
Pre-Clinical Evaluation of the Proteasome Inhibitor Ixazomib against Bortezomib-Resistant Leukemia Cells and Primary Acute Leukemia Cells

 ,
,
Abstract
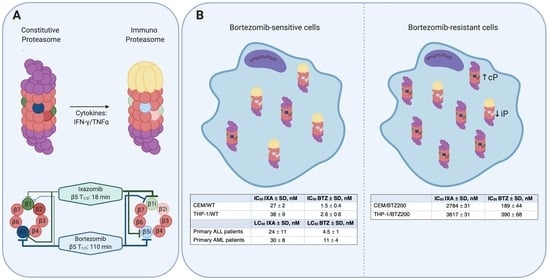
:
1. Introduction
2. Materials and Methods
2.1. Drugs and Materials
2.2. Cell Culture
2.3. Patient Samples
2.4. Proteasome (Subunit) Activity Assay
2.5. Cell Growth Inhibition Assay
2.6. Apoptosis and Cell Cycle Assays (Flow Cytometry)
2.7. Statistics
3. Results
3.1. Mechanism of Action of IXA and BTZ: Subunit Inhibition Profile in (BTZ-Resistant) Leukemia Cell Lines
3.2. Sensitivity of BTZ-Sensitive and BTZ-Resistant ALL, AML, and MM Cell Lines to IXA
3.3. IXA-Induced Apoptosis and Effects on Cell Cycle in Leukemia Cells
3.4. Combination Effects of IXA with DEX and Ara-C
3.5. Sensitivity of Primary ALL and AML Samples to IXA vs. BTZ
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Society, A.C. American Cancer Society. Cancer Facts & Figures 2020. Am. Cancer Soc. J. 2020, 1–52. [Google Scholar]
- Pui, C.H.; Pei, D.; Campana, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Coustan-Smith, E.; Jeha, S.; et al. Improved prognosis for older adolescents with acute lymphoblastic leukemia. J. Clin. Oncol. 2011, 29, 386–391. [Google Scholar] [CrossRef]
- Pui, C.H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006, 354, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Kaspers, G.J. Pediatric acute myeloid leukemia. Expert Rev. Anticancer Ther. 2012, 12, 405–413. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Inaba, H. Childhood acute myeloid leukaemia. Br. J. Haematol. 2012, 159, 259–276. [Google Scholar] [CrossRef]
- Cloos, J.; Roeten, M.S.F.; Franke, N.E.; Van Meerloo, J.; Zweegman, S.; Kaspers, G.J.L.; Jansen, G. (Immuno) proteasomes as therapeutic target in acute leukemia. Cancer Metastasis Rev. 2017, 36, 599–615. [Google Scholar] [CrossRef]
- Aguiar, P.M.; de Mendonça Lima, T.; Colleoni, G.W.B.; Storpirtis, S. Efficacy and safety of bortezomib, thalidomide, and lenalidomide in multiple myeloma: An overview of systematic reviews with meta-analyses. Crit. Rev. Oncol. Hematol. 2017, 113, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Cengiz Seval, G.; Beksac, M. The safety of bortezomib for the treatment of multiple myeloma. Expert Opin. Drug Saf. 2018, 17, 953–962. [Google Scholar] [CrossRef]
- Gavriatopoulou, M.; Musto, P.; Caers, J.; Merlini, G.; Kastritis, E.; van de Donk, N.; Gay, F.; Hegenbart, U.; Hajek, R.; Zweegman, S.; et al. European myeloma network recommendations on diagnosis and management of patients with rare plasma cell dyscrasias. Leukemia 2018, 32, 1883–1898. [Google Scholar] [CrossRef]
- Robak, P.; Robak, T. Bortezomib for the Treatment of Hematologic Malignancies: 15 Years Later. Drugs R&D 2019, 19, 73–92. [Google Scholar] [CrossRef] [Green Version]
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharmacol. 2018, 81, 227–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewerth, D.; Dingjan, I.; Cloos, J.; Jansen, G.; Kaspers, G. Proteasome inhibitors in acute leukemia. Expert Rev. Anticancer Ther. 2013, 13, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Schnerch, D.; Schüler, J.; Follo, M.; Felthaus, J.; Wider, D.; Klingner, K.; Greil, C.; Duyster, J.; Engelhardt, M.; Wäsch, R. Proteasome inhibition enhances the efficacy of volasertib-induced mitotic arrest in AML in vitro and prolongs survival in vivo. Oncotarget 2017, 8, 21153–21166. [Google Scholar] [CrossRef] [Green Version]
