
NLRP3 Inflammasome at the Interface of Inflammation, Endothelial Dysfunction, and Type 2 Diabetes


Abstract
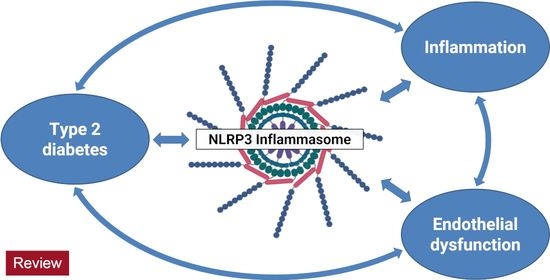
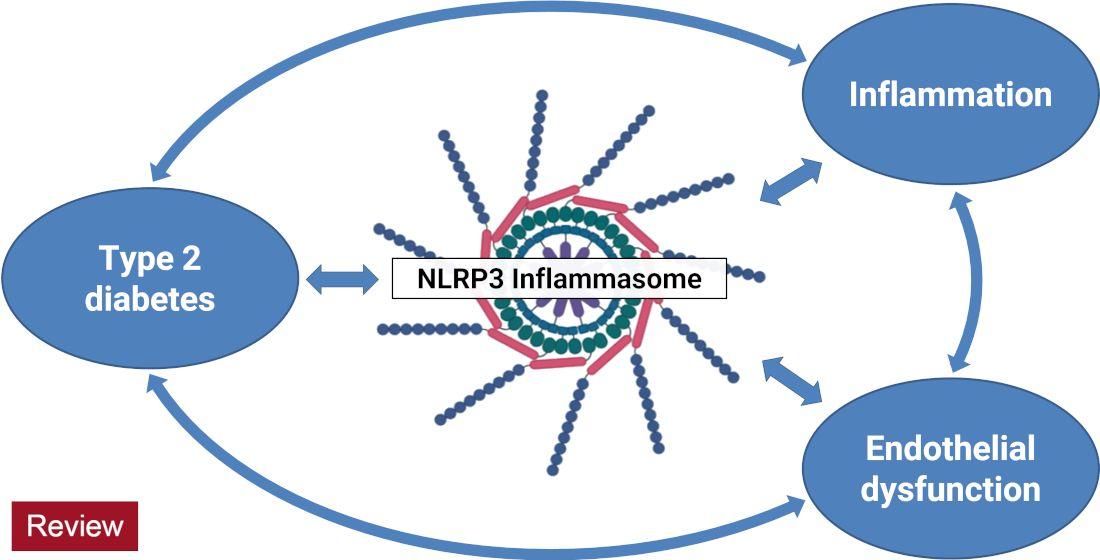
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The NLRP3 Inflammasome and Inflammatory Regulators
2.1. NLRP3 Inflammasome
2.2. Caspase-1
2.3. Interleukin 1β
2.4. Interleukin 18
3. Mechanism of Inflammasome NLRP3 Activation
4. Endothelial Dysfunction as the First Key Step to Activating the Inflammasome
5. Role of NLRP3 Inflammasome in Diabetes and Activation Inflammasome by Metabolic Signals
6. Epigenetic Regulation of NLRP3 Inflammasome by microRNA
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3′UTR | 3′untranslated region |
| 39 UTRs | 39 untranslated regions |
| AGEs | advanced glycation end products |
| AKT | serine/threonine protein kinase B |
| AIM | absentin-melanoma |
| ALRs | absentin-melanoma-like receptors |
| AMPK | adenosine monophosphate-activated protein kinase |
| ASC | apoptosis-associated speck-like protein |
| CARD | C-terminal caspase recruitment domain |
| CRP | C-reactice protein |
| CTLs | C-type lectins |
| DAG | diacylglycerol |
| DAMPs | damage-associated molecular patterns |
| dsDNA | bacterial double-stranded DNA |
| ET-1 | endothelin-1 |
| ER | endoplasmic reticulum |
| ERK1/2 | extracellular signal-regulated kinases 1/2 |
| eNOS | endothelial NO synthase |
| FFAs | saturated free fatty acids |
| G3PDH | glyceraldehyde-3-phosphate dehydrogenase |
| GSH | glutathione |
| GSK3β | glycogen synthase kinase 3 beta |
| GLUT1 | glucose transporter 1 |
| GLUT4 | glucose transporter 4 |
| HFD | high-fat diet |
| IAAP | islet amyloid polipeptyde |
| ICAM-1 | intercellular adhesion molecule 1 |
| ICE | interleukin-1β-converting enzyme |
| IDF | International Diabetes Federation |
| IL | interleukins |
| IRAK1 | IL-1 receptor-associated kinase |
| IRS-1 | insulin receptor substrate-1 |
| JNKs | c-Jun N-terminal kinases |
| LRR | leucine-rich repeats |
| MAPK | mitogen-activated protein kinases |
| MCP1 | monocyte chemoattractant protein 1 |
| MCT1 | monocarboxylate transporter 1 |
| miRNAs | microRNAs |
| mRNAs | messenger RNAs |
| mTOR | mechanistic target of rapamycin |
| mTORC | mechanistic target of rapamycin complex |
| NBD | nucleotide-binding domain |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP | nucleotide-binding oligomerization domain-like receptor with a pyrin domain |
| NLRP3 | protein 3 inflammasome complex |
| NLR | Nod-like receptor |
| NO | nitric oxide |
| NOD | nucleotide-binding oligomerization domain |
| NOD | nucleotide oligomerization domain |
| NPAS3 | neuronal Per-Arnt-Sim domain protein 3 |
| NT | nucleotides |
| ORP8 | oxysterol-binding-protein-related protein 8 |
| PAMPs | pathogen-associated molecular patterns |
| PARP-1 | poly (ADP ribose) polymerase 1 |
| PERK | protein kinase-like endoplasmic reticulum kinase |
| PI3K | phosphatidylinositol 3-kinase |
| PKC | protein kinase C |
| PKCα | protein kinase C alpha |
| PRRs | pattern recognition receptor |
| PYD | pyrin domain |
| RIG-I | retinoic acid inducible gene-I |
| RLRs | RIG-I-like receptors |
| ROS | reactive oxygen species |
| SFAs | saturated fatty acids |
| SGK1 | serum- and glucocorticoid-inducible kinase 1 |
| T2DM | type 2 diabetes mellitus |
| TGF-β | transforming growth factor beta |
| Th | T helper |
| TLRs | toll-like receptors |
| TNFα | tumor necrosis factor alpha |
| TNFR | tumor necrosis factor receptor |
| TORC1 | target of rapamycin complex 1 |
| TRAF6 | TNFR-associated factor 6 |
| TXN | thioredoxin |
| TXNIP | thioredoxin-interacting protein |
| UPR | unfolded protein response |
| VCAM-1 | vascular cell adhesion molecule |
| VEGF | vascular endothelial growth factor |
References
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019. [Google Scholar]
- Guariguata, L.; Whiting, D.R.; Hambleton, I.; Beagley, J.; Linnenkamp, U.; Shaw, J.E. Global Estimates of Diabetes Prevalence for 2013 and Projections For 2035. Diabetes Res. Clin. Pr. 2014, 103, 137–149. [Google Scholar] [CrossRef]
- Leon, B.M.; Maddox, T.M. Diabetes and Cardiovascular Disease: Epidemiology, Biological Mechanisms, Treatment Recommendations and Future Research. World J. Diabetes 2015, 6, 1246–1258. [Google Scholar] [CrossRef]
- Unnikrishnan, R.; Pradeepa, R.; Joshi, S.R.; Mohan, V. Type 2 Diabetes: Demystifying the Global Epidemic. Diabetes 2017, 66, 1432–1442. [Google Scholar] [CrossRef] [Green Version]
- Al-Lawati, J.A. Diabetes Mellitus: A Local and Global Public Health Emergency! Oman Med. J. 2017, 32, 177–179. [Google Scholar] [CrossRef]
- Hameed, I.; Masoodi, S.R.; Mir, S.A.; Nabi, M.; Ghazanfar, K.; Ganai, B.A. Type 2 Diabetes Mellitus: From a Metabolic Disorder to an Inflammatory Condition. World J. Diabetes 2015, 6, 598–612. [Google Scholar] [CrossRef]
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Böni-Schnetzler, M.; Donath, M.Y. The Role of Inflammation in β-cell Dedifferentiation. Sci. Rep. 2017, 7, 6285. [Google Scholar] [CrossRef]
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Frostegård, J. Immune Mechanisms in Atherosclerosis, Especially in Diabetes Type 2. Front. Endocrinol. 2013, 4, 162. [Google Scholar] [CrossRef] [Green Version]
- Dixit, V.D. Nlrp3 Inflammasome Activation in Type 2 Diabetes: Is It Clinically Relevant? Diabetes 2013, 62, 22–24. [Google Scholar] [CrossRef] [Green Version]
- Stutz, A.; Golenbock, D.T.; Latz, E. Inflammasomes: Too Big to Miss. J. Clin. Investig. 2009, 119, 3502–3511. [Google Scholar] [CrossRef] [Green Version]
- Groslambert, M.; Py, B.F. Spotlight on the NLRP3 Inflammasome Pathway. J. Inflamm. Res. 2018, 11, 359–374. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Duewell, P. NLRP3 Inflammasome Activation in Inflammaging. Semin. Immunol. 2018, 40, 61–73. [Google Scholar] [CrossRef]
- Shao, B.Z.; Cao, Q.; Liu, C. Targeting NLRP3 Inflammasome in the Treatment of CNS Diseases. Front. Mol. Neurosci. 2018, 11, 320. [Google Scholar] [CrossRef]
- Benetti, E.; Chiazza, F.; Patel, N.S.; Collino, M. The NLRP3 Inflammasome as a Novel Player of the Intercellular Crosstalk in Metabolic Disorders. Mediat. Inflamm. 2013, 2013, 678627. [Google Scholar] [CrossRef]
- Yin, Y.; Pastrana, J.L.; Li, X.; Huang, X.; Mallilankaraman, K.; Choi, E.T.; Madesh, M.; Wang, H.; Yang, X.F. Inflammasomes: Sensors of Metabolic Stresses for Vascular Inflammation. Front. Biosci. 2013, 18, 638–649. [Google Scholar]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular Mechanisms and Functions of Pyroptosis, Inflammatory Caspases and Inflammasomes in Infectious Diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Conley, S.M.; Abais, J.M.; Boini, K.M.; Li, P.L. Inflammasome Activation in Chronic Glomerular Diseases. Curr. Drug Targets 2017, 18, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Jha, S.; Ting, J.P. Inflammasome-Associated NucleotiDe-binding Domain, Leucine-Rich Repeat Proteins and Inflammatory Diseases. J. Immunol. 2009, 12, 7623–7629. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Liu, R.; Yu, Q.; Bi, Y.; Liu, G. Metabolic Regulation of Inflammasomes in Inflammation. Immunology 2019, 2, 95–109. [Google Scholar] [CrossRef]
