
Cardiac Hypertrophy Changes Compartmentation of cAMP in Non-Raft Membrane Microdomains


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning and Transgenic Mouse Generation
2.2. Transverse Aortic Constriction (TAC)
2.3. Adult Mouse Ventricular Myocyte Isolation and Live Cell Imaging
2.4. Confocal Microscopy
2.5. Histology, Morphometric Analysis, and Echocardiography
2.6. Immunoblot Analysis
2.7. Statistics
3. Results
3.1. E1-CAAX Biosensor Mouse Generation and Characterization
3.2. TAC Model
3.3. Live Cell Imaging of cAMP in Non-Lipid Raft Membrane Domains
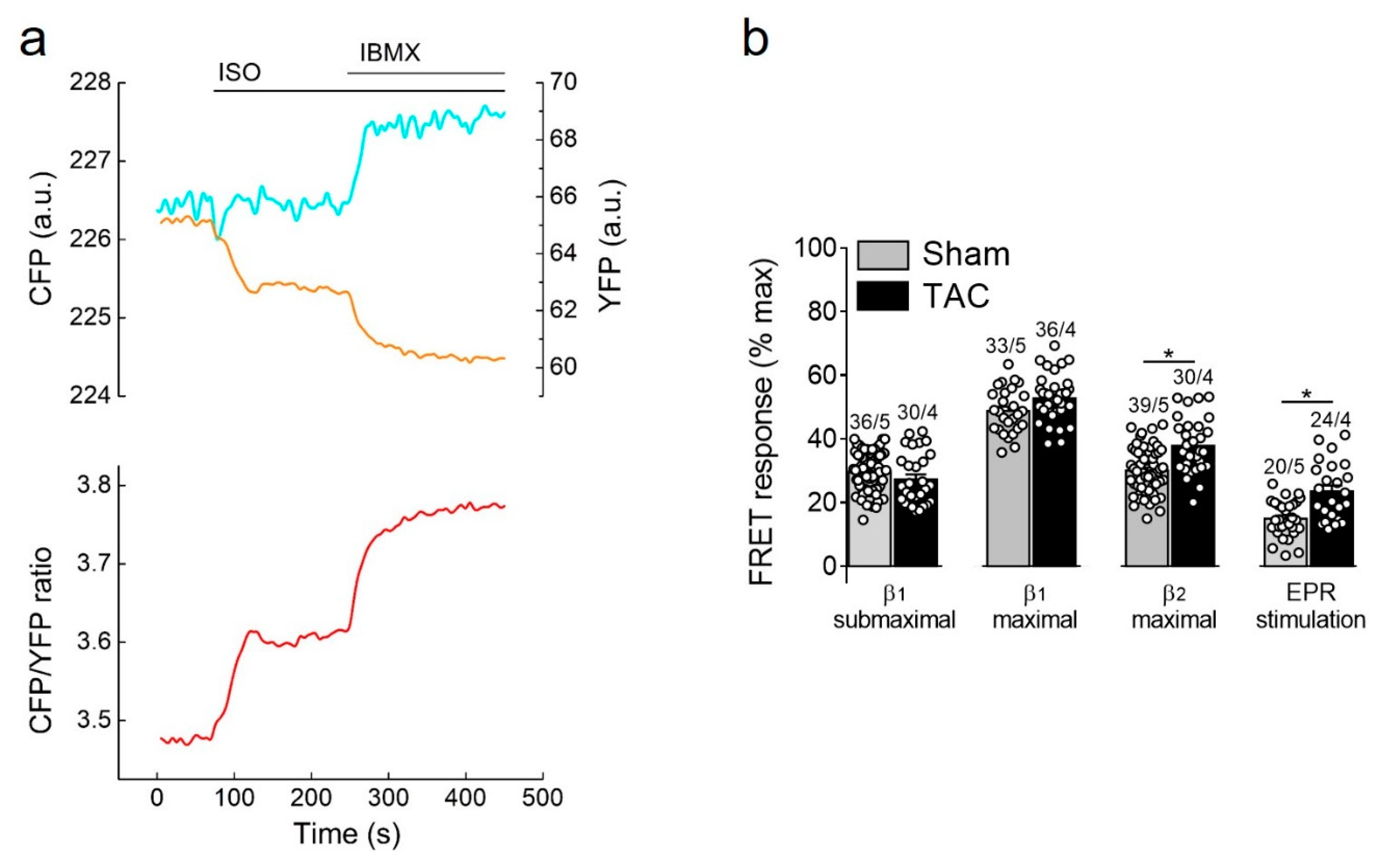
3.3.1. Responses to β-Adrenergic and Prostaglandin Receptor Stimulation
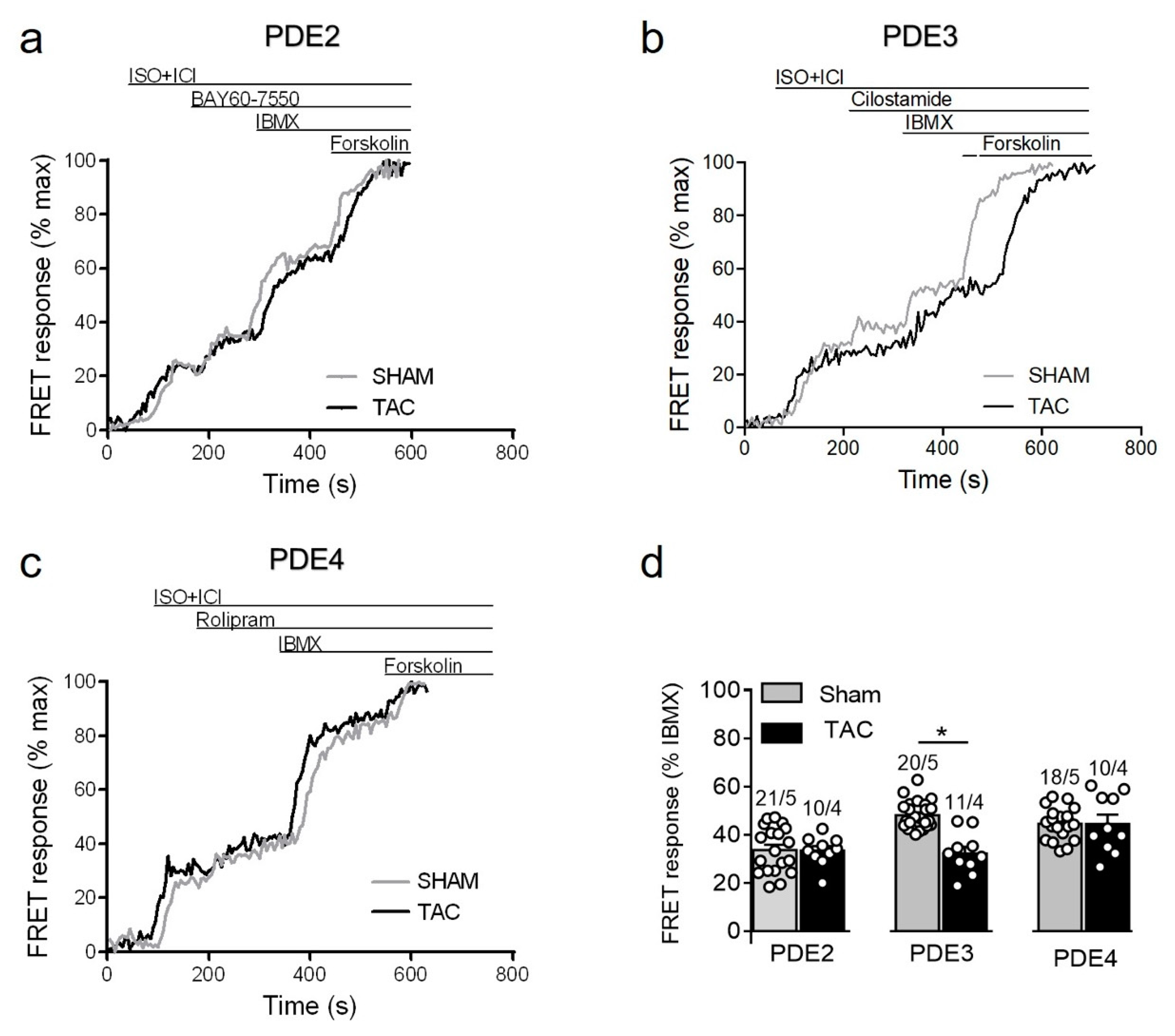
3.3.2. PDE Dependent Regulation of β1-Adrenergic Receptor cAMP Responses
3.3.3. Responses to PDE Inhibitors after β2-Adrenergic Receptor Stimulation
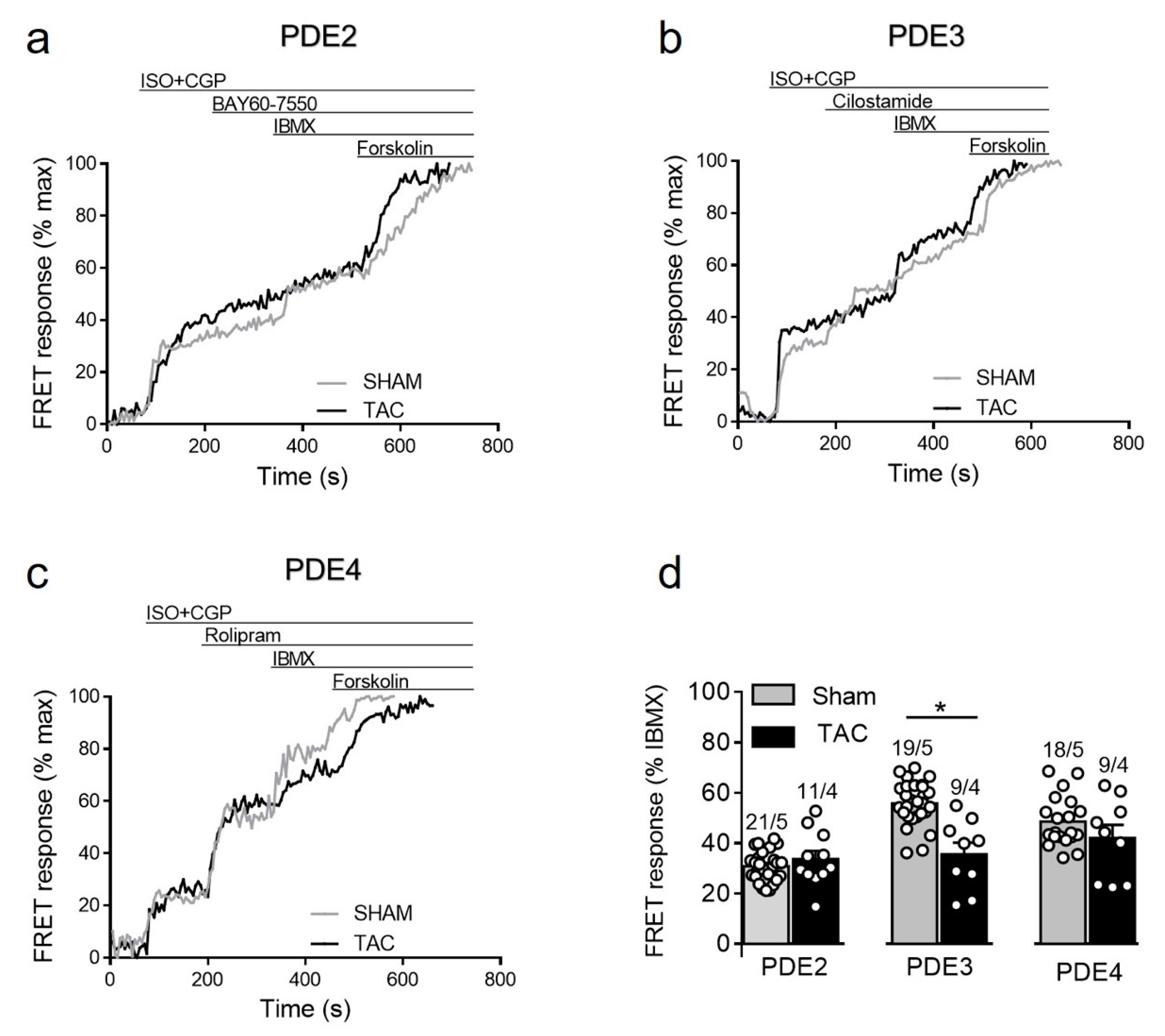
3.3.4. PDE Regulation of Prostaglandin Receptor Responses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brunton, L.L.; Hayes, J.S.; Mayer, S.E. Functional compartmentation of cyclic AMP and protein kinase in heart. Adv. Cycl. Nucleotide Res. 1981, 14, 391–397. [Google Scholar]
- Buxton, I.L.; Brunton, L.L. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem. 1983, 258, 10233–10239. [Google Scholar] [CrossRef]
- Fischmeister, R.; Castro, L.R.; Abi-Gerges, A.; Rochais, F.; Jurevicius, J.; Leroy, J.; Vandecasteele, G. Compartmentation of cyclic nucleotide signaling in the heart: The role of cyclic nucleotide phosphodiesterases. Circ. Res. 2006, 99, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Perera, R.K.; Nikolaev, V.O. Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol. 2013, 207, 650–662. [Google Scholar] [CrossRef]
