoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Culture Reagents
2.3. siRNA Transfection
2.4. Western Blot Analysis
2.5. Subcellular Fractionation Analysis
2.6. Real-Time PCR Analysis
2.7. Quantification of Microscopic Images
2.8. Cell Viability Assays
2.9. Colony Formation Assays
2.10. Zebrafish Lines
2.11. Cell Preparation and Zebrafish Larvae Xenotransplantation
2.12. Zebrafish Larvae Drug Treatment and Efficiency Evaluation
2.13. Gene Expression Analysis
2.14. Statistical Analysis
3. Results
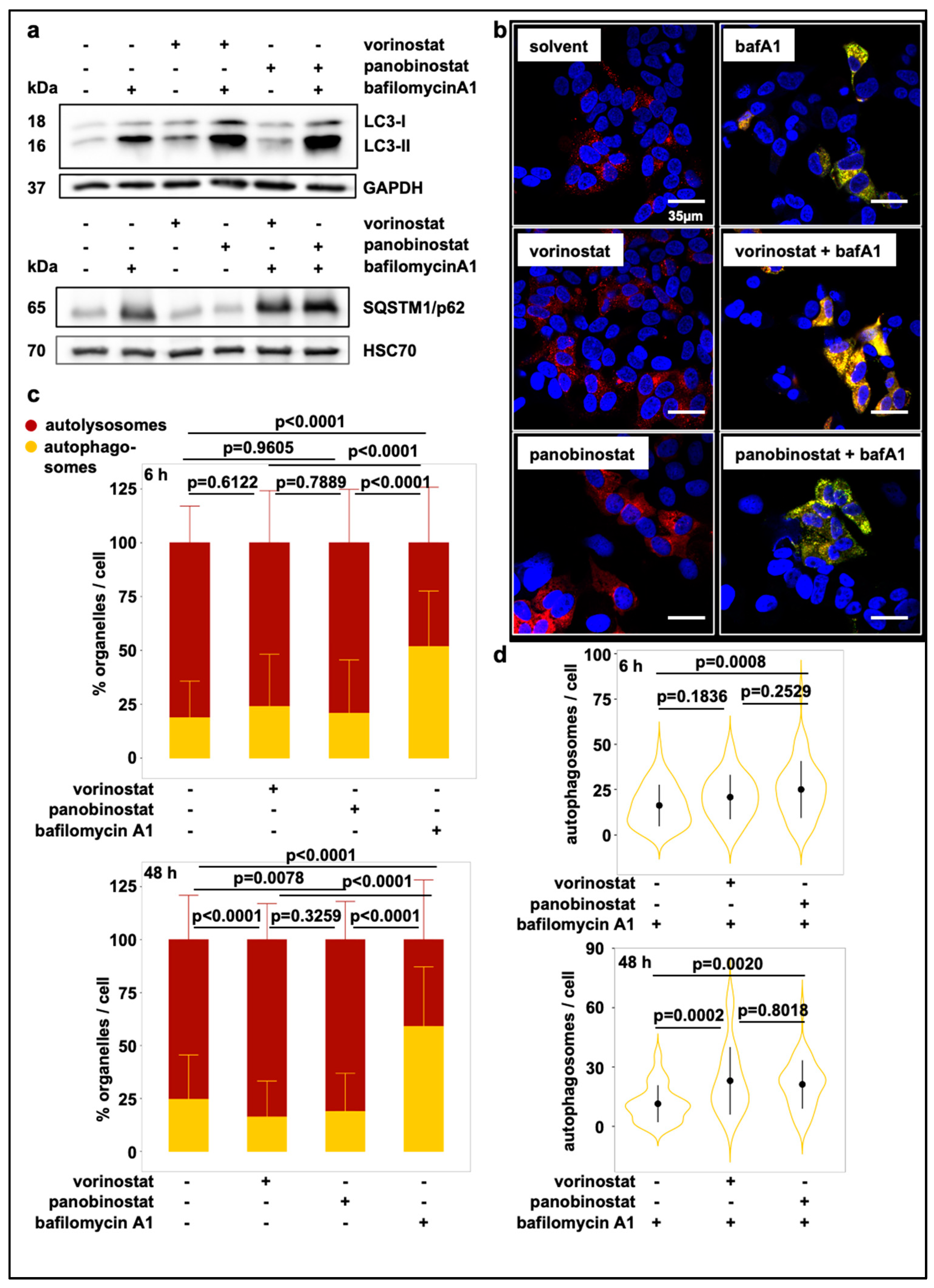
3.1. Broad-Spectrum HDAC Inhibitors Induce Autophagic Flux in SK-N-BE(2)-C Neuroblastoma Cells
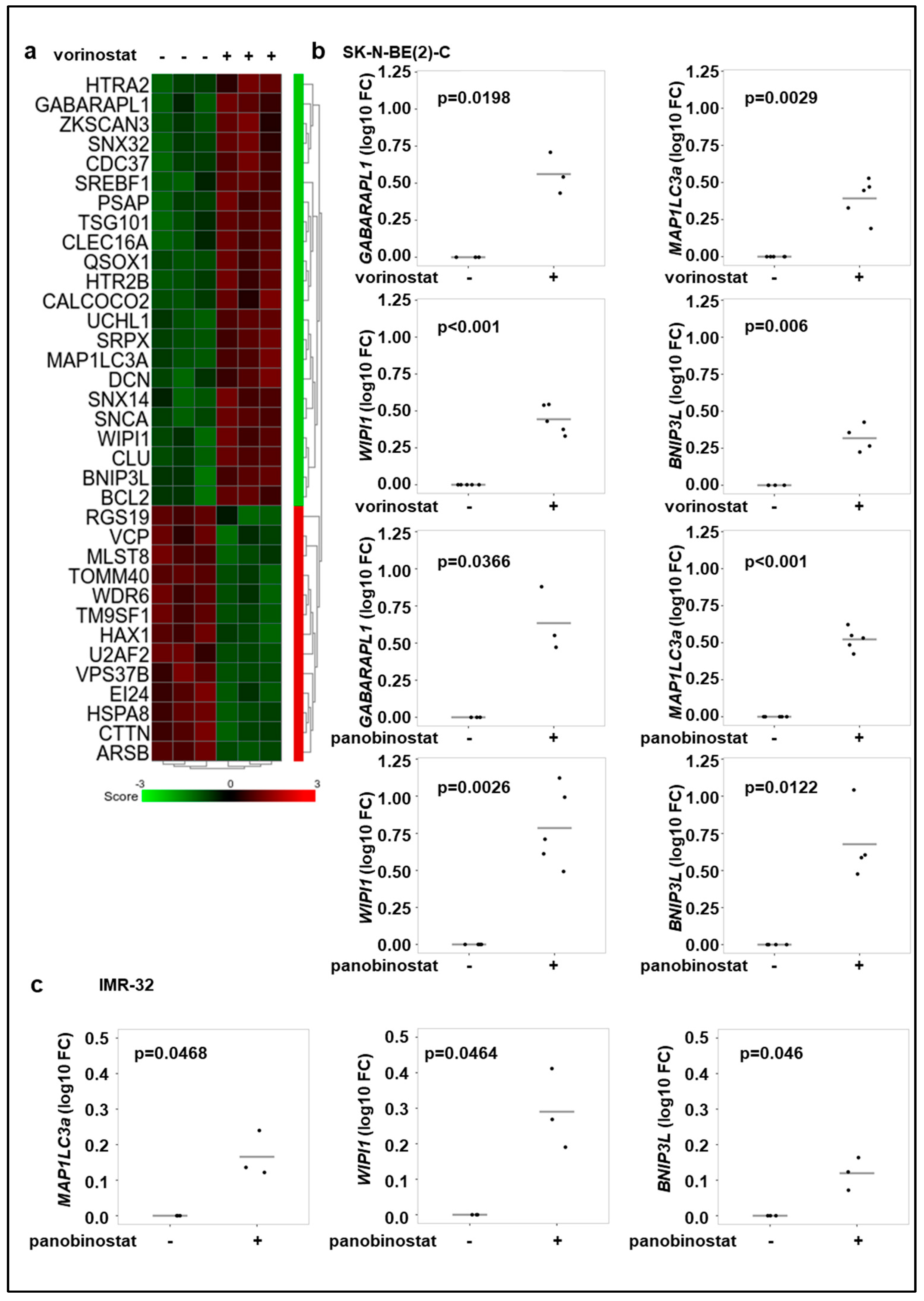
3.2. Broad-Spectrum HDAC Inhibitors Increase the Expression of Autophagy Genes in Neuroblastoma Cell Lines
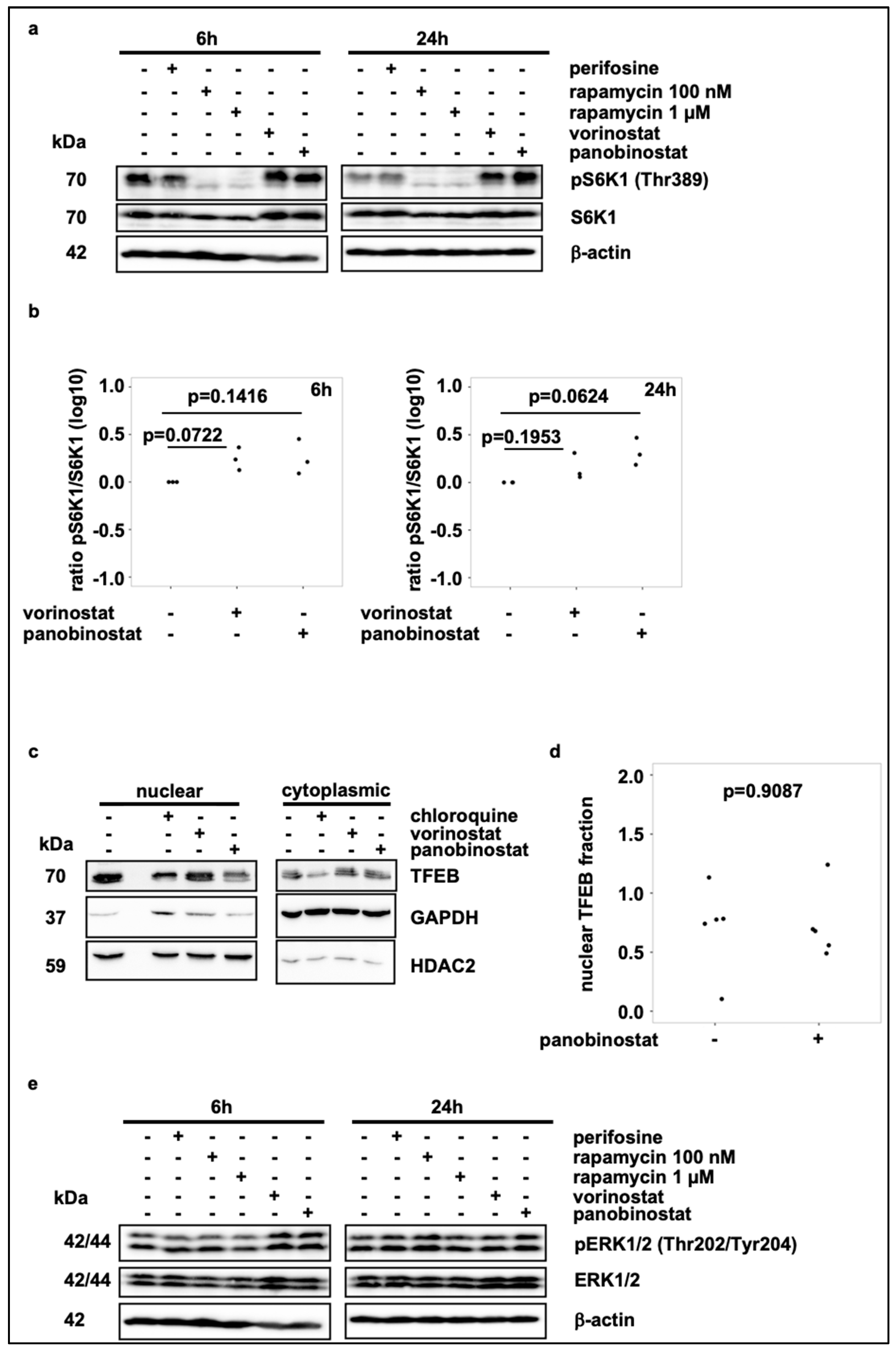
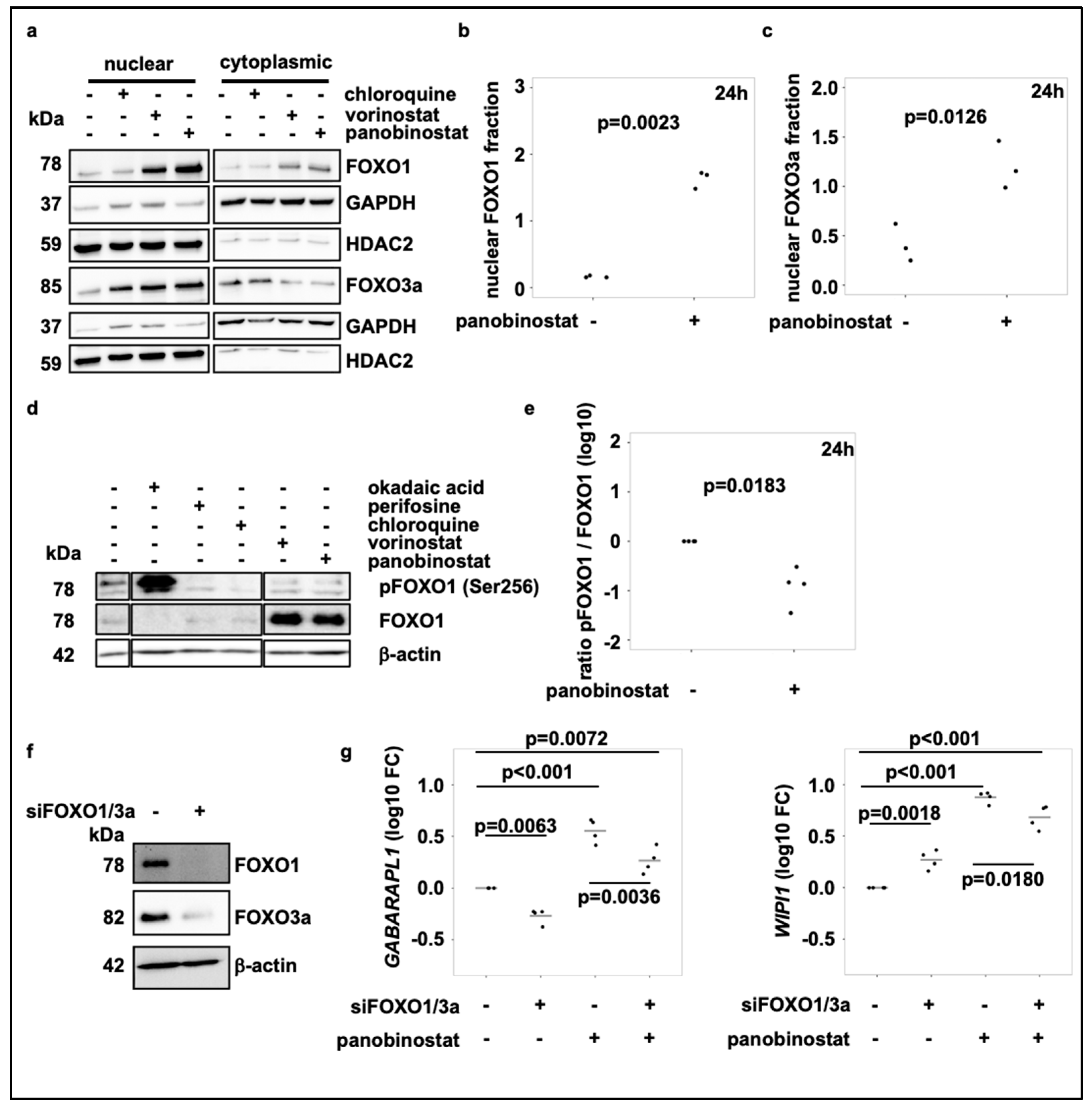
3.3. Broad-Spectrum HDAC Inhibitors Do Not Reduce mTORC1 Activity or Induce TFEB Translocation, But Activate FOXO1 and FOXO3 Transcription Factors in SK-N-BE(2)-C Cells
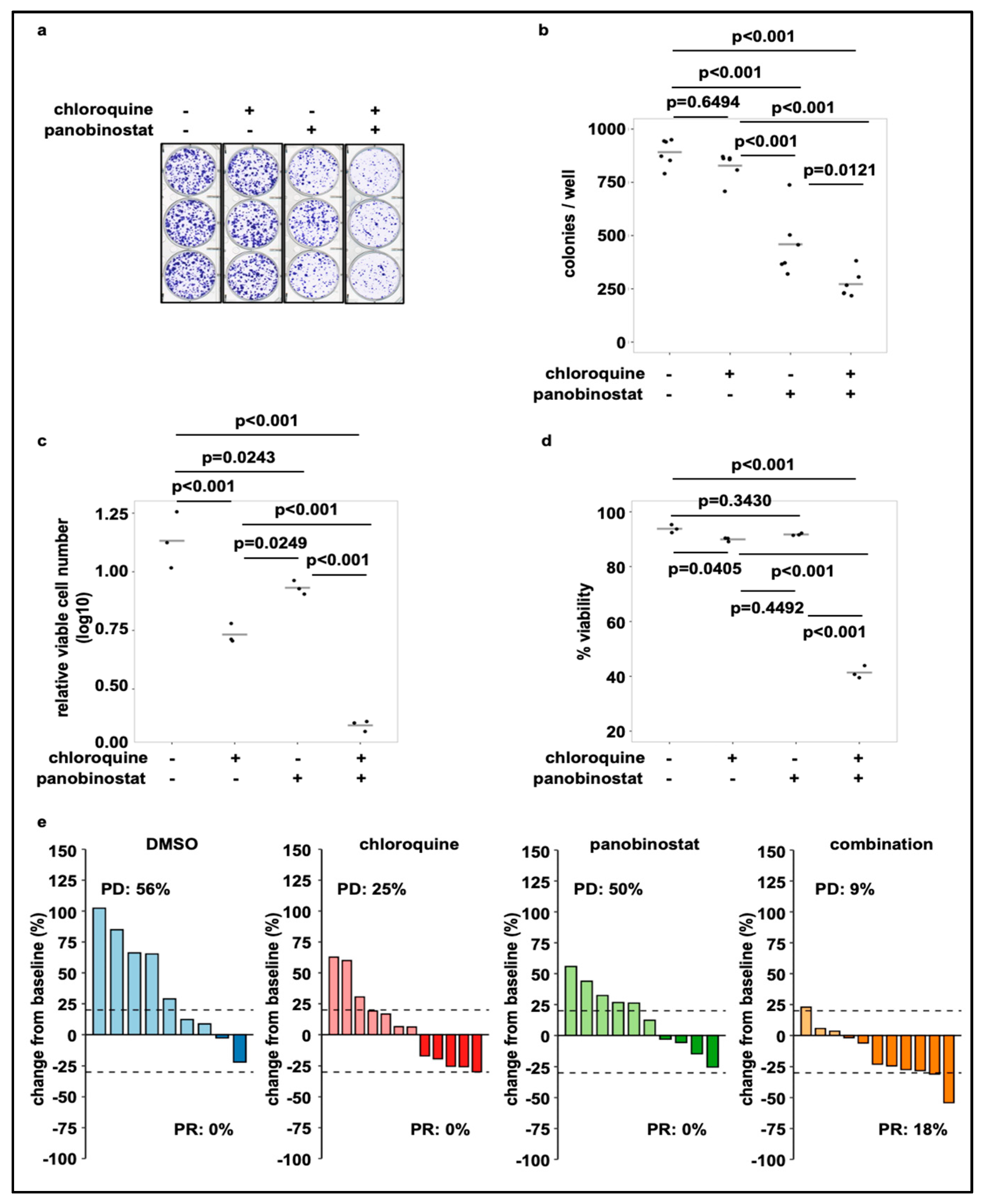
3.4. Treatment of SK-N-BE(2)-C Cells with Panobinostat in Combination with Chloroquine Reduces Cell Viability and Tumor Growth in Cell Culture and In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Smith, M.A.; Seibel, N.L.; Altekruse, S.F.; Ries, L.A.; Melbert, D.L.; O’Leary, M.; Smith, F.O.; Reaman, G.H. Outcomes for children and adolescents with cancer: Challenges for the twenty-first century. J. Clin. Oncol. 