- Ahechu, P.; Zozaya, G.; Martí, P.; Hernández-Lizoáin, J.L.; Baixauli, J.; Unamuno, X.; Frühbeck, G.; Catalán, V. NLRP3 Inflammasome: A Possible Link Between Obesity-Associated Low-Grade Chronic Inflammation and Colorectal Cancer Development. Front. Immunol. 2018, 9, 2918. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, C.; Chen, Z.; Liu, L.; Jiang, J.; Wu, Z.; Zhao, M.; Chen, Y. NLRP3: A Novel Mediator in Cardiovascular Disease. J. Immunol. Res. 2018, 2018, 5702103. [Google Scholar] [CrossRef]
- Conforti-Andreoni, C.; Ricciardi-Castagnoli, P.; Mortellaro, A. The Inflammasomes in Health and Disease: From Genetics to Molecular Mechanisms of Autoinflammation and Beyond. Cell Mol. Immunol. 2011, 2, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, M.V.; Miller, E.A.; Bhardwaj, N. Activation and Measurement of NLRP3 Inflammasome Activity Using IL-1β in Human Monocyte-Derived Dendritic Cells. J. Vis. Exp. 2014, 87, e51284. [Google Scholar]
- Jiang, D.; Chen, S.; Sun, R.; Zhang, X.; Wang, D. The NLRP3 Inflammasome: Role in Metabolic Disorders and Regulation by Metabolic Pathways. Cancer Lett. 2018, 419, 8–19. [Google Scholar] [CrossRef]
- Volpe, C.M.; Anjos, P.M.; Nogueira-Machado, J.A. Inflammasome as a New Therapeutic Target for Diabetic Complications. Recent Pat. Endocr. Metab. Immune Drug Discov. 2016, 10, 56–62. [Google Scholar] [CrossRef]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular Mechanisms Controlling Caspase Activation and Function. Cold Spring Harb. Perspect. Biol. 2013, 5, a008672. [Google Scholar] [CrossRef]
- Kuranaga, E. Beyond Apoptosis: Caspase Regulatory Mechanisms and Functions in Vivo. Genes Cells 2012, 2, 83–97. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase Functions in Cell Death and Disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a008656. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.J.; Henry, C.M.; Cullen, S.P. A Perspective on Mammalian Caspases as Positive and Negative Regulators of Inflammation. Mol. Cell 2012, 4, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.L.G.; Poreba, M.; Snipas, S.J.; Groborz, K.; Drag, M.; Salvesen, G.S. Extensivepeptide and Natural Protein Substrate Screens Reveal That Mouse Caspase-11 Has Much Narrower Substrate Specificity Than Caspase-1. J. Biol. Chem. 2018, 18, 7058–7067. [Google Scholar] [CrossRef] [Green Version]
- Xi, H.; Zhang, Y.; Xu, Y.; Yang, W.Y.; Jiang, X.; Sha, X.; Cheng, X.; Wang, J.; Qin, X.; Yu, Y.; et al. Caspase-1 Inflammasome Activation Mediates Homocysteine-Induced Pyrop-Apoptosis in Endothelial Cells. Circ. Res. 2016, 118, 1525–1539. [Google Scholar] [CrossRef] [Green Version]
- Junger, W.G. Immune Cell Regulation by Autocrine Purinergic Signalling. Nat. Rev. Immunol. 2011, 3, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Fettelschoss, A.; Kistowska, M.; LeibundGut-Landmann, S.; Beer, H.D.; Johansen, P.; Senti, G.; Contassot, E.; Bachmann, M.F.; French, L.E.; Oxenius, A.; et al. Inflammasome Activation and IL-1β Target IL-1α for Secretion as Opposed to Surface expression. Proc. Natl. Acad. Sci. USA 2011, 108, 18055–18060. [Google Scholar] [CrossRef] [Green Version]
- Platnich, J.M.; Muruve, D.A. NOD-Like Receptors and Inflammasomes: A Review of Their Canonical and Non-canonical Signaling Pathways. Arch. Biochem. Biophys. 2019, 18, 30994–30999. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and Chemokines: At the Crossroads of Cell Signalling and Inflammatory Disease. Biochim. Biophys. Acta 2014, 11, 2563–2582. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, N.C.; Shayakhmetov, D.M. Interleukin 1α and the Inflammatory Process. Nat. Immunol. 2016, 8, 906–913. [Google Scholar] [CrossRef] [Green Version]
- Crișan, T.O.; Netea, M.G.; Joosten, L.A. Innate Immune Memory: Implications for Host Responses to Damage-Associated Molecular Patterns. Eur. J. Immunol. 2016, 4, 817–828. [Google Scholar] [CrossRef]
- Krakauer, T. Inflammasomes, Autophagy, and Cell Death: The Trinity of Innate Host Defense against Intracellular Bacteria. Mediat. Inflamm. 2019, 2019, 2471215. [Google Scholar] [CrossRef] [Green Version]
- Boutari, C.; Mantzoros, C.S. Inflammation: A Key Player Linking Obesity with Malignancies. Metabolism 2018, 81, A3–A6. [Google Scholar] [CrossRef]
- Assmann, T.S.; Brondani, L.D.A.; Bouças, A.P.; Canani, L.H.; Crispim, D. Toll-Like Receptor 3 (TLR3) and the Development of Type 1 Diabetes Mellitus. Arch. Endocrinol. Metab. 2015, 1, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 1, 16–34. [Google Scholar] [CrossRef]
- MacLeod, A.S.; Mansbridge, J.N. The Innate Immune System in Acute and Chronic Wounds. Adv. Wound. Care. 2016, 2, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Martín-Sánchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.I.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.; Mortellaro, A.; et al. Inflammasome-Dependent IL-1β Release Depends Upon Membrane Permeabilisation. Cell Death Differ. 2016, 23, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- England, H.; Summersgill, H.R.; Edye, M.E.; Rothwell, N.J.; Brough, D. Release of Interleukin-1α or Interleukin-1β Depends on Mechanism of Cell Death. J. Biol. Chem. 2014, 23, 15942–15950. [Google Scholar] [CrossRef] [Green Version]
- Robbins, G.R.; Wen, H.; Ting, J.P. Inflammasomes and Metabolic Disorders: Old Genes in Modern Diseases. Mol. Cell 2014, 54, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol. Rev. 2018, 1, 8–27. [Google Scholar] [CrossRef]
- Menu, P.; Vince, J.E. The NLRP3 Inflammasome in Health and Disease: The Good, the Bad and the Ugly. Clin. Exp. Immunol. 2011, 1, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Speaker, K.J.; Fleshner, M. Interleukin-1 Beta: A Potential Link between Stress and the Development of Visceral Obesity. BMC Physiol. 2012, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Grant, R.W.; Dixit, V.D. Mechanisms of Disease: Inflammasome Activation and the Development of Type 2 Diabetes. Front. Immunol. 2013, 4, 50. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.; Pessin, J.E. Adipokines Mediate Inflammation and Insulin Resistance. Front. Endocrinol. 2013, 4, 71. [Google Scholar] [CrossRef] [Green Version]
