
Epithelial Cells and Inflammation in Pulmonary Wound Repair


Abstract
:1. Epithelial Roles in Tissue Repair
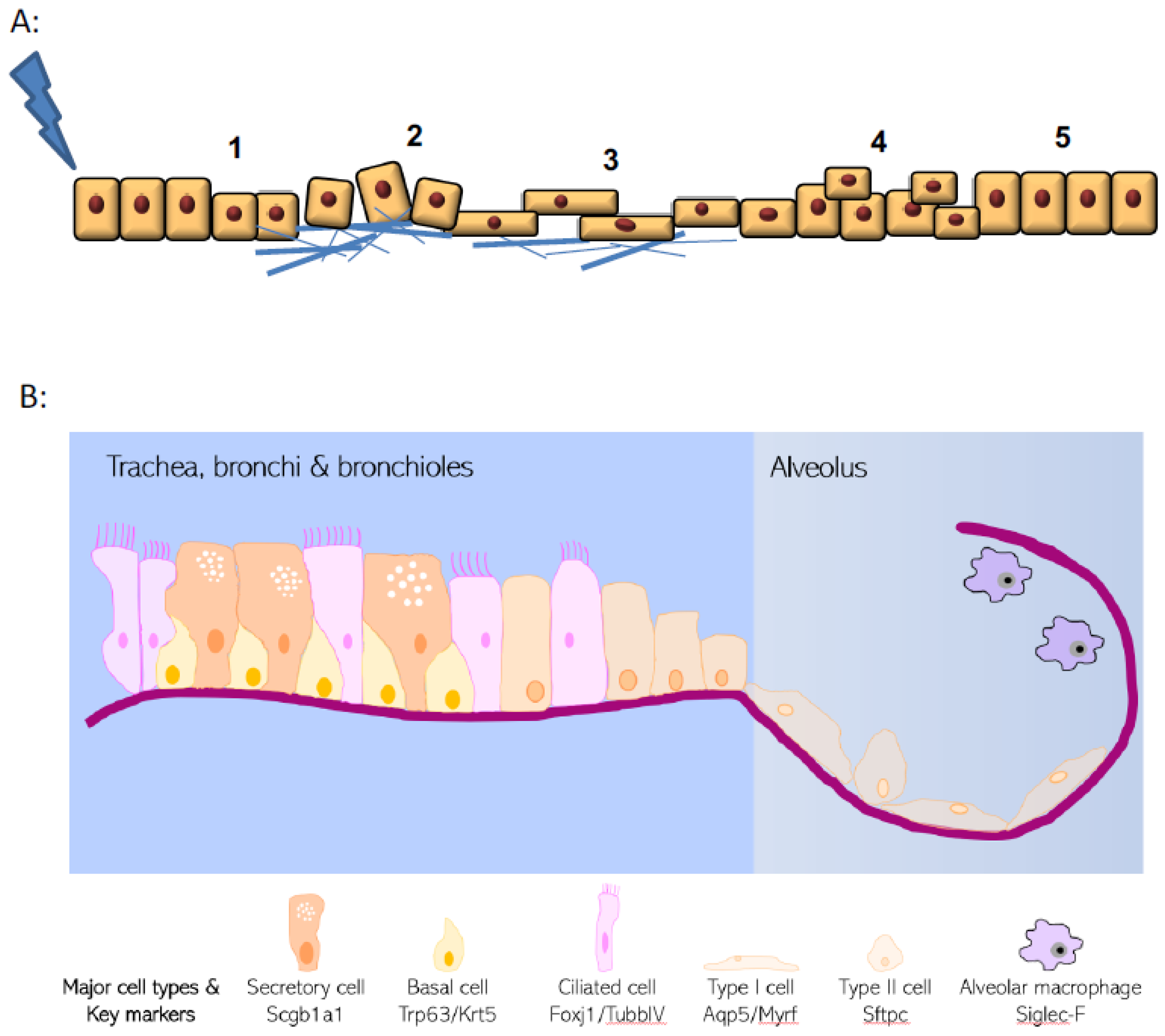
2. Epithelial Structure and Evolving Knowledge on Progenitor Populations
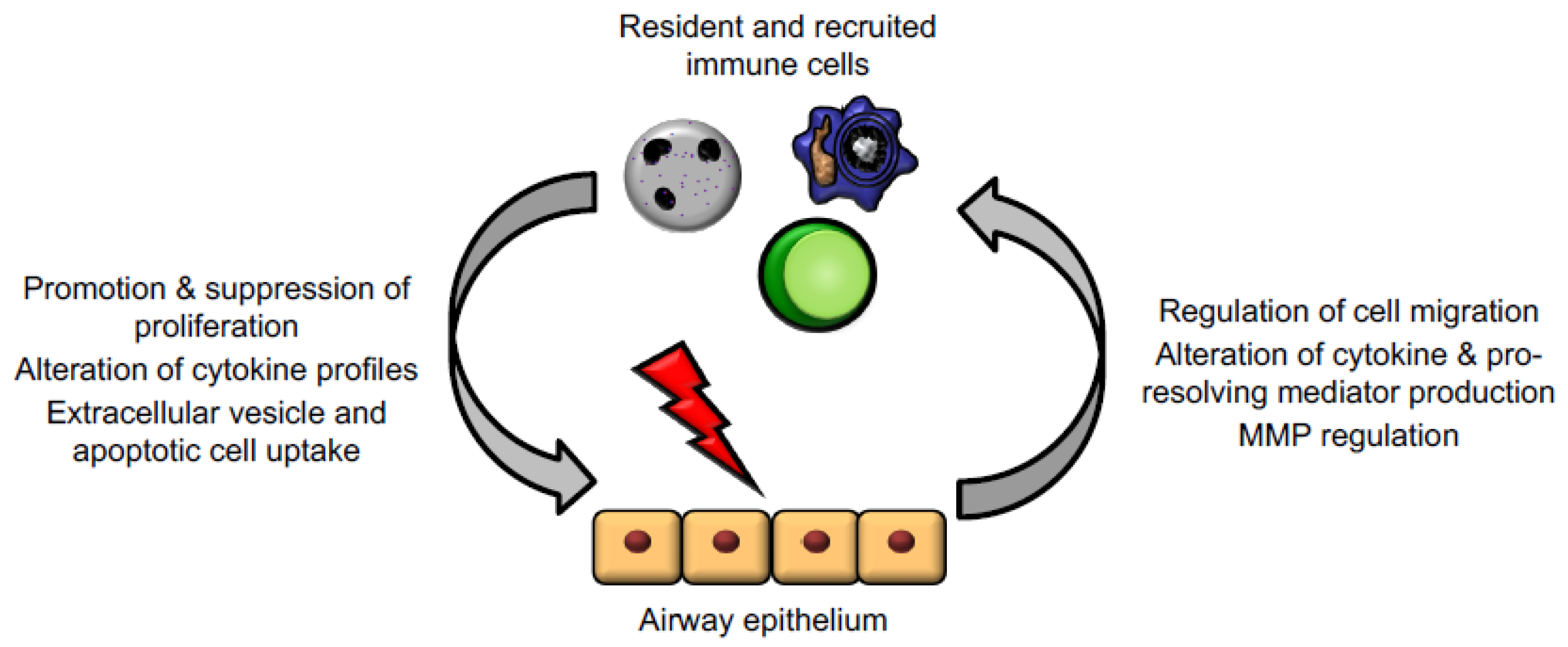
3. Epithelial Cell–Immune Cell Crosstalk
4. Infection Influence on Tissue Repair
5. Mechanisms for Promoting Tissue Repair
5.1. Growth Factors
5.2. Soluble Lipid Mediators
5.3. Cytokines
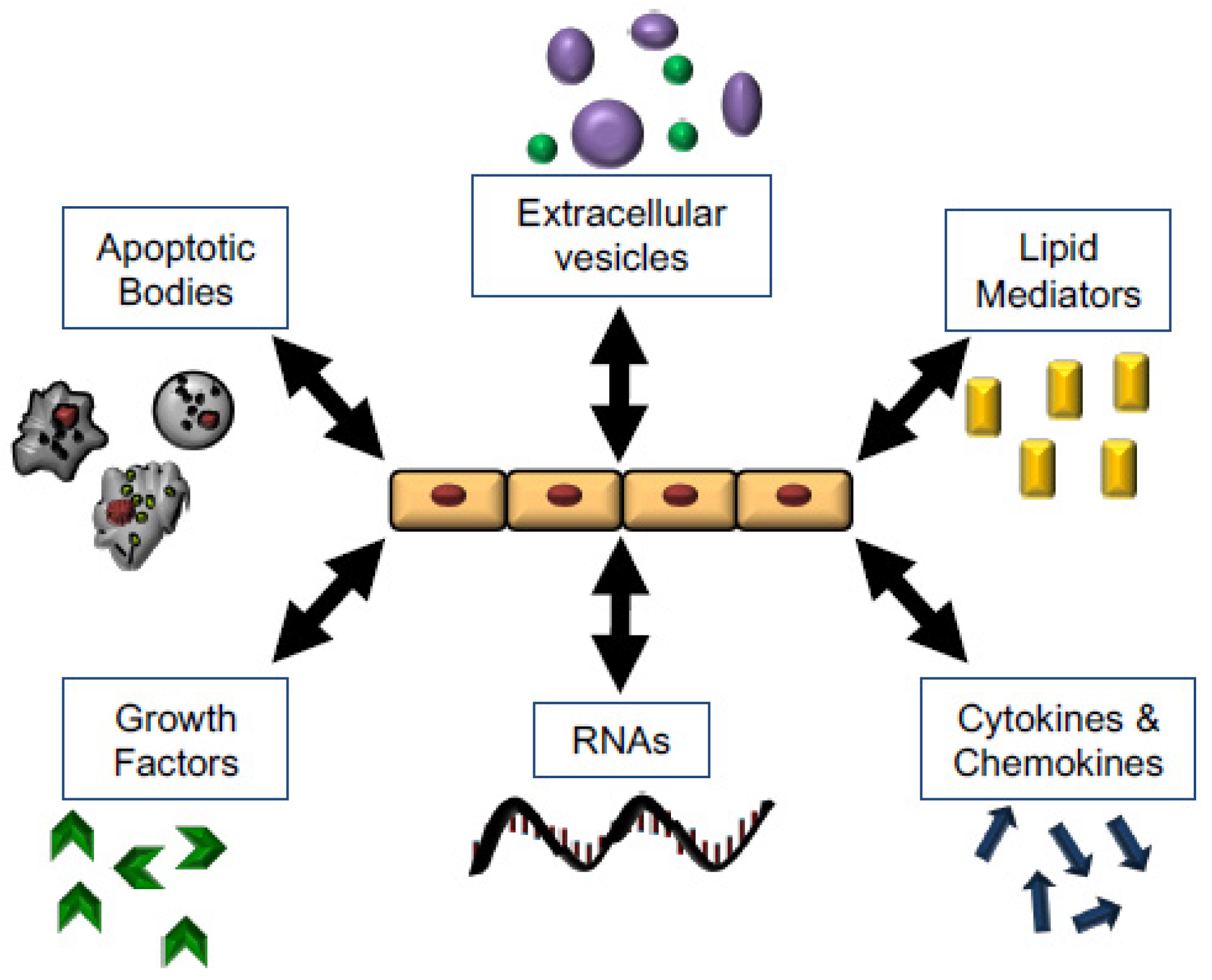
5.4. RNA, Apoptotic Bodies, Microvesicles, and Exosomes
5.5. Secondary Messengers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mediator | Effects on Repair | Implication in Fibrosis | Key References |
|---|---|---|---|
| Growth factors | |||
| EGF | EGF and its receptor upregulated after airway injury. Promotes migration and wound healing of primary airway epithelial cells in vitro. EGF receptor dominant negative mutant impair basal cell proliferation after injury in vivo. | Overexpression of EGF receptor in bronchial epithelium and type 2 pneumocytes of IPF patients. EGFR inhibition by gefitinib results in development of pulmonary fibrosis. | [62,64,66,132,133] |
| IGF | Increases expression of anti-apoptotic proteins in airway epithelial cells. Also associated with increased ECM deposition and fibrosis. | Increased IGF-1 present in IPF tissue and associated with decreased pulmonary function and disease progression. Inhibition of IGF-1R by OSI-906 delayed progression and decreased mortality in murine lung. | [73,74,134] |
| VEGF | Alveolar cell proliferation and enhanced wound healing in vitro | VEGF-A from AT2 cells may play protective role and aid regeneration of wall defects. VEGF-Axxxa family is profibrotic and VEGF-Axxxb is inhibitory. | [79,80,135,136] |
| TGFα | Increased wound healing of alveolar cells in vitro. | Chronic conditional expression of TGFα induces pulmonary fibrosis independently of inflammation in adult murine lung. | [85,137] |
| Lipid mediators | |||
| PGE2 | Enhanced proliferation and wound closure of airway epithelium in vitro. | Inhibition of the PGE2 degrading enzyme, 15-Prostaglandin dehydrogenase, increases PGE2 concentrations and ameliorates lung function and increases proliferation in a bleomycin mouse model of pulmonary fibrosis. Potent downregulator of fibroblast activation. | [94,95,138,139] |
| Lipoxin A4 | Promotes primary alveolar epithelium proliferation and wound closure, inhibits apoptosis and cytokine production in vitro. | Decreased lipoxin A4/LTB4 ratio advances fibrosis. Upregulation of ALX receptor associated with reduced collagen accumulation in vivo. | [100,101,139] |
| RvD3 | Increased epithelial proliferation and reduced inflammation and organ injury after acid-induced lung injury in vivo. | [103] | |
| Cytokines | |||
| CCR3 ligands | Upregulated epithelial proliferation and chemotaxis and enhanced wound repair in vitro. | Lung fibrotic response limited by neutralising CCR3 receptor, expression of profibrotic mediators decreased. | [110,140] |
| IL-22 | Promotes airway epithelial proliferation and protects against lung dysfunction, morbidity, and fibrosis after influenza infection in vivo. | Protective role against severe fibrosis following bacterial infection. | [111,112] |
| Other | |||
| Airway mucin gene (MUC5B) | Attenuates ciliated cell differentiation in repair. MUC5B disrupts alveolar repair by interfering with the interaction between AT2 and the matrix. | Promoter polymorphism is a strong genetic risk for IPF. | [141,142] |
6. Looking Forward: What Lies Next?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Kotton, D.N.; Morrisey, E.E. Lung regeneration: Mechanisms, applications and emerging stem cell populations. Nat. Med. 2014, 20, 822–832. [Google Scholar] [CrossRef] [Green Version]
- Spella, M.; Lilis, I.; Stathopoulos, G.T. Shared epithelial pathways to lung repair and disease. Eur. Respir. Rev. 2017, 26, 170048. [Google Scholar] [CrossRef] [Green Version]
- Kia’I, N.; Bajaj, T. Histology, Respiratory Epithelium; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2020. [Google Scholar]
- Hermanns, M.I.; Unger, R.E.; Kehe, K.; Peters, K.; Kirkpatrick, C.J. Lung epithelial cell lines in coculture with human pulmonary microvascular endothelial cells: Development of an alveolo-capillary barrier in vitro. Lab. Investig. 2004, 84, 736–752. [Google Scholar] [CrossRef] [Green Version]