- Messinger, Y.H.; Gaynon, P.S.; Sposto, R.; van der, G.J.; Eckroth, E.; Malvar, J.; Bostrom, B.C. Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood 2012, 120, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messinger, Y.; Gaynon, P.; Raetz, E.; Hutchinson, R.; Dubois, S.; Glade-Bender, J.; Sposto, R.; Van der, G.J.; Eckroth, E.; Bostrom, B.C. Phase I study of bortezomib combined with chemotherapy in children with relapsed childhood acute lymphoblastic leukemia (ALL): A report from the therapeutic advances in childhood leukemia (TACL) consortium. Pediatr. Blood Cancer 2010, 55, 254–259. [Google Scholar] [CrossRef] [Green Version]
- August, K.J.; Guest, E.M.; Lewing, K.; Hays, J.A.; Gamis, A.S. Treatment of children with relapsed and refractory acute lymphoblastic leukemia with mitoxantrone, vincristine, pegaspargase, dexamethasone, and bortezomib. Pediatr. Blood Cancer 2020, 67, e28062. [Google Scholar] [CrossRef] [PubMed]
- Colunga-Pedraza, J.E.; González-Llano, O.; González-Martinez, C.E.; Gómez-Almaguer, D.; Yáñez-Reyes, J.M.; Jiménez-Antolinez, V.; Colunga-Pedraza, P.R. Outpatient low toxic regimen with bortezomib in relapsed/refractory acute lymphoblastic leukemia in pediatrics and AYA patients: Single-center Mexican experience. Pediatr. Blood Cancer 2020, 67, e28241. [Google Scholar] [CrossRef]
- Niewerth, D.; Franke, N.E.; Jansen, G.; Assaraf, Y.G.; van Meerloo, J.; Kirk, C.J.; Degenhardt, J.; Anderl, J.; Schimmer, A.D.; Zweegman, S.; et al. Higher ratio immune versus constitutive proteasome level as novel indicator of sensitivity of pediatric acute leukemia cells to proteasome inhibitors. Haematologica 2013, 98, 1896–1904. [Google Scholar] [CrossRef]
- Horton, T.M.; Whitlock, J.A.; Lu, X.; O’Brien, M.M.; Borowitz, M.J.; Devidas, M.; Raetz, E.A.; Brown, P.A.; Carroll, W.L.; Hunger, S.P. Bortezomib reinduction chemotherapy in high-risk ALL in first relapse: A report from the Children’s Oncology Group. Br. J. Haematol. 2019, 186, 274–285. [Google Scholar] [CrossRef]
- Aplenc, R.; Meshinchi, S.; Sung, L.; Alonzo, T.; Choi, J.; Fisher, B.; Gerbing, R.; Hirsch, B.; Horton, T.; Khawash, S.; et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: A report from the Children’s Oncology Group. Haematologica 2020, 105, 1879–1886. [Google Scholar] [CrossRef]
- Niewerth, D.; Jansen, G.; Assaraf, Y.G.; Zweegman, S.; Kaspers, G.J.L.; Cloos, J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist. Updates 2015, 18, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Barrio, S.; Stühmer, T.; Da-Viá, M.; Barrio-Garcia, C.; Lehners, N.; Besse, A.; Cuenca, I.; Garitano-Trojaola, A.; Fink, S.; Leich, E.; et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2019, 33, 447–456. [Google Scholar] [CrossRef]
- Lee, M.J.; Bhattarai, D.; Yoo, J.; Miller, Z.; Park, J.E.; Lee, S.; Lee, W.; Driscoll, J.J.; Kim, K.B. Development of Novel Epoxyketone-Based Proteasome Inhibitors as a Strategy to Overcome Cancer Resistance to Carfilzomib and Bortezomib. J. Med. Chem. 2019, 62, 4444–4455. [Google Scholar] [CrossRef]
- Park, J.E.; Miller, Z.; Jun, Y.; Lee, W.; Kim, K.B. Next-generation proteasome inhibitors for cancer therapy. Transl. Res. 2018, 198, 1–16. [Google Scholar] [CrossRef]