2010, 28, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Maris, J.M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 2016, 122, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and recent advances in the treatment of neuroblastoma. Expert Rev. Anticancer. Ther. 2017, 17, 369–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Autin, P.; Blanquart, C.; Fradin, D. Epigenetic Drugs for Cancer and microRNAs: A Focus on Histone Deacetylase Inhibitors. Cancers 2019, 11, 1530. [Google Scholar] [CrossRef] [Green Version]
- Sultana, F.; Manasa, K.L.; Shaik, S.P.; Bonam, S.R.; Kamal, A. Zinc Dependent Histone Deacetylase Inhibitors in Cancer Therapeutics: Recent Update. Curr. Med. Chem. 2019, 26, 7212–7280. [Google Scholar] [CrossRef]
- Hassell, K.N. Histone Deacetylases and their Inhibitors in Cancer Epigenetics. Diseases 2019, 7, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2241. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M. FOXOphagy path to inducing stress resistance and cell survival. Nat. Cell Biol. 2012, 14, 786–788. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [Green Version]
- Hale, A.N.; Ledbetter, D.J.; Gawriluk, T.R.; Rucker, E.B., 3rd. Autophagy: Regulation and role in development. Autophagy 2013, 9, 951–972. [Google Scholar] [CrossRef]
- Settembre, C.; Ballabio, A. TFEB regulates autophagy: An integrated coordination of cellular degradation and recycling processes. Autophagy 2011, 7, 1379–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Debnath, J. Autophagy and tumorigenesis. FEBS Lett. 2010, 584, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaza, N.; Kohli, L.; Roth, K.A. Autophagy in brain tumors: A new target for therapeutic intervention. Brain Pathol. 2012, 22, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Skowron, M.; Berthold, F. Reliable transcript quantification by real-time reverse transcriptase-polymerase chain reaction in primary neuroblastoma using normalization to averaged expression levels of the control genes HPRT1 and SDHA. J. Mol. Diagn. 2005, 7, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Wrobel, J.K.; Najafi, S.; Ayhan, S.; Gatzweiler, C.; Krunic, D.; Ridinger, J.; Milde, T.; Westermann, F.; Peterziel, H.; Meder, B.; et al. Rapid In Vivo Validation of HDAC Inhibitor-Based Treatments in Neuroblastoma Zebrafish Xenografts. Pharmaceuticals 2020, 13, 345. [Google Scholar] [CrossRef] [PubMed]