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.-M.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Schiller, H.B.; Montoro, D.T.; Simon, L.M.; Rawlins, E.L.; Meyer, K.B.; Strunz, M.; Braga, F.A.V.; Timens, W.; Koppelman, G.H.; Budinger, G.R.S.; et al. The Human Lung Cell Atlas: A High-Resolution Reference Map of the Human Lung in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2019, 61, 31–41. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nat. Cell Biol. 2020, 587, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Bils, R.F. Identification of Cells Labeled with Tritiated Thymidine in the Pulmonary Alveolar Walls of the Mouse1,2,3. Am. Rev. Respir. Dis. 1969, 100, 372–378. [Google Scholar] [CrossRef]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.Y.; Nettesheim, P.; Randell, S.H. Growth and differentiation of tracheal epithelial progenitor cells. Am. J. Physiol. Cell. Mol. Physiol. 1994, 266, L296–L307. [Google Scholar] [CrossRef]
- Rawlins, E.L.; Okubo, T.; Xue, Y.; Brass, D.M.; Auten, R.L.; Hasegawa, H.; Wang, F.; Hogan, B.L. The Role of Scgb1a1+ Clara Cells in the Long-Term Maintenance and Repair of Lung Airway, but Not Alveolar, Epithelium. Cell Stem Cell 2009, 4, 525–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, K.U.; Reynolds, S.D.; Giangreco, A.; Hurley, C.M.; Stripp, B.R. Clara Cell Secretory Protein–Expressing Cells of the Airway Neuroepithelial Body Microenvironment Include a Label-Retaining Subset and Are Critical for Epithelial Renewal after Progenitor Cell Depletion. Am. J. Respir. Cell Mol. Biol. 2001, 24, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Tata, P.R.; Mou, H.; Pardo-Saganta, A.; Zhao, R.; Prabhu, M.; Law, B.M.; Vinarsky, V.; Cho, J.L.; Breton, S.; Sahay, A.; et al. Dedifferentiation of com-mitted epithelial cells into stem cells in vivo. Nature 2013, 503, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Adamson, I.Y.; Bowden, D.H. The type 2 cell as progenitor of alveolar epithelial regeneration. A cytodynamic study in mice after exposure to oxygen. Lab. Investig. 1974, 30, 35–42. [Google Scholar]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.E.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.C.; Looney, M.R.; et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nat. Cell Biol. 2015, 517, 621–625. [Google Scholar] [CrossRef]
- Zuo, W.; Zhang, T.; Wu, D.Z.; Guan, S.P.; Liew, A.-A.; Yamamoto, Y.; Wang, X.; Lim, S.J.; Vincent, M.; Lessard, M.; et al. p63+Krt5+ distal airway stem cells are essential for lung regeneration. Nat. Cell Biol. 2015, 517, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.A.; Li, X.; Alexander, J.P.; Brumwell, A.; Lorizio, W.; Tan, K.; Sonnenberg, A.; Wei, Y.; Vu, T.H. Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J. Clin. Investig. 2011, 121, 2855–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathiriya, J.J.; Brumwell, A.N.; Jackson, J.R.; Tang, X.; Chapman, H.A. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell Stem Cell 2020, 26, 346–358. [Google Scholar] [CrossRef]
- Lee, J.-H.; Bhang Dong, H.; Beede, A.; Huang Tian, L.; Stripp, B.R.; Bloch, K.D.; Wagers, A.J.; Tseng, Y.-H.; Ryeom, S.; Kim, C.F.; et al. Lung Stem Cell Differentiation in Mice Directed by Endothelial Cells via a BMP4-NFATc1-Thrombospondin-1 Axis. Cell 2014, 156, 440–455. [Google Scholar] [CrossRef] [Green Version]
- Levy, B.D.; Vachier, I.; Serhan, C.N. Resolution of Inflammation in Asthma. Clin. Chest Med. 2012, 33, 559–570. [Google Scholar] [CrossRef] [Green Version]
- Sumagin, R.; Robin, A.Z.; Nusrat, A.; Parkos, C.A. Transmigrated neutrophils in the intestinal lumen engage ICAM-1 to regulate the epithelial barrier and neutrophil recruitment. Mucosal Immunol. 2014, 7, 905–915. [Google Scholar] [CrossRef]
- Godwin, J.W.; Pinto, A.R.; Rosenthal, N.A. Macrophages are required for adult salamander limb re-generation. Proc. Natl. Acad. Sci. USA 2013, 110, 9415–9420. [Google Scholar] [CrossRef] [Green Version]
- Petrie, T.A.; Strand, N.S.; Yang, C.T.; Rabinowitz, J.S.; Moon, R.T. Macrophages modulate adult zebrafish tail fin regeneration. Development 2014, 141, 2581–2591. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, T.; Kaburagi, Y.; Hamaguchi, Y.; Hasegawa, M.; Takehara, K.; Steeber, D.A.; Tedder, T.F.; Sato, S. Delayed Wound Healing in the Absence of Intercellular Adhesion Molecule-1 or L-Selectin Expression. Am. J. Pathol. 2000, 157, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas Subhra, K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Sinha, M.; Datta, S.; Abas, M.; Chaffee, S.; Sen, C.K.; Roy, S. Monocyte and Macrophage Plasticity in Tissue Repair and Regeneration. Am. J. Pathol. 2015, 185, 2596–2606. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, S.D.; Senior, R.M. Matrix metalloproteinases. Matrix degradation and more. Am. J. Respir. Cell Mol. Biol. 1999, 20, 1100–1102. [Google Scholar] [CrossRef] [PubMed]