- Kupperman, E.; Lee, E.C.; Cao, Y.; Bannerman, B.; Fitzgerald, M.; Berger, A.; Yu, J.; Yang, Y.; Hales, P.; Bruzzese, F.; et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010, 70, 1970–1980. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.G.; Baz, R.; Wang, M.; Jakubowiak, A.J.; Laubach, J.P.; Harvey, R.D.; Talpaz, M.; Berg, D.; Liu, G.; Yu, J.; et al. Phase 1 study of twice-weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood 2014, 124, 1038–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attar, E.C.; De Angelo, D.J.; Supko, J.G.; D’Amato, F.; Zahrieh, D.; Sirulnik, A.; Wadleigh, M.; Ballen, K.K.; McAfee, S.; Miller, K.B.; et al. Phase I and pharmacokinetic study of bortezomib in combination with idarubicin and cytarabine in patients with acute myelogenous leukemia. Clin. Cancer Res. 2008, 14, 1446–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolewski, P.; Rydygier, D. Ixazomib: An investigational drug for the treatment of lymphoproliferative disorders. Expert Opin. Investig. Drugs 2019, 28, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin. Cancer Res. 2011, 17, 5311–5321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teicher, B.A.; Tomaszewski, J.E. Proteasome inhibitors. Biochem. Pharmacol. 2015, 96, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Hanley, M.J.; Xia, C.; Labotka, R.; Harvey, R.D.; Venkatakrishnan, K. Clinical Pharmacology of Ixazomib: The First Oral Proteasome Inhibitor. Clin. Pharmacokinet. 2019, 58, 431–449. [Google Scholar] [CrossRef]
- Richardson, P.G.; Zweegman, S.; O’Donnell, E.K.; Laubach, J.P.; Raje, N.; Voorhees, P.; Ferrari, R.H.; Skacel, T.; Kumar, S.K.; Lonial, S. Ixazomib for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2018, 19, 1949–1968. [Google Scholar] [CrossRef]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; Ghazarian, R.N.; Ou, M.; Luderer, M.J.; Kusdono, H.D.; Azab, A.K. Spotlight on ixazomib: Potential in the treatment of multiple myeloma. Drug Des. Devel. Ther. 2016, 10, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.G.; Kumar, S.; Laubach, J.P.; Paba-Prada, C.; Gupta, N.; Berg, D.; van de Velde, H.; Moreau, P. New developments in the management of relapsed/refractory multiple myeloma—The role of ixazomib. J. Blood Med. 2017, 8, 107–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Laplant, B.; Reeder, C.; Roy, V.; Buadi, F.; Gertz, M.; Laumann, K.; Bergsagel, P.; Dispenzieri, A.; Kapoor, P.; et al. Randomized phase 2 trial of two different doses of ixazomib in patients with relapsed multiple myeloma not refractory to bortezomib. Blood 2015, 126, 3050. [Google Scholar] [CrossRef]
- Suarez-Kelly, L.P.; Kemper, G.M.; Duggan, M.C.; Stiff, A.; Noel, T.C.; Markowitz, J.; Luedke, E.A.; Yildiz, V.O.; Yu, L.; Jaime-Ramirez, A.C.; et al. The combination of MLN2238 (ixazomib) with interferon-alpha results in enhanced cell death in melanoma. Oncotarget 2016, 7, 81172–81186. [Google Scholar] [CrossRef] [Green Version]
- Advani, A.S.; Cooper, B.; Visconte, V.; Elson, P.; Chan, R.; Carew, J.; Wei, W.; Mukherjee, S.; Gerds, A.; Carraway, H.; et al. A phase I/II trial of MEC (mitoxantrone, etoposide, cytarabine) in combination with ixazomib for relapsed refractory acute myeloid leukemia. Clin. Cancer Res. 2019, 25, 4231–4237. [Google Scholar] [CrossRef] [Green Version]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef] [Green Version]