- Fior, R.; Povoa, V.; Mendes, R.V.; Carvalho, T.; Gomes, A.; Figueiredo, N.; Ferreira, M.G. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc. Natl. Acad. Sci. USA 2017, 114, E8234–E8243. [Google Scholar] [CrossRef] [Green Version]
- Wertman, J.; Veinotte, C.J.; Dellaire, G.; Berman, J.N. The Zebrafish Xenograft Platform: Evolution of a Novel Cancer Model and Preclinical Screening Tool. Adv. Exp. Med. Biol. 2016, 916, 289–314. [Google Scholar] [CrossRef]
- Wickham, H. Programming with ggplot2. In ggplot2: Elegant Graphics for Data Analysis; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Tweddle, D.A.; Malcolm, A.J.; Bown, N.; Pearson, A.D.; Lunec, J. Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res. 2001, 61, 8–13. [Google Scholar] [PubMed]
- Gogolin, S.; Ehemann, V.; Becker, G.; Brueckner, L.M.; Dreidax, D.; Bannert, S.; Nolte, I.; Savelyeva, L.; Bell, E.; Westermann, F. CDK4 inhibition restores G(1)-S arrest in MYCN-amplified neuroblastoma cells in the context of doxorubicin-induced DNA damage. Cell Cycle 2013, 12, 1091–1104. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Naiditch, J.; Czurylo, M.; Jie, C.; Lautz, T.; Clark, S.; Jafari, N.; Qiu, Y.; Chu, F.; Madonna, M.B. Differential effect of long-term drug selection with doxorubicin and vorinostat on neuroblastoma cells with cancer stem cell characteristics. Cell Death Dis. 2013, 4, e740. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Vinod, V.; Padmakrishnan, C.J.; Vijayan, B.; Gopala, S. ‘How can I halt thee?’ The puzzles involved in autophagic inhibition. Pharmacol. Res. 2014, 82, 1–8. [Google Scholar] [CrossRef]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef] [Green Version]
- Lopez, G.; Torres, K.; Liu, J.; Hernandez, B.; Young, E.; Belousov, R.; Bolshakov, S.; Lazar, A.J.; Slopis, J.M.; McCutcheon, I.E.; et al. Autophagic survival in resistance to histone deacetylase inhibitors: Novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res. 2011, 71, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.H.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.M. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [Green Version]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebecca, V.W.; Amaravadi, R.K. Emerging strategies to effectively target autophagy in cancer. Oncogene 2016, 35, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belounis, A.; Nyalendo, C.; Le Gall, R.; Imbriglio, T.V.; Mahma, M.; Teira, P.; Beaunoyer, M.; Cournoyer, S.; Haddad, E.; Vassal, G.; et al. Autophagy is associated with chemoresistance in neuroblastoma. BMC Cancer 2016, 16, 891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Invest. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [Green Version]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Oehme, I.; Linke, J.P.; Bock, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [Green Version]
- Ridinger, J.; Koeneke, E.; Kolbinger, F.R.; Koerholz, K.; Mahboobi, S.; Hellweg, L.; Gunkel, N.; Miller, A.K.; Peterziel, H.; Schmezer, P.; et al. Dual role of HDAC10 in lysosomal exocytosis and DNA repair promotes neuroblastoma chemoresistance. Sci. Rep. 2018, 8, 10039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Adachi, S.; Matsubara, H.; Imai, T.; Yui, Y.; Mizushima, Y.; Hiraumi, Y.; Watanabe, K.; Kamitsuji, Y.; Toyokuni, S.Y.; et al. Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int. J. Cancer 2009, 124, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Zatloukal, K. Molecular mechanism leading to SAHA-induced autophagy in tumor cells: Evidence for a p53-dependent pathway. Cancer Cell Int. 2016, 16, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, R.; Lei, Y.; Wang, K.; Lau, Q.C.; Xie, N.; Zhou, S.; Nie, C.; Chen, L.; Wei, Y.; et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells. Autophagy 2010, 6, 711–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giacomo, V.; Di Valerio, V.; Rapino, M.; Bosco, D.; Cacciatore, I.; Ciulla, M.; Marrazzo, A.; Fiorito, J.; Di Stefano, A.; Cataldi, A. MRJF4, a novel histone