- Legrand, C.; Gilles, C.; Zahm, J.M.; Polette, M.; Buisson, A.C.; Kaplan, H.; Birembaut, P.; Tournier, J.M. Airway epithelial cell migration dynamics. MMP-9 role in cell-extracellular matrix remodeling. J. Cell Biol. 1999, 146, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef]
- Murray, P.J. The primary mechanism of the IL-10-regulated antiinflammatory response is to se-lectively inhibit transcription. Proc. Natl. Acad. Sci. USA 2005, 102, 8686–8691. [Google Scholar] [CrossRef] [Green Version]
- Minutti, C.M.; Knipper, J.A.; Allen, J.E.; Zaiss, D.M. Tissue-specific contribution of macrophages to wound healing. Semin. Cell Dev. Biol. 2017, 61, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-Wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.; Waisman, A.; Ranjan, R.; Roes, J.; Krieg, T.; Müller, W.; Roers, A.; Eming, S.A. Differential Roles of Macrophages in Diverse Phases of Skin Repair. J. Immunol. 2010, 184, 3964–3977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbings, S.L.; Thomas, S.M.; Atif, S.M.; McCubbrey, A.L.; Desch, A.N.; Danhorn, T.; Leach, S.M.; Bratton, D.L.; Henson, P.M.; Janssen, W.J.; et al. Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am. J. Respir. Cell Mol. Biol. 2017, 57, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleki, F.; Farahani, N.; Hayat, S.M.G.; Pirro, M.; Bianconi, V.; Barreto, G.E.; Sahebkar, A. The Role of Efferocytosis in Autoimmune Diseases. Front. Immunol. 2018, 9, 1645. [Google Scholar] [CrossRef] [Green Version]
- Bosurgi, L.; Cao, Y.G.; Cabeza-Cabrerizo, M.; Tucci, A.; Hughes, L.D.; Kong, Y.; Weinstein, J.S.; Licona-Limon, P.; Schmid, E.T.; Pelorosso, F.; et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356, 1072–1076. [Google Scholar] [CrossRef] [Green Version]
- Divekar, R.; Kita, H. Recent advances in epithelium-derived cytokines (IL-33, IL-25, and thymic stromal lymphopoietin) and allergic inflammation. Curr. Opin. Allergy Clin. Immunol. 2015, 15, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Hams, E.; Armstrong, M.E.; Barlow, J.L.; Saunders, S.P.; Schwartz, C.; Cooke, G.; Fahy, R.J.; Crotty, T.B.; Hirani, N.; Flynn, R.J.; et al. IL-25 and type 2 in-nate lymphoid cells induce pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Luzina, I.G.; Kopach, P.; Lockatell, V.; Kang, P.H.; Nagarsekar, A.; Burke, A.P.; Hasday, J.D.; Todd, N.W.; Atamas, S.P. Interleukin-33 poten-tiates bleomycin-induced lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 999–1008. [Google Scholar] [CrossRef]
- Jung, M.Y.; Smrž, D.; Desai, A.; Bandara, G.; Ito, T.; Iwaki, S.; Kang, J.H.; Andrade, M.V.; Hilderbrand, S.C.; Brown, J.M.; et al. IL-33 induces a hyporesponsive phe-notype in human and mouse mast cells. J. Immunol. 2013, 190, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, K.; Siebeck, M.; Gropp, R. The route to pathologies in chronic inflammatory diseases characterized by T helper type 2 immune cells. Clin. Exp. Immunol. 2014, 178, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Havran, W.L.; Jameson, J.M.; Witherden, D.A. Epithelial Cells and Their Neighbors. III. Interactions between intraepithelial lymphocytes and neighboring epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G627–G630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Zhang, C.; Liu, J.; Miao, Q. Regulatory T-cells promote pulmonary repair by modulating T helper cell immune responses in lipopolysaccharide-induced acute respiratory distress syndrome. Immunology 2019, 157, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Smith, C.W.; Zhang, W.; Burns, A.R.; Li, Z. NK Cells Modulate the Inflammatory Response to Corneal Epithelial Abrasion and Thereby Support Wound Healing. Am. J. Pathol. 2012, 181, 452–462. [Google Scholar] [CrossRef] [Green Version]
- McAleer, J.P.; Kolls, J.K. Directing traffic: IL-17 and IL-22 coordinate pulmonary immune defense. Immunol. Rev. 2014, 260, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Wirsdörfer, F.; Jendrossek, V. The Role of Lymphocytes in Radiotherapy-Induced Adverse Late Effects in the Lung. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monticelli, L.A.; Sonnenberg, G.F.; Artis, D. Innate lymphoid cells: Critical regulators of allergic in-flammation and tissue repair in the lung. Curr. Opin. Immunol. 2012, 24, 284–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avishai, E.; Yeghiazaryan, K.; Golubnitschaja, O. Impaired wound healing: Facts and hypotheses for multi-professional considerations in predictive, preventive and personalised medicine. EPMA J. 2017, 8, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; DiPietro, L.A. Factors Affecting Wound Healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
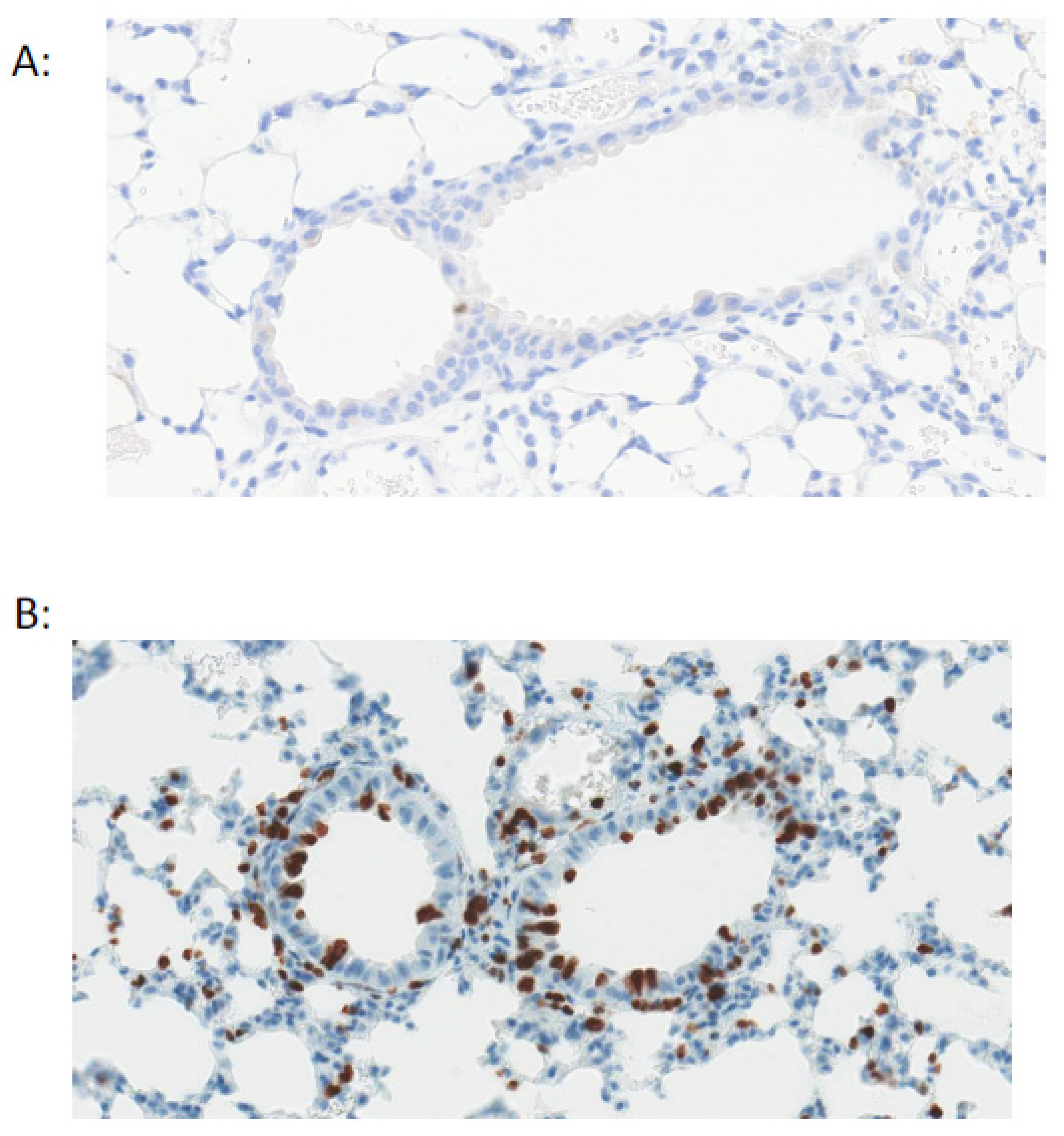