- Franke, N.E.; Niewerth, D.; Assaraf, Y.G.; van Meerloo, J.; Vojtekova, K.; van Zantwijk, C.H.; Zweegman, S.; Chan, E.T.; Kirk, C.J.; Geerke, D.P.; et al. Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012, 26, 757–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewerth, D.; Kaspers, G.J.L.; Assaraf, Y.G.; van Meerloo, J.; Kirk, C.J.; Anderl, J.; Blank, J.L.; van de Ven, P.M.; Zweegman, S.; Jansen, G.; et al. Interferon-γ-induced upregulation of immunoproteasome subunit assembly overcomes bortezomib resistance in human hematological cell lines. J. Hematol. Oncol. 2014, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- van Meerloo, J.; Kaspers, G.J.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Bijnsdorp, I.V.; Giovannetti, E.; Peters, G.J. Analysis of drug interactions. Methods Mol. Biol. 2011, 731, 421–434. [Google Scholar] [CrossRef]
- Zweegman, S.; Stege, C.A.M.; Haukas, E.; Schjesvold, F.H.; Levin, M.-D.; Waage, A.; Leys, R.B.L.; Klein, S.K.; Szatkowski, D.; Axelsson, P.; et al. Ixazomib-Thalidomide-low dose dexamethasone induction followed by maintenance therapy with ixazomib or placebo in newly diagnosed multiple myeloma patients not eligible for autologous stem cell transplantation; results from the randomized phase II HOVON-1. Haematologica 2020, 105, 2879–2882. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibaudeau, T.A.; Smith, D.M. A practical review of proteasome pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, S.; Cai, C.Y.; Assaraf, Y.G.; Guo, H.Q.; Cui, Q.; Wei, L.; Huang, J.J.; Ashby, C.R.; Chen, Z.S. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resist. Updates 2020, 48, 100663. [Google Scholar] [CrossRef]
- Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 1994, 79, 13–21. [Google Scholar] [CrossRef]
- Parlati, F.; Lee, S.J.; Aujay, M.; Suzuki, E.; Levitsky, K.; Lorens, J.B.; Micklem, D.R.; Ruurs, P.; Sylvain, C.; Lu, Y.; et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009, 114, 3439–3447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewerth, D.; Kaspers, G.J.; Jansen, G.; van Meerloo, J.; Zweegman, S.; Jenkins, G.; Whitlock, J.A.; Hunger, S.P.; Lu, X.; Alonzo, T.A.; et al. Proteasome subunit expression analysis and chemosensitivity in relapsed paediatric acute leukaemia patients receiving bortezomib-containing chemotherapy. J. Hematol. Oncol. 2016, 9, 1756–8722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewerth, D.; van Meerloo, J.; Jansen, G.; Assaraf, Y.G.; Hendrickx, T.C.; Kirk, C.J.; Anderl, J.L.; Zweegman, S.; Kaspers, G.J.L.; Cloos, J. Anti-leukemic activity and mechanisms underlying resistance to the novel immunoproteasome inhibitor PR-924. Biochem. Pharmacol. 2014, 89, 43–51. [Google Scholar] [CrossRef]
- Garcia, J.S.; Huang, M.; Medeiros, B.C.; Mitchell, B.S. Selective Toxicity of Investigational Ixazomib for Human Leukemia Cells Expressing Mutant Cytoplasmic NPM1: Role of Reactive Oxygen Species. Clin. Cancer Res. 2016, 22, 1978–1988. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Halasi, M.; Patel, A.; Schultz, R.; Kalakota, N.; Chen, Y.H.; Aardsma, N.; Liu, L.; Crispino, J.D.; Mahmud, N.; et al. FOXM1 contributes to treatment failure in acute myeloid leukemia. JCI Insight 2018, 3, e121583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsas, P.; Galan-Malo, P.; Marzo, I.; Naval, J. Bortezomib resistance in a myeloma cell line is associated to PSMβ5 overexpression and polyploidy. Leuk. Res. 2012, 36, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Ri, M.; Iida, S.; Nakashima, T.; Miyazaki, H.; Mori, F.; Ito, A.; Inagaki, A.; Kusumoto, S.; Ishida, T.; Komatsu, H.; et