deacetylase inhibitor, induces p21 mediated autophagy in PC3 prostate cancer cells. Cell Mol. Biol. (Noisy-le-Grand) 2015, 61, 17–23. [Google Scholar]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audesse, A.J.; Dhakal, S.; Hassell, L.A.; Gardell, Z.; Nemtsova, Y.; Webb, A.E. FOXO3 directly regulates an autophagy network to functionally regulate proteostasis in adult neural stem cells. PLoS Genet. 2019, 15, e1008097. [Google Scholar] [CrossRef] [PubMed]
- Fullgrabe, J.; Ghislat, G.; Cho, D.H.; Rubinsztein, D.C. Transcriptional regulation of mammalian autophagy at a glance. J. Cell Sci. 2016, 129, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Diab, H.I.; Lishu, L.; Jeong, A.L.; Patange, S.; Raben, N.; Puertollano, R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 2014, 7, ra9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, Y.; Liu, K.W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrakovcic, M.; Frohlich, L.F. Molecular Determinants of Cancer Therapy Resistance to HDAC Inhibitor-Induced Autophagy. Cancers 2019, 12, 109. [Google Scholar] [CrossRef] [Green Version]
- Stankov, M.V.; El Khatib, M.; Kumar Thakur, B.; Heitmann, K.; Panayotova-Dimitrova, D.; Schoening, J.; Bourquin, J.P.; Schweitzer, N.; Leverkus, M.; Welte, K.; et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia 2014, 28, 577–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khoury, V.; Pierson, S.; Szwarcbart, E.; Brons, N.H.; Roland, O.; Cherrier-De Wilde, S.; Plawny, L.; Van Dyck, E.; Berchem, G. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B-cell chronic lymphocytic leukemia. Leukemia 2014, 28, 1636–1646. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Körholz, K.; Ridinger, J.; Krunic, D.; Najafi, S.; Gerloff, X.F.; Frese, K.; Meder, B.; Peterziel, H.; Vega-Rubin-de-Celis, S.; Witt, O.; et al. Broad-Spectrum HDAC Inhibitors Promote Autophagy through FOXO Transcription Factors in Neuroblastoma. Cells 2021, 10, 1001. https://doi.org/10.3390/cells10051001
Körholz K, Ridinger J, Krunic D, Najafi S, Gerloff XF, Frese K, Meder B, Peterziel H, Vega-Rubin-de-Celis S, Witt O, et al. Broad-Spectrum HDAC Inhibitors Promote Autophagy through FOXO Transcription Factors in Neuroblastoma. Cells. 2021; 10(5):1001. https://doi.org/10.3390/cells10051001
Chicago/Turabian StyleKörholz, Katharina, Johannes Ridinger, Damir Krunic, Sara Najafi, Xenia F. Gerloff, Karen Frese, Benjamin Meder, Heike Peterziel, Silvia Vega-Rubin-de-Celis, Olaf Witt, and et al. 2021. "Broad-Spectrum HDAC Inhibitors Promote Autophagy through FOXO Transcription Factors in Neuroblastoma" Cells 10, no. 5: 1001. https://doi.org/10.3390/cells10051001