- Dorward, D.A.; Russell, C.D.; Um, I.H.; Elshani, M.; Armstrong, S.D.; Penrice-Randal, R.; Millar, T.; Lerpiniere, C.E.B.; Tagliavini, G.; Hartley, C.S.; et al. Tissue-specific tolerance in fatal Covid-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with Covid-19—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Ruffin, M.; Brochiero, E. Repair Process Impairment by Pseudomonas aeruginosa in Epithelial Tissues: Major Features and Potential Therapeutic Avenues. Front. Cell. Infect. Microbiol. 2019, 9, 182. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Li, Z.; Maitz, P.K. Pseudomonas pyocyanin inhibits wound repair by inducing prema-ture cellular senescence: Role for p38 mitogen-activated protein kinase. Burns 2009, 35, 500–508. [Google Scholar] [CrossRef]
- Cott, C.; Thuenauer, R.; Landi, A.; Kühn, K.; Juillot, S.; Imberty, A.; Madl, J.; Eierhoff, T.; Römer, W. Pseudomonas aeruginosa lectin LecB inhibits tissue repair processes by triggering β-catenin degradation. Biochim. Biophys. Acta BBA Bioenerg. 2016, 1863, 1106–1118. [Google Scholar] [CrossRef] [Green Version]
- Tzilas, V.; Bouros, E.; Barbayianni, I.; Karampitsakos, T.; Kourtidou, S.; Ntassiou, M.; Ninou, I.; Aidinis, V.; Bouros, D.; Tzouvelekis, A.; et al. Vitamin D prevents experimental lung fibrosis and predicts survival in patients with idiopathic pulmonary fi-brosis. Pulm. Pharmacol. Ther. 2019, 55, 17–24. [Google Scholar] [CrossRef]
- Li, S.-R.; Tan, Z.-X.; Chen, Y.-H.; Hu, B.; Zhang, C.; Wang, H.; Zhao, H.; Xu, D.-X. Vitamin D deficiency exacerbates bleo-mycin-induced pulmonary fibrosis partially through aggravating TGF-β/Smad2/3-mediated epitheli-al-mesenchymal transition. Respir. Res. 2019, 20, 266. [Google Scholar] [CrossRef]
- Belderbos, M.E.; Houben, M.L.; Wilbrink, B.; Lentjes, E.; Bloemen, E.M.; Kimpen, J.L.L.; Rovers, M.; Bont, L. Cord Blood Vitamin D Deficiency Is Associated With Respiratory Syncytial Virus Bronchiolitis. Pediatrics 2011, 127, e1513–e1520. [Google Scholar] [CrossRef]
- Tesfaigzi, Y.; Johnson, N.F.; Lechner, J.F. Induction of EGF receptor and erbB-2 during endotoxin-induced alveolar type II cell proliferation in the rat lung. Int. J. Exp. Pathol. 1996, 77, 143–154. [Google Scholar] [CrossRef]
- Van Winkle, L.S.; Isaac, J.M.; Plopper, C.G. Distribution of epidermal growth factor receptor and lig-ands during bronchiolar epithelial repair from naphthalene-induced Clara cell injury in the mouse. Am. J. Pathol. 1997, 151, 443–459. [Google Scholar]
- Heijink, I.H.; van Oosterhout, A.; Kapus, A. Epidermal growth factor receptor signalling contrib-utes to house dust mite-induced epithelial barrier dysfunction. Eur. Respir. J. 2010, 36, 1016–1026. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; McKinnis, V.S.; Nawrocki, A.; White, S.R. Stimulation of Migration and Wound Repair of Guinea-Pig Airway Epithelial Cells in Response to Epidermal Growth Factor. Am. J. Respir. Cell Mol. Biol. 1998, 18, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Sydlik, U.; Bierhals, K.; Soufi, M.; Abel, J.; Schins, R.P.F.; Unfried, K. Ultrafine carbon particles induce apoptosis and proliferation in rat lung epithelial cells via specific signaling pathways both using EGF-R. Am. J. Physiol. Cell. Mol. Physiol. 2006, 291, L725–L733. [Google Scholar] [CrossRef]
- Brechbuhl, H.M.; Li, B.; Smith, R.W.; Reynolds, S.D. Epidermal growth factor receptor activity is nec-essary for mouse basal cell proliferation. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L800–L810. [Google Scholar] [CrossRef] [Green Version]
- Puddicombe, S.M.; Polosa, R.; Richter, A.; Krishna, M.T.; Howarth, P.H.; Holgate, S.T.; Davies, D.E. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 2000, 14, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Hirota, N.; Risse, P.A.; Novali, M.; McGovern, T.; Al-Alwan, L.; McCuaig, S.; Proud, D.; Hayden, P.; Hamid, Q.; Martin, J.G.; et al. Histamine may induce airway remodeling through release of epidermal growth factor receptor ligands from bronchial epi-thelial cells. FASEB J. 2012, 26, 1704–1716. [Google Scholar] [CrossRef] [PubMed]
- Schnackenberg, B.J.; Jones, S.M.; Pate, C.; Shank, B.; Sessions, L.; Pittman, L.M.; Cornett, L.E.; Kurten, R.C. The beta-agonist isoproterenol attenuates EGF-stimulated wound closure in human airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L485–L491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinh, N.T.; Privé, A.; Kheir, L.; Bourret, J.C.; Hijazi, T.; Amraei, M.G.; Noël, J.; Brochiero, E. Involvement of KATP and KvLQT1 K+ channels in EGF-stimulated alveolar epithelial cell repair processes. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L870–L882. [Google Scholar] [CrossRef] [Green Version]