- McGillicuddy, F.C.; Harford, K.A.; Reynolds, C.M.; Oliver, E.; Claessens, M.; Mills, K.H.; Roche, H.M. Lack of Interleukin-1 Receptor I (Il-1ri) Protects Mice from High-Fat Diet-Induced Adipose Tissue Inflammation Coincident with Improved Glucose Homeostasis. Diabetes 2011, 6, 1688–1698. [Google Scholar] [CrossRef] [Green Version]
- Peiró, C.; Lorenzo, Ó.; Carraro, R.; Sánchez-Ferrer, C.F. IL-1β Inhibition in Cardiovascular Complications Associated to Diabetes Mellitus. Front. Pharmacol. 2017, 8, 363. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Li, Y.; Shu, T.; Wang, J. Cytokines and Inflammation in a Dipogenesis: An Updated Review. Front. Med. 2019, 13, 314–329. [Google Scholar] [CrossRef]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-Induced β Cell Production of IL-1β Contributes to Glucotoxicity in Human Pancreatic Islets. J. Clin. Investig. 2017, 127, 1589. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-Cells in Type 1 and Type 2 Diabetes Mellitus: Different Pathways to Failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of Pancreatic Beta-Cell Death in Type 1 and Type 2 Diabetes: Many Differences, Few Similarities. Diabetes 2005, 54, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, Inflammation, and Metabolic Disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Dobrian, A.D.; Morris, M.A.; Nadler, J.L. Islet Inflammation: A Unifying Target for Diabetes Treatment? Trends Endocrinol. Metab. 2013, 24, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Cieślak, M.; Wojtczak, A.; Cieślak, M. Role of Pro-Inflammatory Cytokines of Pancreatic Islets and Prospects of Elaboration of New Methods for the Diabetes Treatment. Acta Biochim. Pol. 2015, 62, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules 2019, 9, 850. [Google Scholar] [CrossRef] [Green Version]
- Erion, D.M.; Park, H.J.; Lee, H.Y. The Role of Lipids in the Pathogenesis and Treatment of Type 2 Diabetes and Associated Co-morbidities. BMB Rep. 2016, 49, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The Metabolic Syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Yasuda, K.; Nakanishi, K.; Tsutsui, H. Interleukin-18 in Health and Disease. Int. J. Mol. Sci. 2019, 20, 649. [Google Scholar] [CrossRef] [Green Version]
- Pradeep, A.R.; Hadge, P.; Chowdhry, S.; Patel, S.; Happy, D. Exploring the Role of Th1 Cytokines: Interleukin-17 and Interleukin-18 in Periodontal Health and Disease. J. Oral. Sci. 2009, 51, 261–266. [Google Scholar] [CrossRef] [Green Version]
- Tone, M.; Thompson, S.A.; Tone, Y.; Fairchild, P.J.; Waldmann, H. Regulation of IL-18 (IFN-Gamma-Inducing Factor) Gene Expression. J. Immunol. 1997, 159, 6156–6163. [Google Scholar]
- Ververs, F.A.; Kalkhoven, E.; Van’t Land, B.; Boes, M.; Schipper, H.S. Immunometabolic Activation of Invariant Natural Killer T Cells. Front. Immunol. 2018, 9, 1192. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Cho, D.H.; Park, H.J. IL-18 and Cutaneous Inflammatory Diseases. Int. J. Mol. Sci. 2015, 12, 29357–29369. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Novel Targets for Interleukin 18 Binding Protein. Ann. Rheum. Dis. 2001, 60, 18–24. [Google Scholar]
- Bellora, F.; Castriconi, R.; Doni, A.; Cantoni, C.; Moretta, L.; Mantovani, A.; Moretta, A.; Bottino, C. M-CSF Induces the Expression of a Membrane-Bound Form of IL-18 in a Subset of Human Monocytes Differentiating in Vitro toward Macrophages. Eur. J. Immunol. 2012, 42, 1618–1626. [Google Scholar] [CrossRef]
- Do, D.V.; Ong, C.T.; Khoo, Y.T.; Carbone, A.; Lim, C.P.; Wang, S.; Mukhopadhyay, A.; Cao, X.; Cho, D.H.; Wei, X.Q.; et al. Interleukin-18 System Plays an Importantrole in Keloid Pathogenesis via Epithelial-Mesenchymal Interactions. Br. J. Dermatol. 2012, 166, 1275–1288. [Google Scholar] [CrossRef]
- Yamaoka-Tojo, M.; Tojo, T.; Wakaume, K.; Kameda, R.; Nemoto, S.; Takahira, N.; Masuda, T.; Izumi, T. Circulating Interleukin-18: A Specific Biomarker for Atherosclerosis-Prone Patients with Metabolic Syndrome. Nutr. Metab. 2011, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Akira, S. The Role of IL-18 in Innate Immunity. Curr. Opin. Immunol. 2000, 1, 59–63. [Google Scholar] [CrossRef]
- Slaats, J.; Ten Oever, J.; van de Veerdonk, F.L.; Netea, M.G. IL-1β/IL-6/CRP and IL-18/ferritin: Distinct Inflammatory Programs in Infections. PLoS Pathogens 2016, 12, e1005973. [Google Scholar] [CrossRef]
- Okamura, H.; Tsutsui, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a New Cytokine That Induces IFN-γ Production by T Cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef]
- Wedell-Neergaard, A.S.; Krogh-Madsen, R.; Petersen, G.L.; Hansen, Å.M.; Pedersen, B.K.; Lund, R.; Bruunsgaard, H. Cardiorespiratory Fitness and the Metabolic Syndrome: Roles of Inflammation and Abdominal Obesity. PLoS ONE. 2018, 13, e0194991. [Google Scholar] [CrossRef]
- Krogh-Madsen, R.; Plomgaard, P.; Møller, K.; Mittendorfer, B.; Pedersen, B.K. Influence of TNF-Alpha and IL-6 Infusions on Insulin Sensitivity and Expression of IL-18 in Humans. Am. J. Physiol. Endocrinol. Metab. 2006, 291, 108–114. [Google Scholar] [CrossRef]
- Lindegaard, B.; Matthews, V.B.; Brandt, C.; Hojman, P.; Allen, T.L.; Estevez, E.; Watt, M.J.; Bruce, C.R.; Mortensen, O.H.; Syberg, S.; et al. Interleukin-18 Activates Skeletal Muscle AMPK and Reduces Weight Gain and Insulin Resistance in Mice. Diabetes 2013, 62, 3064–3074. [Google Scholar] [CrossRef] [Green Version]
- Esposito, K.; Nappo, F.; Giugliano, F.; Di Palo, C.; Ciotola, M.; Barbieri, M.; Paolisso, G.; Giugliano, D. Meal Modulation of Circulating Interleukin 18 and Adiponectin Concentrations in Healthy Subjects and in Patients with Type 2 Diabetes Mellitus. Am. J. Clin. Nutr. 2003, 78, 1135–1140. [Google Scholar] [CrossRef] [Green Version]
- Thorand, B.; Kolb, H.; Baumert, J.; Koenig, W.; Chambless, L.; Meisinger, C.; Illig, T.; Martin, S.; Herder, C. Elevated Levels of Interleukin-18 Predict the Development of Type 2 Diabetes: Results from the MONICA/KORA Augsburg Study, 1984–2002. Diabetes 2005, 54, 2932–2938. [Google Scholar] [CrossRef]
- Zhuang, H.; Han, J.; Cheng, L.; Liu, S.-L. A Positive Causal Influence of IL-18 Levels on the Risk of t2dm: A Mendelian Randomization Study. Front. Genet. 2019, 10, 295. [Google Scholar] [CrossRef] [Green Version]