- Bers, D.M.; Xiang, Y.K.; Zaccolo, M. Whole-Cell cAMP and PKA Activity are Epiphenomena, Nanodomain Signaling Matters. Physiology 2019, 34, 240–249. [Google Scholar] [CrossRef]
- Bastug-Özel, Z.; Wright, P.T.; Kraft, A.E.; Pavlovic, D.; Howie, J.; Froese, A.; Fuller, W.; Gorelik, J.; Shattock, M.J.; Nikolaev, V.O. Heart failure leads to altered beta2-adrenoceptor/cyclic adenosine monophosphate dynamics in the sarcolemmal phospholemman/Na,K ATPase microdomain. Cardiovasc Res. 2019, 115, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, A.; Mika, D. cAMP/PKA signaling compartmentalization in cardiomyocytes: Lessons from FRET-based biosensors. J. Mol. Cell. Cardiol. 2019, 131, 112–121. [Google Scholar] [CrossRef]
- Iancu, R.V.; Ramamurthy, G.; Warrier, S.; Nikolaev, V.O.; Lohse, M.J.; Jones, S.W.; Harvey, R.D. Cytoplasmic cAMP concentrations in intact cardiac myocytes. Am. J. Physiol. Cell Physiol. 2008, 295, C414–C422. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Bünemann, M.; Schmitteckert, E.; Lohse, M.J.; Engelhardt, S. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ. Res. 2006, 99, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef]
- Harvey, R.D.; Calaghan, S.C. Caveolae create local signalling domains through their distinct protein content, lipid profile and morphology. J. Mol. Cell. Cardiol. 2012, 52, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Head, B.P.; Patel, H.H.; Roth, D.M.; Lai, N.C.; Niesman, I.R.; Farquhar, M.G.; Insel, P.A. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J. Biol. Chem. 2005, 280, 31036–31044. [Google Scholar] [CrossRef] [Green Version]