- Moodley, S.; Derouet, M.; Bai, X.H.; Xu, F.; Kapus, A.; Yang, B.B.; Liu, M. Stimulus-dependent dissociation between XB130 and Tks5 scaffold proteins promotes airway epithelial cell migration. Oncotarget 2016, 7, 76437–76452. [Google Scholar] [CrossRef] [Green Version]
- Lallemand, A.V.; Ruocco, S.M.; Joly, P.M.; Gaillard, D.A. In vivo localization of the insulin-like growth factors I and II (IGF I and IGF II) gene expression during human lung development. Int. J. Dev. Biol. 1995, 39, 529–537. [Google Scholar] [PubMed]
- Uh, S.-T.; Inoue, Y.; King, T.E.; Chan, E.D.; Newman, L.S.; Riches, D.W.H. Morphometric Analysis of Insulin-like Growth Factor-I Localization in Lung Tissues of Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 1998, 158, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Chand, H.S.; Woldegiorgis, Z.; Schwalm, K.; McDonald, J.; Tesfaigzi, Y. Acute Inflammation Induces Insulin-like Growth Factor-1 to Mediate Bcl-2 and Muc5ac Expression in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2012, 47, 784–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasaraju, T.A.; Chen, H.; Weng, T.; Bhaskaran, M.; Jin, N.; Chen, J.; Chen, Z.; Chinoy, M.R.; Liu, L. Expression profile of IGF system during lung injury and recovery in rats exposed to hyperoxia: A possible role of IGF-1 in alveolar epithelial cell proliferation and differentiation. J. Cell. Biochem. 2006, 97, 984–998. [Google Scholar] [CrossRef] [PubMed]
- Mu, M.; Gao, P.; Yang, Q.; He, J.; Wu, F.; Han, X.; Guo, S.; Qian, Z.; Song, C. Alveolar Epithelial Cells Promote IGF-1 Production by Alveolar Macrophages Through TGF-β to Suppress Endogenous Inflammatory Signals. Front. Immunol. 2020, 11, 1585. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.C.; Gorantla, V.; Makena, P.S.; Luellen, C.; Sinclair, S.E.; Schwingshackl, A.; Waters, C.M. Insulin-like growth factor-I stimulates differentiation of ATII cells to ATI-like cells through activation of Wnt5a. Am. J. Physiol. Cell. Mol. Physiol. 2013, 305, L222–L228. [Google Scholar] [CrossRef] [Green Version]
- Tonkin, J.; Temmerman, L.; Sampson, R.D.; Gallego-Colon, E.; Barberi, L.; Bilbao, D.; Schneider, M.D.; Musaro’, A.; Rosenthal, N. Monocyte/Macrophage-derived IGF-1 Orchestrates Murine Skeletal Muscle Regeneration and Modulates Autocrine Polarization. Mol. Ther. 2015, 23, 1189–1200. [Google Scholar] [CrossRef] [Green Version]
- Varet, J.; Douglas, S.K.; Gilmartin, L.; Medford, A.R.L.; Bates, D.O.; Harper, S.J.; Millar, A.B. VEGF in the lung: A role for novel isoforms. Am. J. Physiol. Cell. Mol. Physiol. 2010, 298, L768–L774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.R.; Perkins, G.D.; Fujisawa, T.; Pettigrew, K.A.; Gao, F.; Ahmed, A. Vascular endothelial growth factor promotes physical wound repair and is anti-apoptotic in primary distal lung epithelial and A549 cells. Crit. Care Med. 2007, 35, 2164–2170. [Google Scholar] [CrossRef]
- Karmpaliotis, D.; Kosmidou, I.; Ingenito, E.P.; Hong, K.; Malhotra, A.; Sunday, M.E.; Haley, K.J. Angiogenic growth factors in the pathophysiology of a murine model of acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2002, 283, L585–L595. [Google Scholar] [CrossRef] [Green Version]
- Schliwa, M. Action of cytochalasin D on cytoskeletal networks. J. Cell Biol. 1982, 92, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.; Rossiter, H.B.; Wagner, P.D.; Breen, E.C. Lung-targeted VEGF inactivation leads to an emphysema phenotype in mice. J. Appl. Physiol. 2004, 97, 1559–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strandjord, T.P.; Clark, J.G.; E Guralnick, D.; Madtes, D.K. Immunolocalization of Transforming Growth Factor-α, Epidermal Growth Factor (EGF), and EGF-Receptor in Normal and Injured Developing Human Lung. Pediatr. Res. 1995, 38, 851–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheradmand, F.; Folkesson, H.G.; Shum, L.; Derynk, R.; Pytela, R.; Matthay, M.A. Transforming growth factor-alpha enhances alveolar epithelial cell repair in a new in vitro model. Am. J. Physiol. Cell. Mol. Physiol. 1994, 267, L728–L738. [Google Scholar] [CrossRef] [PubMed]
- Howat, W.J.; Holgate, S.T.; Lackie, P.M. TGF-beta isoform release and activation during in vitro bronchial epithelial wound repair. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L115–L123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, B.C.; Borok, Z. TGF-beta-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [Green Version]
- Bartram, U.; Speer, C.P. The role of transforming growth factor beta in lung development and disease. Chest 2004, 125, 754–765. [Google Scholar] [CrossRef]