- Campden, R.I.; Zhang, Y. The Role of Lysosomal Cysteine Cathepsins in NLRP3 Inflammasome Activation. Arch. Biochem. Biophys. 2019, 670, 32–42. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent Advances in the Mechanisms of NLRP3 Inflammasome Activation and Its Inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Yabal, M.; Calleja, D.J.; Simpson, D.S.; Lawlor, K.E. Stressing Out the Mitochondria: Mechanistic Insights into NLRP3 Inflammasome Activation. J. Leukoc. Biol. 2019, 105, 377–399. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and Regulation of the Inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Lee, S.; Suh, G.Y.; Ryter, S.W.; Choi, A.M. Regulation and Function of the Nucleotide Binding Domain Leucine-Rich Repeat-Containing Receptor, Pyrin Domain-Containing-3 Inflammasome in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Próchnicki, T.; Mangan, M.S.; Latz, E. Recent Insights into the Molecular Mechanisms of the NLRP3 Inflammasome Activation. F1000Research 2016, 4692016. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Chai, J. Structural Mechanisms in NLR Inflammasome Assembly and Signaling. Curr. Top. Microbiol. Immunol. 2016, 397, 23–42. [Google Scholar]
- Chen, Y.; Wang, L.; Pitzer, A.L.; Li, X.; Li, P.L.; Zhang, Y. Contribution of Redox-Dependent Activation of Endothelial NLRP3 Inflammasomes to Hyperglycemia-Induced Endothelial Dysfunction. J. Mol. Med. 2016, 94, 1335–1347. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends. Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.; Schilling, J.D. Lysosomes Integrate Metabolic-Inflammatory Cross-Talk in Primary Macrophage Inflammasome Activation. J. Biol. Chem. 2014, 289, 9158–9171. [Google Scholar] [CrossRef] [Green Version]
- Shim, D.W.; Lee, K.H. Posttranslational Regulation of the NLR Family Pyrin Domain-Containing 3 Inflammasome. Front. Immunol. 2018, 9, 1054. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Zhang, X.; Xiao, Y.; Zhang, W.; Wang, Y.; Ma, W.; Wang, X.; Song, P.; Lai, L.; Chen, H.; et al. ABRO1 Promotes NLRP3 Inflammasome Activation through Regulation of NLRP3 Deubiquitination. EMBO J. 2019, 38, e100376. [Google Scholar] [CrossRef]
- Cersosimo, E.; DeFronzo, R.A. Insulin Resistance and Endothelial Dysfunction: The Road Map to Cardiovascular Diseases. Diabetes Metab. Res. Rev. 2006, 22, 423–436. [Google Scholar] [CrossRef]
- Murakami, M.; Simons, M. Regulation of Vascular Integrity. J. Mol. Med. 2009, 87, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial Dysfunction and Platelet Hyperactivity in Type 2 Diabetes Mellitus: Molecular Insights and Therapeutic Strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef]
- Avogaro, A.; Albiero, M.; Menegazzo, L.; de Kreutzenberg, S.; Fadini, G.P. Endothelial Dysfunction in Diabetes: The Role of Reparatory Mechanisms. Diabetes Care 2011, 34, S285–S290. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The Vascular Endothelium and Human Diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The Sterile Inflammatory Response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef] [Green Version]
- Land, W.G. The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I—Promoting Inflammation and Immunity. Sultan Qaboos Univ. Med. J. 2015, 15, e9–e21. [Google Scholar]
- Land, W.G. The Role of Damage-Associated Molecular Patterns (DAMPs) in Human Diseases: Part II: DAMPs as Diagnostics, Prognostics and Therapeutics in Clinical Medicine. Sultan Qaboos Univ. Med. J. 2015, 15, e157–e170. [Google Scholar]
- Meza, C.A.; La Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef] [Green Version]
- Smolders, V.F.; Zodda, E.; Quax, P.H.A.; Carini, M.; Barberà, J.A.; Thomson, T.M.; Tura-Ceide, O.; Cascante, M. Metabolic Alterations in Cardiopulmonary Vascular Dysfunction. Front. Mol. Biosci. 2019, 5, 120. [Google Scholar] [CrossRef]
- Park, K.H.; Park, W.J. Endothelial Dysfunction: Clinical Implications in Cardiovascular Disease and Therapeutic Approaches. J. Korean Med. Sci. 2015, 30, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Sánchez, A.; Madrigal-Santillán., E.; Bautista., M.; Esquivel-Soto, J.; Morales-González, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, Oxidative Stress, and Obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef] [Green Version]
- Francisqueti, F.V.; Chiaverini, L.C.; Santos, K.C.; Minatel, I.O.; Ronchi, C.B.; Ferron, A.J.; Ferreira, A.L.; Corrêa, C.R. The Role of Oxidative Stress on the Pathophysiology of Metabolic Syndrome. Rev. Assoc. Med. Bras. 2017, 63, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, N.; Ozono, R.; Higashi, Y.; Maeda, R.; Kihara, Y. Association of Insulin Resistance, Plasma Glucose Level, and Derum Insulin Level with Hypertension in a Population with Different Stages of Impaired Glucose Metabolism. J. Am. Heart. Assoc. 2020, 9, e015546. [Google Scholar] [CrossRef]
- Wang, F.; Guo, X.; Shen, X.; Kream, R.M.; Mantione, K.J.; Stefano, G.B. Vascular Dysfunction Associated with Type 2 Diabetes and Alzheimer’s Disease: A Potential Etiological Linkage. Med. Sci. Monit. Basic Res. 2014, 20, 118–129. [Google Scholar]
- Adams, V.; Linke, A. Impact of Exercise Training on Cardiovascular Disease and Risk. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 728–734. [Google Scholar] [CrossRef]
- Hadi, H.A.; Suwaidi, J.A. Endothelial Dysfunction in Diabetes Mellitus. Vasc. Health Risk Manag. 2007, 3, 853–876. [Google Scholar]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Forouhi, N.G.; Misra, A.; Mohan, V.; Taylor, R.; Yancy, W. Dietary and Nutritional Approaches for Prevention and Management of Type 2 Diabetes. BMJ 2018, 361, k2234. [Google Scholar] [CrossRef] [Green Version]
- Paneni, F.; Beckman, J.A.; Creager, M.A.; Cosentino, F. Diabetes and Vascular Disease: Pathophysiology, Clinical Consequences, and Medical Therapy: Part I. Heart J. 2013, 34, 2436–2443. [Google Scholar] [CrossRef]
- Henning, R.J. Type-2 Diabetes Mellitus and Cardiovascular Disease. Future Cardiol. 2018, 14, 491–509. [Google Scholar] [CrossRef]
- Bakker, W.; Eringa, E.C.; Sipkema, P.; van Hinsbergh, V.W.M. Endothelial Dysfunction and Diabetes: Roles of Hyperglycemia, Impaired Insulin Signaling and Obesity. Cell Tissue Res. 2009, 335, 165. [Google Scholar] [CrossRef] [Green Version]
- Lteif, A.; Vaishnava, P.; Baron, A.D.; Mather, K.J. Endothelin Limits Insulin Action in Obese/Insulin-Resistant Humans. Diabetes 2007, 56, 728–734. [Google Scholar] [CrossRef] [Green Version]
- Mather, K.; Anderson, T.J.; Verma, S. Insulin Action in the Vasculature: Physiology and Pathophysiology. J. Vasc. Res. 2001, 38, 415–422. [Google Scholar] [CrossRef]
- Vincent, M.A.; Montagnani, M.; Quon, M.J. Molecular and Physiologic Actions of Insulin Related to Production of Nitric Oxide in Vascular Endothelium. Curr. Diab. Rep. 2003, 3, 279–288. [Google Scholar] [CrossRef]
- Muniyappa, R.; Montagnani, M.; Koh, K.K.; Quon, M.J. Cardiovascular Actions of Insulin. Endocr. Rev. 2007, 28, 463–491. [Google Scholar] [CrossRef]
- Janus, A.; Szahidewicz-Krupska, E.; Mazur, G.; Doroszko, A. Insulin Resistance and Endothelial Dysfunction Constitute a Common Therapeutic Target in Cardiometabolic Disorders. Mediat. Inflamm. 2016, 2016, 3634948. [Google Scholar] [CrossRef] [Green Version]