- Insel, P.A.; Head, B.P.; Ostrom, R.S.; Patel, H.H.; Swaney, J.S.; Tang, C.M.; Roth, D.M. Caveolae and lipid rafts: G protein-coupled receptor signaling microdomains in cardiac myocytes. Ann. N. Y. Acad. Sci. 2005, 1047, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.R.; Yang, P.C.; Rice, M.; Singer, C.A.; Nikolaev, V.O.; Lohse, M.J.; Clancy, C.E.; Harvey, R.D. Role of membrane microdomains in compartmentation of cAMP signaling. PLoS ONE 2014, 9, e95835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.R.; Gratwohl, J.; Cozad, M.; Yang, P.C.; Clancy, C.E.; Harvey, R.D. Compartmentalized cAMP Signaling Associated with Lipid Raft and Non-raft Membrane Domains in Adult Ventricular Myocytes. Front. Pharmacol. 2018, 9, 332. [Google Scholar] [CrossRef]
- Perera, R.K.; Sprenger, J.U.; Steinbrecher, J.H.; Hubscher, D.; Lehnart, S.E.; Abesser, M.; Schuh, K.; El-Armouche, A.; Nikolaev, V.O. Microdomain Switch of cGMP-Regulated Phosphodiesterases Leads to ANP-Induced Augmentation of beta-Adrenoceptor-Stimulated Contractility in Early Cardiac Hypertrophy. Circ. Res. 2015, 116, 1304–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaev, V.O.; Bünemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 2004, 279, 37215–37218. [Google Scholar] [CrossRef] [Green Version]
- Zacharias, D.A.; Violin, J.D.; Newton, A.C.; Tsien, R.Y. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 2002, 296, 913–916. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Zhang, D.; Swenson, L.; Chakrabarti, G.; Abel, E.D.; Litwin, S.E. Minimally invasive aortic banding in mice: Effects of altered cardiomyocyte insulin signaling during pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1261–H1269. [Google Scholar] [CrossRef] [Green Version]