- Peters-Golden, M.; Feyssa, A. Transcellular eicosanoid synthesis in cocultures of alveolar epithelial cells and macrophages. Am. J. Physiol. Cell. Mol. Physiol. 1993, 264, L438–L447. [Google Scholar] [CrossRef]
- Jame, A.J.; Lackie, P.M.; Cazaly, A.M.; Sayers, I.; Penrose, J.F.; Holgate, S.T.; Sampson, A.P. Human bronchial epithelial cells express an active and inducible biosynthetic pathway for leukotrienes B4and C4. Clin. Exp. Allergy 2007, 37, 880–892. [Google Scholar] [CrossRef]
- Moore, B.B.; Peters-Golden, M.; Christensen, P.J.; Lama, V.; Kuziel, W.A.; Paine, R., 3rd; Toews, G.B. Alveolar epithelial cell inhibition of fibroblast proliferation is regulated by MCP-1/CCR2 and mediated by PGE2. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L342–L349. [Google Scholar] [CrossRef] [Green Version]
- Hangai, S.; Ao, T.; Kimura, Y.; Matsuki, K.; Kawamura, T.; Negishi, H.; Nishio, J.; Kodama, T.; Taniguchi, T.; Yanai, H. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc. Natl. Acad. Sci. USA 2016, 113, 3844–3849. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, L.M.; Belvisi, M.G.; Bode, K.A.; Bauer, J.; Schmidt, C.; Suchy, M.-T.; Tsikas, D.; Scheuerer, J.; Lasitschka, F.; Gröne, H.-J.; et al. Bronchial Epithelial Cell-Derived Prostaglandin E2 Dampens the Reactivity of Dendritic Cells. J. Immunol. 2011, 186, 2095–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madanayake, T.W.; Fidler, T.P.; Fresquez, T.M.; Bajaj, N.; Rowland, A.M. Cytochrome P450 2S1 Depletion Enhances Cell Proliferation and Migration in Bronchial Epithelial Cells, in Part, through Modulation of Prostaglandin E2 Synthesis. Drug Metab. Dispos. 2012, 40, 2119–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savla, U.; Appel, H.J.; Sporn, P.H.S.; Waters, C.M. Prostaglandin E2regulates wound closure in airway epithelium. Am. J. Physiol. Cell. Mol. Physiol. 2001, 280, L421–L431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnans, C.; Gras, D.; Chavis, C.; Mainprice, B.; Vachier, I.; Godard, P.; Chanez, P. Synthesis and anti-inflammatory effect of lipoxins in human airway epithelial cells. Biomed. Pharm. 2007, 61, 261–267. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, H.-M.; Thatcher, T.H.; Levy, E.P.; Fulton, R.A.; Owens, K.M.; Phipps, R.P.; Sime, P.J. Resolvin D1 Attenuates Polyinosinic-Polycytidylic Acid–Induced Inflammatory Signaling in Human Airway Epithelial Cells via TAK1. J. Immunol. 2014, 193, 4980–4987. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Abbott, J.; Cheng, L.; Colby, J.K.; Lee, J.W.; Levy, B.D.; Matthay, M.A. Human Mesenchymal Stem (Stromal) Cells Promote the Resolution of Acute Lung Injury in Part through Lipoxin A4. J. Immunol. 2015, 195, 875–881. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; D’Souza, V.K.; Bartis, D.; Dancer, R.C.; Parekh, D.; Naidu, B.; Gao-Smith, F.; Wang, Q.; Jin, S.; Lian, Q.; et al. Lipoxin A4promotes lung epithelial repair whilst inhibiting fibroblast proliferation. ERJ Open Res. 2016, 2, 00079–02015. [Google Scholar] [CrossRef]
- Bonnans, C.; Fukunaga, K.; Levy, M.A.; Levy, B.D. Lipoxin A4 Regulates Bronchial Epithelial Cell Responses to Acid Injury. Am. J. Pathol. 2006, 168, 1064–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, G.; Buchanan, P.; Perriere, M.; Al-Alawi, M.; Costello, R.W.; Verriere, V.; McNally, P.; Harvey, B.J.; Urbach, V. Activation of P2RY11 and ATP Release by Lipoxin A4 Restores the Airway Surface Liquid Layer and Epithelial Repair in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 51, 178–190. [Google Scholar]
- Colby, J.K.; Abdulnour, R.-E.E.; Sham, H.P.; Dalli, J.; Colas, R.A.; Winkler, J.W.; Hellmann, J.; Wong, B.; Cui, Y.; El-Chemaly, S.; et al. Resolvin D3 and Aspirin-Triggered Resolvin D3 Are Protective for Injured Epithelia. Am. J. Pathol. 2016, 186, 1801–1813. [Google Scholar] [CrossRef] [Green Version]
- Grumbach, Y.; Quynh, N.V.T.; Chiron, R.; Urbach, V. LXA4 stimulates ZO-1 expression and transepithelial electrical resistance in human airway epithelial (16HBE14o-) cells. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L101–L108. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; He, S.; Yuan, J.; Miao, S.; Gao, H.; Zhang, J.; Li, Y.; Peng, W.; Wu, P. Lipoxin A4 attenuates LPS-induced mouse acute lung injury via Nrf2-mediated E-cadherin expression in airway epithelial cells. Free. Radic. Biol. Med. 2016, 93, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, H.; Wang, Y.; Xia, H.; Gong, J.; Li, B.; Yao, S.; Shang, Y. Maresin 1 Maintains the Permeability of Lung Epithelial Cells In Vitro and In Vivo. Inflammation 2016, 39, 1981–1989. [Google Scholar] [CrossRef]