- Muniyappa, R.; Sowers, J.R. Role of Insulin Resistance in Endothelial Dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef]
- Siegelaar, S.E.; Holleman, F.; Hoekstra, J.B.; DeVries, J.H. Glucose Variability; Does It Matter? Endocr. Rev. 2010, 31, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M. Reactive Oxygen and Nitrogen Species in Pathogenesis of Vascular Complications of Diabetes. Diabetes Metab. J. 2012, 36, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Takeda, Y.; Matoba, K.; Sekiguchi, K.; Nagai, Y.; Yokota, T.; Utsunomiya, K.; Nishimura, R. Endothelial Dysfunction in Diabetes. Biomedicines 2020, 8, 182. [Google Scholar] [CrossRef]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of Reactive Oxygen Species in the Progression of Type 2 Diabetes and Atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef] [Green Version]
- Widlansky, M.E.; Gutterman, D.D. Regulation of Endothelial Function by Mitochondrial Reactive Oxygen Species. Antioxid. Redox Signal. 2011, 15, 1517–1530. [Google Scholar] [CrossRef] [Green Version]
- Maamoun, H.; Benameur, T.; Pintus, G.; Munusamy, S.; Agouni, A. Crosstalk between Oxidative Stress and Endoplasmic Reticulum (ER) Stress in Endothelial Dysfunction and Aberrant Angiogenesis Associated with Diabetes: A Focus on the Protective Roles of Heme Oxygenase (HO)-1. Front. Physiol. 2019, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, The Dark Side of Glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Maessen, D.E.; Stehouwer, C.D.; Schalkwijk, C.G. The Role of Methylglyoxal and the Glyoxalase System in Diabetes and Other Age-Related Diseases. Clin. Sci. 2015, 128, 839–861. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, J. New Insights of µ-Calpain in the Pathogenesis of Diabetic Vascular Injury. Diabetes 2015, 64, 693–695. [Google Scholar] [CrossRef] [Green Version]
- Booth, A.A.; Khalifah, R.G.; Todd, P.; Hudson, B.G. In Vitro Kinetic Studies of Formation of Antigenic Advanced Glycation End Products (AGEs) Novel Inhibition of Post-Amadori Glycation Pathways. J. Biol. Chem. 1997, 272, 5430–5437. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M.; Vlassara, H.; Cerami, A. Nonenzymatic Glycosylation and the Pathogenesis of Diabetic Complications. Ann. Intern. Med. 1984, 101, 527–537. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thallas, V.; Thomas, M.C.; Founds, H.W.; Burns, W.C.; Jerums, G.; Cooper, M.E. The Breakdown of Preexisting Advanced Glycation End Products Is Associated with Reduced Renal Fibrosis in Experimental Diabetes. FASEB J. 2003, 17, 1762–1764. [Google Scholar] [CrossRef]
- Litwinoff, E.; Hurtado Del Pozo, C.; Ramasamy, R.; Schmidt, A.M. Emerging Targets for Therapeutic Development in Diabetes and Its Complications: The RAGE Signaling Pathway. Clin. Pharmacol. Ther. 2015, 98, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-Associated Sustained Activation of the Transcription Factor Nuclear Factor-kappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef] [Green Version]
- Younce, C.W.; Wang, K.; Kolattukudy, P.E. Hyperglycaemia-Induced Cardiomyocyte Death Is Mediated via MCP-1 Production and Induction of a Novel Zinc-Finger Protein MCPIP. Cardiovasc. Res. 2010, 87, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Primer, K.R.; Psaltis, P.J.; Tan, J.T.M.; Bursill, C.A. The Role of High-Density Lipoproteins in Endothelial Cell Metabolism and Diabetes-Impaired Angiogenesis. Int. J. Mol. Sci. 2020, 21, 3633. [Google Scholar] [CrossRef]
- Beleznai, T.; Bagi, Z. Activation of Hexosamine Pathway Impairs Nitric Oxide (NO)-Dependent Arteriolar Dilations by Increased Protein O-GlcNAcylation. Vascul. Pharmacol. 2012, 56, 115–1121. [Google Scholar] [CrossRef] [Green Version]
- Beyer, A.M.; Weihrauch, D. Hexosamine Pathway Activation and O-Linked-N-Acetylglucosamine: Novel Mediators of Endothelial Dysfunction in Hyper-Glycemia and Diabetes. Vascul. Pharmacol. 2012, 56, 113–114. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Hummasti, S.; Hotamisligil, G.S. Endoplasmic Reticulum Stress and Inflammation in Obesity and Diabetes. Circ. Res. 2010, 107, 579–591. [Google Scholar] [CrossRef]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Özcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Flamment, M.; Hajduch, E.; Ferré, P.; Foufelle, F. New Insights into ER Stress-Induced Insulin Resistance. Trends Endocrinol. Metab. 2012, 23, 381–390. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The Endoplasmic Reticulum: Structure, Function and Response to Cellular Signaling. Cell Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Marciniak, S.J.; Ron, D. Endoplasmic Reticulum Stress Signaling in Disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. The Mammalian Unfolded Protein Response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Basha, B.; Samuel, S.M.; Triggle, C.R.; Ding, H. Endothelial Dysfunction in Diabetes Mellitus: Possible Involvement of Endoplasmic Reticulum Stress? Exp. Diabetes Res. 2012, 2012, 481840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooradian, A.D.; Haas, M.J. Glucose-Induced Endoplasmic Reticulum Stress Is Independent of Oxidative Stress: A Mechanistic Explanation for the Failure of Antioxidant Therapy in Diabetes. Free Radic. Biol. Med. 2011, 50, 1140–1143. [Google Scholar] [CrossRef] [PubMed]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int. J. Mol. Sci. 2019, 20, 1658. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Fernandes, C.; Liu, Y.; Wu, Y.; Wu, H.; Brophy, M.L.; Deng, L.; Song, K.; Wen, A.; Wong, S.; et al. Role of Endoplasmic Reticulum Stress Signalling in Diabetic Endothelial Dysfunction and Atherosclerosis. Diabetes Vasc. Dis. Res. 2017, 14, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Villalobos-Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, L. Endoplasmic Reticulum Stress and Development of Insulin Resistance in Adipose, Skeletal, Liver, and Foetoplacental Tissue in Diabesity. Mol. Asp. Med. 2019, 66, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Contreras, A.G.; Dormond, O.; Edelbauer, M.; Calzadilla, K.; Hoerning, A.; Pal, S.; Briscoe, D.M. mTOR-Understanding the Clinical Effects. Transplant. Proc. 2008, 40, S9–S12. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Gravel, S.P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR Coordinates Protein Synthesis, Mitochondrial Activity and Proliferation. Cell Cycle. 2015, 14, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Zhang, W. Role of mTOR in Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2018, 19, 2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, M.S. The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Reho, J.J.; Guo, D.F.; Rahmouni, K. Mechanistic Target of Rapamycin Complex 1 Signaling Modulates Vascular Endothelial Function through Reactive Oxygen Species. J. Am. Heart. Assoc. 2019, 8, e010662. [Google Scholar] [CrossRef]