al. Bortezomib-resistant myeloma cell lines: A role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010, 24, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Yang, J.; Song, X.; Gong, S.; Zhou, H.; Guo, L.; Song, N.; Bao, X.; Chen, P.; Wang, J. Point mutation of the proteasome beta5 subunit gene is an important mechanism of bortezomib resistance in bortezomib-selected variants of Jurkat T cell lymphoblastic lymphoma/leukemia line. J. Pharmacol. Exp. Ther. 2008, 326, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wilt, L.H.; Jansen, G.; Assaraf, Y.G.; Van, M.J.; Cloos, J.; Schimmer, A.D.; Chan, E.T.; Kirk, C.J.; Peters, G.J.; Kruyt, F.A. Proteasome-based mechanisms of intrinsic and acquired bortezomib resistance in non-small cell lung cancer. Biochem. Pharmacol. 2012, 83, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Kale, A.J.; Moore, B.S. Molecular Mechanisms of Acquired Proteasome Inhibitor Resistance. J. Med. Chem. 2012, 55, 10317–10327. [Google Scholar] [CrossRef]
- Ruckrich, T.; Kraus, M.; Gogel, J.; Beck, A.; Ovaa, H.; Verdoes, M.; Overkleeft, H.S.; Kalbacher, H.; Driessen, C. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia 2009, 23, 1098–1105. [Google Scholar] [CrossRef] [Green Version]
- Brünnert, D.; Kraus, M.; Stühmer, T.; Kirner, S.; Heiden, R.; Goyal, P.; Driessen, C.; Bargou, R.C.; Chatterjee, M. Novel cell line models to study mechanisms and overcoming strategies of proteasome inhibitor resistance in multiple myeloma. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1666–1676. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; den Dulk, H.; Overkleeft, H.S.; et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tundo, G.R.; Sbardella, D.; Santoro, A.M.; Coletta, A.; Oddone, F.; Grasso, G.; Milardi, D.; Lacal, P.M.; Marini, S.; Purrello, R.; et al. The proteasome as a druggable target with multiple therapeutic potentialities: Cutting and non-cutting edges. Pharmacol. Ther. 2020, 213, 107579. [Google Scholar] [CrossRef]
- Zaal, E.A.; Wu, W.; Jansen, G.; Zweegman, S.; Cloos, J.; Berkers, C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017, 5, 7. [Google Scholar] [CrossRef]
- Gonzalez-Santamarta, M.; Quinet, G.; Reyes-Garau, D.; Sola, B.; Roué, G.; Rodriguez, M.S. Resistance to the Proteasome Inhibitors: Lessons from Multiple Myeloma and Mantle Cell Lymphoma. In Proteostasis and Disease: From Basic Mechanisms to Clinics; Barrio, R., Sutherland, J.D., Rodriguez, M.S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 153–174. ISBN 978-3-030-38266-7. [Google Scholar]
- Franke, N.E.; Kaspers, G.L.; Assaraf, Y.G.; van Meerloo, J.; Niewerth, D.; Kessler, F.L.; Poddighe, P.J.; Kole, J.; Smeets, S.J.; Ylstra, B.; et al. Exocytosis of polyubiquitinated proteins in bortezomib-resistant leukemia cells: A role for MARCKS in acquired resistance to proteasome inhibitors. Oncotarget 2016, 7, 74779–74796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbrugge, S.E.; Assaraf, Y.G.; Dijkmans, B.A.; Scheffer, G.L.; Al, M.; Den Uyl, D.; Oerlemans, R.; Chan, E.T.; Kirk, C.J.; Peters, G.J.; et al. Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: Ex vivo efficacy of (immuno)proteasome inhibitors in mononuclear blood cells from patients with. J. Pharmacol. Exp. Ther. 2012, 341, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Moreau, P.; Karamanesht, I.I.; Domnikova, N.; Kyselyova, M.Y.; Vilchevska, K.V.; Doronin, V.A.; Schmidt, A.; Hulin, C.; Leleu, X.; Esseltine, D.L.; et al. Pharmacokinetic, pharmacodynamic and covariate analysis of subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma. Clin. Pharmacokinet. 2012, 51, 823–829. [Google Scholar] [CrossRef]