- Meng, F.; Mambetsariev, I.; Tian, Y.; Beckham, Y.; Meliton, A.; Leff, A.; Gardel, M.L.; Allen, M.J.; Birukov, K.G.; Birukova, A.A. Attenuation of Lipopolysaccharide-Induced Lung Vascular Stiffening by Lipoxin Reduces Lung Inflammation. Am. J. Respir. Cell Mol. Biol. 2015, 52, 152–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Park, M.K.; Lee, E.J.; Lee, C.H. Resolvin D1 inhibits TGF-β1-induced epithelial mesenchymal transition of A549 lung cancer cells via lipoxin A4 receptor/formyl peptide receptor 2 and GPR32. Int. J. Biochem. Cell Biol. 2013, 45, 2801–2807. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, R.; Chen, L.; Tan, W.; Sun, Z.; Xia, H.; Li, B.; Yu, Y.; Gong, J.; Tang, M.; et al. Maresin 1 Inhibits Epithelial-to-Mesenchymal Transition in Vitro and Attenuates Bleomycin Induced Lung Fibrosis in Vivo. Shock 2015, 44, 496–502. [Google Scholar] [CrossRef]
- Beck, L.A.; Tancowny, B.; Brummet, M.E.; Asaki, S.Y.; Curry, S.L.; Penno, M.B.; Foster, M.; Bahl, A.; Stellato, C. Functional Analysis of the Chemokine Receptor CCR3 on Airway Epithelial Cells. J. Immunol. 2006, 177, 3344–3354. [Google Scholar] [CrossRef]
- Pociask, D.A.; Scheller, E.V.; Mandalapu, S.; McHugh, K.J.; Enelow, R.I.; Fattman, C.L.; Kolls, J.K.; Alcorn, J.F. IL-22 Is Essential for Lung Epithelial Repair following Influenza Infection. Am. J. Pathol. 2013, 182, 1286–1296. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Thakar, M.S.; Ouyang, W.; Malarkannan, S. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol. 2013, 6, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.S.; Choi, H.; Jiang, X.; Yin, L.; Seet, J.E.; Patzel, V.; Engelward, B.P.; Chow, V.T.K. Micro-RNAs in regenerating lungs: An integrative systems biology analysis of murine influenza pneumonia. BMC Genom. 2014, 15, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Ganesh, K.; Khanna, S.; Sen, C.K.; Roy, S. Engulfment of Apoptotic Cells by Macrophages: A Role of MicroRNA-21 in the Resolution of Wound Inflammation. J. Immunol. Baltim. 1950 2014, 192, 1120–1129. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Thomson, J.M.; Wong, H.Y.F.; Hammond, S.M.; Hogan, B. Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev. Biol. 2007, 310, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Zhang, Y.; Hurd, L.; Hannenhalli, S.; Liu, F.; Lu, M.M.; Morrisey, E.E. Regulation of lung endoderm progenitor cell behavior by miR302/367. Development 2011, 138, 1235–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McVey, M.; Tabuchi, A.; Kuebler, W.M. Microparticles and acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L364–L381. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.J.; Speth, J.M.; Peters-Golden, M. Signed, Sealed, Delivered: Microenvironmental Modulation of Extracellular Vesicle-Dependent Immunoregulation in the Lung. Front. Cell Dev. Biol. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogarty, C.E.; Bergmann, A. Killers creating new life: Caspases drive apoptosis-induced proliferation in tissue repair and disease. Cell Death Differ. 2017, 24, 1390–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogarty, C.E.; Diwanji, N.; Lindblad, J.L.; Tare, M.; Amcheslavsky, A.; Makhijani, K.; Brückner, K.; Fan, Y.; Bergmann, A. Extracellular Reactive Oxygen Species Drive Apoptosis-Induced Proliferation via Drosophila Macrophages. Curr. Biol. 2016, 26, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Zhang, D.; Lee, H.; Menon, A.A.; Wu, J.; Hu, K. Macrophage-derived apoptotic bodies promote the proliferation of the recipient cells via shuttling microRNA-221/222. J. Leukoc. Biol. 2017, 101, 1349–1359. [Google Scholar] [CrossRef] [Green Version]
- Collino, F.; Pomatto, M.; Bruno, S.; Lindoso, R.S.; Tapparo, M.; Sicheng, W.; Quesenberry, P.; Camussi, G. Exosome and Microvesicle-Enriched Fractions Isolated from Mesenchymal Stem Cells by Gradient Separation Showed Different Molecular Signatures and Functions on Renal Tubular Epithelial Cells. Stem Cell Rev. Rep. 2017, 13, 226–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.-Y.; Tran, J.A.; Chang, J.-H.; Azar, D.T.; Zieske, J.D. Potential role of corneal epithelial cell-derived exosomes in corneal wound healing and neovascularization. Sci. Rep. 2017, 7, srep40548. [Google Scholar] [CrossRef] [Green Version]