- Decker, B.; Pumiglia, K. mTORc1 Activity Is Necessary and Sufficient for Phosphorylation of eNOSS1177. Physiol. Rep. 2018, 6, e13733. [Google Scholar] [CrossRef] [PubMed]
- Lei-Leston, A.C.; Murphy, A.G.; Maloy, K.J. Epithelial Cell Inflammasomes in Intestinal Immunity and Inflammation. Front. Immunol. 2017, 8, 1168. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Boini, K.M.; Pitzer, A.L.; Gulbins, E.; Zhang, Y.; Li, P.L. Endothelial NLRP3 Inflammasome Activation Associated with Lysosomal Destabilization during Coronary Arteritis. Biochim. Biophys. Acta 2015, 1853, 396–408. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, Y.; Li, X.; Zhang, Y.; Gulbins, E.; Zhang, Y. Enhancement of Endothelial Permeability by Free Fatty Acid through Lysosomal Cathepsin B-Mediated NLRP3 Inflammasome Activation. Oncotarget 2016, 7, 73229–73241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Z.; Fan, Y.; Liu, X.; Xue, J.; Han, Z.; Zhu, C.; Wang, X. NLRP3 Inflammasome Promotes Diabetes-Induced Endothelial Inflammation and Atherosclerosis. Diabetes, Metab. Syndr. Obesity Targets Ther. 2019, 12, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, M.; Sahraoui, A.; Høyem, M.; Øgaard, J.; Lien, E.; Aukrust, P.; Yndestad, A.; Ranheim, T.; Scholz, H. NLRP3 Inflammasome Mediates Oxidative Stress-Induced Pancreatic Islet Dysfunction. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E912–E923. [Google Scholar] [CrossRef]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a Link between Obesity, Metabolic Syndrome and Type 2 Diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N. Innate Immune Activation in Obesity. Mol. Aspects Med. 2013, 34, 12–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanti, J.F.; Ceppo, F.; Jager, J.; Berthou, F. Implication of Inflammatory Signalingpathways in Obesity-Induced Insulin Resistance. Front. Endocrinol. 2013, 3, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar]
- Ringling, R.E.; Gastecki, M.L.; Woodford, M.L.; Lum-Naihe, K.J.; Grant, R.W.; Pulakat, L.; Vieira-Potter, V.J.; Padilla, J. Loss of Nlrp3 Does Not Protect Mice from Western Diet-Induced Adipose Tissue Inflammation and Glucose Intolerance. PLoS ONE 2016, 11, e0161939. [Google Scholar] [CrossRef] [PubMed]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflamm. 2013, 2013, 139239. [Google Scholar] [CrossRef] [Green Version]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Esser, N.; L’homme, L.; De Roover, A.; Kohnen, L.; Scheen, A.J.; Moutschen, M.; Piette, J.; Legrand-Poels, S.; Paquot, N. Obesity Phenotype Is Related to nlrp3 Inflammasome Activity and Immunological Profile of Visceral Adipose Tissue. Diabetologia 2013, 56, 2487–2497. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Deng, T.; Peterson, L.E.; Yu, R.; Lin, J.; Hamilton, D.J.; Reardon, P.R.; Sherman, V.; Winnier, G.E.; Zhan, M.; et al. Transcriptome Analysis of Human Adipocytes Implicates the NOD-Like Receptor Pathway in Obesity-Induced Adipose Inflam-Mation. Mol. Cell Endocrinol. 2014, 394, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; He, G.; Peng, Y.; Zhong, W.; Wang, Y.; Zhang, B. Sodium Butyrate Alleviatesadipocyte Inflammation by Inhibiting NLRP3 Pathway. Sci. Rep. 2015, 5, 12676. [Google Scholar] [CrossRef] [Green Version]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N.; et al. Monounsaturated Fatty Acid-Enriched High-Fat Diets Impede Adipose NLRP3 Inflammasome-Mediated IL-1β Secretion and Insulin Resistance despite Obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, C.; Yin, H.; Zhang, X.; Li, Y. NLRP3 Inflammasome: A Potential Alternative Therapy Target for Atherosclerosis. Evid. Based Complement. Alternat. Med. 2020, 15, 0718. [Google Scholar] [CrossRef] [PubMed]
- Arend, W.P.; Palmer, G.; Gabay, C. IL-1, IL-18, and IL-33 Families of Cytokines. Immunol. Rev. 2008, 223, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, A.K.; Ortis, F.; Storling, J.; Feng, Y.M.; Rasschaert, J.; Tonnesen, M.; Van Eylen, F.; Mandrup-Poulsen, T.; Herchuelz, A.; Eizirik, D.L. Cytokines Downregulate the Sarcoendoplasmic Reticulum Pump Ca2+ Atpase 2b and Deplete Endoplasmic Reticulum Ca2+, Leading to Induction of Endoplasmic Reticulum Stress in Pancreatic Beta-Cells. Diabetes 2005, 54, 452–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, G.; Datta, M. IL-1beta Induces ER Stress in a JNK Dependent Manner that Determines Cell Death in Human Pancreatic Epithelial MIA PaCa-2 Cells. Apoptosis 2010, 15, 864–876. [Google Scholar] [CrossRef]
- Banerjee, M.; Saxena, M. Interleukin-1 (IL-1) Family of Cytokines: Role in Type 2 Diabetes. Clin. Chim. Acta 2012, 413, 1163–1170. [Google Scholar] [CrossRef]
- Bitto, A.; Altavilla, D.; Pizzino, G.; Irrera, N.; Pallio, G.; Colonna, M.R.; Squadrito, F. Inhibition of Inflammasome Activation Improves the Impaired Pattern of Healing in Genetically Diabetic Mice. Br. J. Pharmacol. 2014, 171, 2300–2307. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Wang, W.; Okla, M.; Kang, I.; Moreau, R.; Chung, S. Suppression of NLRP3 Inflammasome by γ-Tocotrienol Ameliorates Type 2 Diabetes. J. Lipid. Res. 2016, 57, 66–76. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 Directly Targets the NLRP3 ATP-Hydrolysis Motif for Inflammasome Inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide Inhibits the Cryopyrin/NALP3 Inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 Inflammasome Activation in Patients with Type 2 Diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Henriksbo, B.D.; Lau, T.C.; Cavallari, J.F.; Denou, E.; Chi, W.; Lally, J.S.; Crane, J.D.; Duggan, B.M.; Foley, K.P.; Fullerton, M.D.; et al. Fluvastatin Causes NLRP3 Inflammasome-Mediated Adipose Insulin Resistance. Diabetes 2014, 63, 3742–3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dangwal, S.; Stratmann, B.; Bang, C.; Lorenzen, J.M.; Kumarswamy, R.; Fiedler, J.; Falk, C.S.; Scholz, C.S.; Thum, T.; Tschoepe, D. Impairment of Wound Healing in Patients with Type 2 Diabetes Mellitus Influences Circulating MicroRNA Patterns via in-Flammatory Cytokines. Arter. Thromb. Vasc. Biol. 2015, 35, 1480–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, R.; Siracusa, R.; Genovese, T.; Cuzzocrea, S.; Di Paola, R. Focus on the Role of NLRP3 Inflammasome in Diseases. Int. J. Mol. Sci. 2020, 21, 4223. [Google Scholar] [CrossRef] [PubMed]
- Sepehri, Z.; Kiani, Z.; Afshari, M.; Kohan, F.; Dalvand, A.; Ghavami, S. Inflammasomes and Type 2 Diabetes: An Updated Systematic Review. Immunol. Lett. 2017, 24, 97–103. [Google Scholar] [CrossRef]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 Inflammasome: From a Danger Signal Sensor to a Regulatory Node of Oxidative Stress and Inflammatory Diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef]