- Börner, S.; Schwede, F.; Schlipp, A.; Berisha, F.; Calebiro, D.; Lohse, M.J.; Nikolaev, V.O. FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat. Protoc. 2011, 6, 427–438. [Google Scholar] [CrossRef]
- Sprenger, J.U.; Perera, R.K.; Gotz, K.R.; Nikolaev, V.O. FRET microscopy for real-time monitoring of signaling events in live cells using unimolecular biosensors. J. Vis. Exp. 2012, e4081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, E.; Lauterbach, M.A.; Kohl, T.; Westphal, V.; Williams, G.S.; Steinbrecher, J.H.; Streich, J.H.; Korff, B.; Tuan, H.T.; Hagen, B.; et al. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ. Res. 2012, 111, 402–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V.O. In vivo model with targeted cAMP biosensor reveals changes in receptor-microdomain communication in cardiac disease. Nat. Commun. 2015, 6, 6965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, K.; Schmitt, J.P.; Schmitteckert, E.M.; Lohse, M.J. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat. Med. 2009, 15, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gao, E.; Chan, T.O.; Zhang, X.Q.; Song, J.; Shang, X.; Koch, W.J.; Feldman, A.M.; Cheung, J.Y. Induced overexpression of Na(+)/Ca(2+) exchanger does not aggravate myocardial dysfunction induced by transverse aortic constriction. J. Card Fail. 2013, 19, 60–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xu, J.; Wang, Q.; Brainard, R.E.; Watson, L.J.; Jones, S.P.; Epstein, P.N. Reduced cardiac