- Tomasoni, S.; Longaretti, L.; Rota, C.; Morigi, M.; Conti, S.; Gotti, E.; Capelli, C.; Introna, M.; Remuzzi, G.; Benigni, A. Transfer of Growth Factor Receptor mRNA Via Exosomes Unravels the Regenerative Effect of Mesenchymal Stem Cells. Stem Cells Dev. 2013, 22, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Borges, F.T.; Melo, S.A.; Özdemir, B.C.; Kato, N.; Revuelta, I.; Miller, C.A.; Ii, V.H.G.; LeBleu, V.S.; Kalluri, R. TGF-β1–Containing Exosomes from Injured Epithelial Cells Activate Fibroblasts to Initiate Tissue Regenerative Responses and Fibrosis. J. Am. Soc. Nephrol. 2012, 24, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.-G.; Cao, Y.; Yang, J.; Lee, J.H.; Choi, H.S.; Jin, Y. Lung epithelial cell-derived extracellular vesicles activate macrophage-mediated inflammatory responses via ROCK1 pathway. Cell Death Dis. 2015, 6, e2016. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Zhang, D.; Zhu, Z.; Cruz, C.S.D.; Jin, Y. Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci. Rep. 2016, 6, 35250. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Q.; Xiong, W.; Yang, C.; Gagnon, C.; Hardy, P. Lymphocyte-derived microparticles induce bronchial epithelial cells’ pro-inflammatory cytokine production and apoptosis. Mol. Immunol. 2013, 55, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Xu, K.; Zhang, J.; Kumar, A.; Yu, F.-S.X. Wound-induced ATP release and EGF receptor activation in epithelial cells. J. Cell Sci. 2007, 120, 815–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen-Gipson, D.S.; Spurzem, K.; Kolm, N.; Spurzem, J.R.; Wyatt, T.A. Adenosine Promotion of Cellular Migration in Bronchial Epithelial Cells Is Mediated by the Activation of Cyclic Adenosine Monophosphate-Dependent Protein Kinase A. J. Investig. Med. 2007, 55, 378–385. [Google Scholar] [CrossRef]
- Salathe, M. Effects of β-agonists on airway epithelial cells. J. Allergy Clin. Immunol. 2002, 110, S275–S281. [Google Scholar] [CrossRef]
- Baughman, R.P.; Lower, E.E.; Miller, M.A.; Bejarano, P.A.; Heffelfinger, S.C. Overexpression of transforming growth factor-alpha and epidermal growth factor-receptor in idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 1999, 16, 57–61. [Google Scholar]
- Tzouvelekis, A.; Ntolios, P.; Karameris, A.; Vilaras, G.; Boglou, P.; Koulelidis, A.; Archontogeorgis, K.; Kaltsas, K.; Zacharis, G.; Sarikloglou, E.; et al. Increased Expression of Epidermal Growth Factor Receptor (EGF-R) in Patients with Different Forms of Lung Fibrosis. Biomed Res. Int. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, D.M.; Kang, J.H.; Choudhury, M.; Andrianifahanana, M.; Yin, X.; Limper, A.H.; Leof, E.B. IPF pathogenesis is dependent upon TGFβ induction of IGF-1. FASEB J. 2020, 34, 5363–5388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barratt, S.L.; Flower, V.A.; Pauling, J.D.; Millar, A.B. VEGF (Vascular Endothelial Growth Factor) and Fibrotic Lung Disease. Int. J. Mol. Sci. 2018, 19, 1269. [Google Scholar] [CrossRef] [Green Version]
- Barratt, S.; Blythe, T.; Ourradi, K.; Jarrett, C.E.; Welsh, G.; Bates, D.O.; Millar, A.B. Effects of hypoxia and hyperoxia on the differential expression of VEGF-A isoforms and receptors in Idiopathic Pulmonary Fibrosis (IPF). Respir. Res. 2018, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Hardie, W.D.; Le Cras, T.D.; Jiang, K.; Tichelaar, J.W.; Azhar, M.; Korfhagen, T.R. Conditional expression of transforming growth factor-α in adult mouse lung causes pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2004, 286, L741–L749. [Google Scholar] [CrossRef] [PubMed]
- Bärnthaler, T.; Theiler, A.; Zabini, D.; Trautmann, S.; Stacher-Priehse, E.; Lanz, I.; Klepetko, W.; Sinn, K.; Flick, H.; Scheidl, S.; et al. Inhibiting eicosanoid degradation exerts antifibrotic effects in a pulmonary fibrosis mouse model and human tissue. J. Allergy Clin. Immunol. 2020, 145, 818–833.e11. [Google Scholar] [CrossRef] [Green Version]
- Castelino, F.V. Lipids and eicosanoids in fibrosis: Emerging targets for therapy. Curr. Opin. Rheumatol. 2012, 24. [Google Scholar] [CrossRef]
- Huaux, F.; Gharaee-Kermani, M.; Liu, T.; Morel, V.; McGarry, B.; Ullenbruch, M.; Kunkel, S.L.; Wang, J.; Xing, Z.; Phan, S.H. Role of Eotaxin-1 (CCL11) and CC Chemokine Receptor 3 (CCR3) in Bleomycin-Induced Lung Injury and Fibrosis. Am. J. Pathol. 2005, 167, 1485–1496. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.V.; Fingerlin, T.E.; Evans, C.M.; Schwarz, M.I.; Schwartz, D.A. MUC5B and Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. 2), S193–S199. [Google Scholar]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Croasdell Lucchini, A.; Gachanja, N.N.; Rossi, A.G.; Dorward, D.A.; Lucas, C.D. Epithelial Cells and Inflammation in Pulmonary Wound Repair. Cells 2021, 10, 339. https://doi.org/10.3390/cells10020339
Croasdell Lucchini A, Gachanja NN, Rossi AG, Dorward DA, Lucas CD. Epithelial Cells and Inflammation in Pulmonary Wound Repair. Cells. 2021; 10(2):339. https://doi.org/10.3390/cells10020339
Chicago/Turabian StyleCroasdell Lucchini, Amanda, Naomi N. Gachanja, Adriano G. Rossi, David A. Dorward, and Christopher D. Lucas. 2021. "Epithelial Cells and Inflammation in Pulmonary Wound Repair" Cells 10, no. 2: 339. https://doi.org/10.3390/cells10020339