- Boden, G. Obesity, Insulin Resistance and Free Fatty Acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Engin, A. Fat Cell and Fatty Acid Turnover in Obesity. Adv. Exp. Med. Biol. 2017, 960, 135–160. [Google Scholar]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty Acid-Induced NLRP3-ASC Inflammasome Activation Interferes with Insulin Signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kien, C.L.; Bunn, J.Y.; Fukagawa, N.K.; Anathy, V.; Matthews, D.E.; Crain, K.I.; Ebenstein, D.B.; Tarleton, E.K.; Pratley, R.E.; Poynter, M.E. Lipidomic Evidence That Lowering the Typical Dietary Palmitate to Oleate Ratio in Humans Decreases the Leukocyte Production of Proinflammatory Cytokines and Muscle Expression of Redox-Sensitive Genes. J. Nutr. Biochem. 2015, 26, 1599–1606. [Google Scholar] [CrossRef] [Green Version]
- L’homme, L.; Esser, N.; Riva, L.; Scheen, A.; Paquot, N.; Piette, J.; Legrand-Poels, S. Unsaturated Fatty Acids Prevent Activation of NLRP3 Inflammasome in Human Monocytes/Macrophages. J. Lipid Res. 2013, 54, 2998–3008. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.K.; Cheung, S.W.; Cheng, K.K. NLRP3 Inflammasome Activation in Adipose Tissues and Its Implications on Metabolic Diseases. Int. J. Mol. Sci. 2020, 11, 4184. [Google Scholar] [CrossRef]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela- Arispe, M.L.; De Caterina, R. Endothelial Permeability, LDL Deposition, and Cardiovascular Risk Factors-A Review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M. Inflammasomes in the Gastrointestinal Tract: Infection, Cancer and Gut Microbiota Homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 12, 721–737. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Chan, K.L.; Zhang, S.; Mejdani, M.; Jacobson, M.R.; Ducos, A.; Bilan, P.J.; Niu, W.; Klip, A. Saturated Fatty Acids Activate Caspase-4/5 in Human Monocytes, Triggering IL-1β and IL-18 Release. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E825–E835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolowska, E.; Blachnio-Zabielska, A. The Role of Ceramides in Insulin Resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.; Chawla, S.; Guchhait, P. Type-2 Diabetes: Current Understanding and Future Perspectives. IUBMB Life 2015, 67, 506–513. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dalmas, É.; Sauter, N.S.; Böni-Schnetzler, M. Inflammation in Obesity and Diabetes: Islet Dysfunction and Therapeutic Opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Koenen, T.B.; Stienstra, R.; van Tits, L.J.; de Graaf, J.; Stalenhoef, A.F.; Joosten, L.A.; Tack, C.J.; Netea, M.G. Hyperglycemia Activates Caspase-1 and TXNIP-Mediated IL-1beta Transcription in Human Adipose Tissue. Diabetes 2011, 60, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yang, B.; Chen, M.; Klein, J.D.; Sands, J.M.; Chen, G. Activation of Protein Kinase C-α and Src Kinase Increases Urea Transporter a1 α-2, 6 Sialylation. J. Am. Soc. Nephrol. 2015, 26, 926–934. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Gu, J.; Gou, F.; Huang, W.; Gao, C.; Chen, G.; Long, Y.; Zhou, X.; Yang, M.; Liu, S.; et al. High Glucose and Lipopolysaccharide Prime NLRP3 Inflammasome via ROS/TXNIP Pathway in Mesangial Cells. J. Diabetes Res. 2016, 2016, 6973175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, X.; Lu, A.L.; Yao, X.M.; Hua, Q.; Li, X.Y.; Qin, L.; Zhang, H.M.; Meng, G.X.; Su, Q. Activation of NLRP3 Inflammasome by Advanced Glycation End Products Promotes Pancreatic Islet Damage. Oxid. Med. Cell Longev. 2017, 2017, 9692546. [Google Scholar]
- Chutkow, W.A.; Birkenfeld, A.L.; Brown, J.D.; Lee, H.Y.; Frederick, D.W.; Yoshioka, J.; Patwari, P.; Kursawe, R.; Cushman, S.W.; Plutzky, J.; et al. Deletion of the Alpha-Arrestin Protein Txnip in Mice Promotes Adiposity and Adipogenesis While Preserving Insulin Sensitivity. Diabetes 2010, 59, 1424–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in Metabolic Regulation: Physiological Role and Therapeutic Outlook. Curr. Drug. Targets 2017, 18, 1095–1103. [Google Scholar] [CrossRef] [Green Version]
- Lane, T.; Flam, B.; Lockey, R.; Kolliputi, N. TXNIP Shuttling: Missing Link between Oxidative Stress and Inflammasome Activation. Front. Physiol. 2013, 4, 50–53. [Google Scholar] [CrossRef] [Green Version]
- Braga, T.T.; Forni, M.F.; Correa-Costa, M.; Ramos, R.N.; Barbuto, J.A.; Branco, P.; Castoldi, A.; Hiyane, M.I.; Davanso, M.R.; Latz, E.; et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017, 7, 39884. [Google Scholar] [CrossRef]
- Ghaemi-Oskouie, F.; Shi, Y. The Role of Uric Acid as an Endogenous Danger Signal in Immunity and Inflammation. Curr. Rheumatol. Rep. 2011, 13, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Makri, P.; Liakopoulos, V.; Stefanidis, I. Urate Crystals Induce NLRP3 Inflammasome-Dependent IL-1β Secretion and Proliferation in Isolated Primary Human T-Cells. Hippokratia 2015, 19, 41–46. [Google Scholar]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 Inflammasome by Islet Amyloid Polypeptide Provides a Mechanism for Enhanced IL-1b in Type 2 Diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Costes, S. Targeting Protein Misfolding to Protect Pancreatic Beta-Cells in Type 2 Diabetes. Curr. Opin. Pharmacol. 2018, 43, 104–110. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morales-Scheihing, D.; Salvadores, N.; Moreno-Gonzalez, I.; Gonzalez, C.; Taylor-Presse, K.; Mendez, N.; Shahnawaz, M.; Gaber, A.O.; Sabek, O.M.; et al. Induction of IAPP Amyloid Deposition and Associated Diabetic Abnormalities by a Prion-like Mechanism. J. Exp. Med. 2017, 214, 2591–2610. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chang, Y.; Zhang, K.; Chen, H.; Tao, S.; Zhang, Z. Implication of the Gut Microbiome Composition of Type 2 Diabetic Patients from Northern China. Sci. Rep. 2020, 10, 5450. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of Diet on the Gut Microbiome and Implications for Human Health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [Green Version]
- Yu, A.I.; Zhao, L.; Eaton, K.A.; Ho, S.; Chen, J.; Poe, S.; Becker, J.; Gonzalez, A.; McKinstry, D.; Hasso, M.; et al. Gut Microbiota Modulate CD8 T Cell Responses to Influence Colitis- Associated Tumorigenesis. Cell Rep. 2020, 31, 107471. [Google Scholar] [CrossRef]
- Cervantes, J.; Nagata, T.; Uchijima, M.; Shibata, K.; Koide, Y. Intracytosolic Listeria Monocytogenes Induces Cell Death through Caspase-1 Activation in Murine Macrophages. Cell Microbiol. 2008, 10, 41–52. [Google Scholar] [CrossRef]