- Gupta, N.; Zhao, Y.; Hui, A.M.; Esseltine, D.L.; Venkatakrishnan, K. Switching from body surface area-based to fixed dosing for the investigational proteasome inhibitor ixazomib: A population pharmacokinetic analysis. Br. J. Clin. Pharmacol. 2015, 79, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.-Y.; Zhang, Y.-P.; Zhang, J.; Zhang, S.-B.; Li, D.; Huang, Z.-Z.; Xin, W.-J. The possible involvement of JNK activation in the spinal dorsal horn in bortezomib-induced allodynia: The role of TNF-α and IL-1β. J. Anesth. 2016, 30, 55–63. [Google Scholar] [CrossRef]
- Xie, J.D.; Chen, S.R.; Chen, H.; Pan, H.L. Bortezomib induces neuropathic pain through protein kinase C-mediated activation of presynaptic NMDA receptors in the spinal cord. Neuropharmacology 2017, 123, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Cavaletti, G.; Bruna, J.; Kyritsis, A.P.; Kalofonos, H.P. Bortezomib-induced peripheral neurotoxicity: An update. Arch. Toxicol. 2014, 88, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Horton, T.M.; Gannavarapu, A.; Blaney, S.M.; D’Argenio, D.Z.; Plon, S.E.; Berg, S.L. Bortezomib interactions with chemotherapy agents in acute leukemia in vitro. Cancer Chemother. Pharmacol. 2006, 58, 13–23. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 IXA ± SD, nM (RF) | IC50 BTZ ± SD, nM (RF) * | |
|---|---|---|
| Wild-type cell lines | ||
| CEM | 27 ± 2 | 1.5 ± 0.4 |
| THP-1 | 38 ± 9 | 2.6 ± 0.6 |
| 8226 # | 22 ± 2 | 2.6 ± 0.3 |
| BTZ low resistance sublines | ||
| CEM/BTZ7 | 291 ± 29 (11) | 12.4 ± 5.8 (10) |
| THP-1/BTZ7 | 277 ± 26 (7) | 70 ± 10 (27) |
| 8226/BTZ7 # | 100 ± 1 (5) | 12.1 ± 0.7 (5) |
| BTZ high resistance sublines | ||
| CEM/BTZ200 | 2784 ± 31 (103) | 189 ± 44 (170) |
| THP-1/BTZ200 | 3817 ± 31 (103) | 390 ± 68 (153) |
| 8226/BTZ100 # | 2332 ± 105 (106) | 106 ± 15 (40) |
| Combination Index DEX | Combination Index Ara-C | |
|---|---|---|
| T-ALL cells | ||
| CEM/WT | 1.22 ± 0.1 | - |
| CEM/BTZ7 | 0.77 ± 0.3 | - |
| CEM/BTZ200 | 1.31 ± 0.2 | - |
| AML cells | ||
| THP-1/WT | - | 2.16 ± 0.3 |
| THP-1/BTZ7 | - | 2.39 ± 1.8 |
| THP-1/BTZ200 | - | 1.62 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roeten, M.S.F.; van Meerloo, J.; Kwidama, Z.J.; ter Huizen, G.; Segerink, W.H.; Zweegman, S.; Kaspers, G.J.L.; Jansen, G.; Cloos, J. Pre-Clinical Evaluation of the Proteasome Inhibitor Ixazomib against Bortezomib-Resistant Leukemia Cells and Primary Acute Leukemia Cells. Cells 2021, 10, 665. https://doi.org/10.3390/cells10030665
Roeten MSF, van Meerloo J, Kwidama ZJ, ter Huizen G, Segerink WH, Zweegman S, Kaspers GJL, Jansen G, Cloos J. Pre-Clinical Evaluation of the Proteasome Inhibitor Ixazomib against Bortezomib-Resistant Leukemia Cells and Primary Acute Leukemia Cells. Cells. 2021; 10(3):665. https://doi.org/10.3390/cells10030665
Chicago/Turabian StyleRoeten, Margot S.F., Johan van Meerloo, Zinia J. Kwidama, Giovanna ter Huizen, Wouter H. Segerink, Sonja Zweegman, Gertjan J.L. Kaspers, Gerrit Jansen, and Jacqueline Cloos. 2021. "Pre-Clinical Evaluation of the Proteasome Inhibitor Ixazomib against Bortezomib-Resistant Leukemia Cells and Primary Acute Leukemia Cells" Cells 10, no. 3: 665. https://doi.org/10.3390/cells10030665