fructose 2,6 bisphosphate increases hypertrophy and decreases glycolysis following aortic constriction. PLoS ONE 2013, 8, e53951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.R.; Clancy, C.E.; Harvey, R.D. Mechanisms Restricting Diffusion of Intracellular cAMP. Sci. Rep. 2016, 6, 19577. [Google Scholar] [CrossRef]
- Froese, A.; Nikolaev, V.O. Imaging alterations of cardiomyocyte cAMP microdomains in disease. Front. Pharmacol. 2015, 6, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudokas, M.W.; Post, J.P.; Sataray-Rodriguez, A.; Sherpa, R.T.; Moshal, K.S.; Agarwal, S.R.; Harvey, R.D. Compartmentation of beta2 -Adrenergic Receptor Stimulated cAMP Responses by Phosphodiesterases Type 2 and 3 in Cardiac Ventricular Myocytes. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.S.; Brunton, L.L.; Mayer, S.E. Selective activation of particulate cAMP-dependent protein kinase by isoproterenol and prostaglandin E1. J. Biol. Chem. 1980, 255, 5113–5119. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Zoccarato, A.; Lissandron, V.; Terrin, A.; Li, X.; Houslay, M.D.; Baillie, G.S.; Zaccolo, M. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ. Res. 2008, 103, 836–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Three Months Old | Six Months Old | |||
|---|---|---|---|---|
| Parameter | Wild-Type | E1-CAAX | Wild-Type | E1-CAAX |
| IVDd (mm) | 0.98 ± 0.04 | 1.00 ± 0.03 | 1.02 ± 0.04 | 1.03 ± 0.04 |
| LVIDd (mm) | 3.65 ± 0.06 | 3.60 ± 0.08 | 3.54 ± 0.11 | 3.50 ± 0.08 |
| LVPWd (mm) | 0.88 ± 0.04 | 0.96 ± 0.05 | 1.04 ± 0.08 | 1.10 ± 0.06 |
| LV mass/BW (mg/g) | 4.28 ± 0.18 | 4.39 ± 0.121 | 4.23 ± 0.18 | 4.19 ± 0.17 |
| FS (%) | 34.7 ± 0.8 | 36.7 ± 1.2 | 32.9 ± 1.9 | 36.0 ± 1.5 |
| EF (%) | 64.7 ± 1.1 | 67.3 ± 1.7 | 61.9 ± 2.9 | 66.5 ± 2.0 |
| Stroke volume (μL) | 37.3 ± 1.3 | 35.8 ± 1.7 | 33.7 ± 2.9 | 34.6 ± 2.3 |
| Cardiac Output (mL/min) | 16.9 ± 0.8 | 15.5 ± 0.8 | 14.8 ± 1.4 | 15.7 ± 1.2 |
| HR (bpm) | 467.4 ± 8.5 | 455.6 ± 4.3 | 436.4 ± 9.7 | 454.5 ± 18.6 |
| n (number of mice) | 14 | 17 | 9 | 9 |
| Parameter | Sham | TAC |
|---|---|---|
| Pressure gradient (mmHg) | 4.3 ± 0.4 | 63.7 ± 7.6 * |
| IVDd (mm) | 1.01 ± 0.08 | 1.25 ± 0.04 * |
| LVIDd (mm) | 3.10 ± 0.26 | 3.34 ± 0.16 |
| LVPWd (mm) | 1.20 ± 0.13 | 1.35 ± 0.10 |
| LV mass/BW (mg/g) | 4.89± 0.40 | 7.12 ± 0.43 * |
| FS (%) | 43.4 ± 6.2 | 32.5 ± 2.7 |
| EF (%) | 73.6 ± 5.6 | 61.6 ± 4.0 |
| Stroke volume (μL) | 30.2 ± 5.6 | 29.1 ± 3.3 |
| Cardiac Output (mL/min) | 15.8 ± 2.1 | 13.6 ± 1.7 |
| HR (bpm) | 463.8 ± 21.8 | 466.5 ± 6.9 |
| n (number of mice) | 5 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlaki, N.; De Jong, K.A.; Geertz, B.; Nikolaev, V.O.; Froese, A. Cardiac Hypertrophy Changes Compartmentation of cAMP in Non-Raft Membrane Microdomains. Cells 2021, 10, 535. https://doi.org/10.3390/cells10030535
Pavlaki N, De Jong KA, Geertz B, Nikolaev VO, Froese A. Cardiac Hypertrophy Changes Compartmentation of cAMP in Non-Raft Membrane Microdomains. Cells. 2021; 10(3):535. https://doi.org/10.3390/cells10030535
Chicago/Turabian StylePavlaki, Nikoleta, Kirstie A. De Jong, Birgit Geertz, Viacheslav O. Nikolaev, and Alexander Froese. 2021. "Cardiac Hypertrophy Changes Compartmentation of cAMP in Non-Raft Membrane Microdomains" Cells 10, no. 3: 535. https://doi.org/10.3390/cells10030535