- Ng, J.; Hirota, S.A.; Gross, O.; Li, Y.; Ulke-Lemee, A.; Potentier, M.S.; Schenck, L.P.; Vilaysane, A.; Seamone, M.E.; Feng, H.; et al. Clostridium Difficile Toxin-Induced Inflammation and Intestinal Injury Are Mediated by the Inflammasome. Gastroenterology 2010, 2, 542–552. [Google Scholar] [CrossRef]
- Boxberger, N.; Hecker, M.; Zettl, U.K. Dysregulation of Inflammasome Priming and Activation by MicroRNAs in Human Immune-Mediated Diseases. J. Immunol. 2019, 202, 2177–2187. [Google Scholar] [CrossRef] [Green Version]
- Dehaini, H.; Awada, H.; El-Yazbi, A.; Zouein, F.A.; Issa, K.; Eid, A.A.; Ibrahim, M.; Badran, A.; Baydoun, E.; Pintus, G.; et al. MicroRNAs as Potential Pharmaco-Targets in Ischemia-Reperfusion Injury Compounded by Diabetes. Cells 2019, 8, 152. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yan, Y.; Xv, W.; Qian, G.; Li, C.; Zou, H.; Li, Y. A New Insight into the Roles of MiRNAs in Metabolic Syndrome. Biomed Res. Int. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.H.; Shen, C.A.; Zhu, B.W.; An, H.Y.; Zheng, B.; Xu, S.B.; Sun, J.C.; Sun, P.C.; Zhang, W.; Wang, J.; et al. Insight into miRNAs Related with Glucometabolic Disorder. Biomed. Pharmacother. 2019, 111, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Lorente-Cebrián, S.; González-Muniesa, P.; Milagro, F.I.; Martínez, J.A. MicroRNAs And Other Non-coding RNAs in Adipose Tissue and Obesity: Emerging Roles as Biomarkers and Therapeutic Targets. Clin. Sci. 2019, 133, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Zamani, P.; Oskuee, R.K.; Atkin, S.L.; Navashenaq, J.G.; Sahebkar, A. MicroRNAs as Important Regulators of the NLRP3 Inflammasome. Prog. Biophys. Mol. Biol. 2020, 150, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Guo, F. MicroRNAs and Type 2 Diabetes. ExRNA 2019, 36, 1. [Google Scholar] [CrossRef] [Green Version]
- Nemecz, M.; Alexandru, N.; Tanko, G.; Georgescu, A. Role of MicroRNA in Endothelial Dysfunction and Hypertension. Curr. Hypertens. Rep. 2016, 18, 87. [Google Scholar] [CrossRef]
- Olivieri, F.; Rippo, M.R.; Prattichizzo, F.; Babini, L.; Graciotti, L.; Recchioni, R.; Procopio, A.D. Toll Like Receptor Signaling in “inflammaging”: MicroRNA as New Players. Immun. Ageing 2013, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Jing, Z.; Cheng, G. MicroRNAs: New Regulators of Toll-like Receptor Signalling Pathways. BioMed Res. Int. 2014, 2014, 945169. [Google Scholar] [CrossRef] [Green Version]
- Haneklaus, M.; Gerlic, M.; O’Neill, L.A.; Masters, S.L. miR-223: Infection, Inflammation and Cancer. J. Intern. Med. 2013, 274, 215–226. [Google Scholar] [CrossRef]
- Kim, D.; Song, J.; Ahn, C.; Kang, Y.; Chun, C.H.; Jin, E.J. Peroxisomal Dysfunction Is Associated with Up-Regulation of Apoptotic Cell Death via miR-223 Induction in Knee Osteoarthritis Patients with Type 2 Diabetes Mellitus. Bone 2014, 64, 124–131. [Google Scholar] [CrossRef]
- Chuang, T.Y.; Wu, H.L.; Chen, C.C.; Gamboa, G.M.; Layman, L.C.; Diamond, M.P.; Azziz, R.; Chen, Y.H. MicroRNA-223 Expression is Upregulated in Insulin Resistant Human Adipose Tissue. J. Diabetes Res. 2015, 94, 3659. [Google Scholar] [CrossRef] [Green Version]
- Wen, D.; Qiao, P.; Wang, L. Circulating MicroRNA-223 as a Potential Biomarker for Obesity. Obes. Res. Clin. Pract. 2015, 9, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Kiechl, S.; Drozdov, I.; Willeit, P.; Mayr, U.; Prokopi, M.; Mayr, A.; Weger, S.; Oberhollenzer, F.; Bonora, E.; et al. Plasma MicroRNA Profiling Reveals Loss of Endothelial miR-126 and Other MicroRNAs in Type 2 Diabetes. Circ. Res. 2010, 107, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Mirra, P.; Nigro, C.; Prevenzano, I.; Leone, A.; Raciti, G.A.; Formisano, P.; Beguinot, F.; Miele, C. The Destiny of Glucose from a MicroRNA Perspective. Front. Endocrinol. 2018, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Buchan, R.J.; Cook, S.A. MicroRNA-223 Regulates Glut4 Expression and Cardiomyocyte Glucose Metabolism. Cardiovasc. Res. 2010, 86, 410–420. [Google Scholar] [CrossRef]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O’Neill, L.A.; Masters, S.L. Cutting Edge: miR-223 and EBV miR-BART15 Regulate the NLRP3 Inflammasome and IL-1β Production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, G.; Meng, C.; Guo, X.; Cheruku, P.S.; Shi, L.; Xu, H.; Li, H.; Wang, G.; Evans, A.R.; Safe, S.; et al. A Novel Regulator of Macrophage Activation: Mir-223 in Obesity-Associated Adipose Tissue Inflammation. Circulation 2012, 125, 2892–2903. [Google Scholar] [CrossRef] [Green Version]
- Shukla, G.C.; Singh, J.; Barik, S. MicroRNAs: Processing, Maturation, Target Recognition and Regulatory Functions. Mol. Cell Pharmacol. 2011, 3, 83–92. [Google Scholar]
- Bauernfeind, F.; Rieger, A.; Schildberg, F.A.; Knolle, P.A.; Schmid-Burgk, J.L.; Hornung, V. NLRP3 Inflammasome Activity Is Negatively Controlled by miR-223. J. Immunol. 2012, 189, 4175–4181. [Google Scholar] [CrossRef] [Green Version]
- LaPierre, M.P.; Stoffel, M. MicroRNAs as Stress Regulators in Pancreatic Beta Cells and Diabetes. Mol. Metab. 2017, 6, 1010–1023. [Google Scholar] [CrossRef]
- Xu, H.; Guo, S.; Li, W.; Yu, P. The Circular RNA Cdr1as, via miR-7 and Its Targets, Regulates Insulin Transcription and Secretion in Islet Cells. Sci. Rep. 2015, 5, 12453. [Google Scholar] [CrossRef]
- Ramachandran, D.; Roy, U.; Garg, S.; Ghosh, S.; Pathak, S.; Kolthur-Seetharam, U. Sirt1 and mir-9 Expression Is Regulated during Glucose-Stimulated Insulin Secretion in Pancreatic β-Islets. FEBS J. 2011, 278, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Plaisance, V.; Abderrahmani, A.; Perret-Menoud, V.; Jacquemin, P.; Lemaigre, F.; Regazzi, R. MicroRNA-9 Controls the Expression of Granuphilin/slp4 and the Secretory Response of Insulin-Producing Cells. J. Biol. Chem. 2006, 281, 26932–26942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumarswamy, R.; Volkmann, I.; Thum, T. Regulation and Function of miRNA-21 in Health and Disease. RNA Biol. 2011, 8, 706–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.J.; Hwang, S.J.; Bae, Y.C.; Jung, J.S. MiR-21 Regulates Adipogenic Differentiation through the Modulation of TGF-Beta Signaling in Mesenchymal Stem Cells Derived from Human Adipose Tissue. Stem Cells 2009, 27, 3093–3102. [Google Scholar] [PubMed]
- Loboda, A.; Sobczak, M.; Jozkowicz, A.; Dulak, J. TGF-β1/Smads and miR-21 in Renal Fibrosis and Inflammation. Mediat. Inflamm. 2016, 2016, 8319283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Sala, L.; Mrakic-Sposta, S.; Micheloni, S.; Prattichizzo, F.; Ceriello, A. Glucose-Sensing microRNA-21 Disrupts ROS Homeostasis and Impairs Antioxidant Responses in Cellular Glucose Variability. Cardiovasc. Diabetol. 2018, 17, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Huang, B.; Wang, H.Y.; Chang, A.; Zheng, X.F.S. Emerging Role of MicroRNAs in mTOR Signaling. Cell. Mol. Life Sci. 2017, 74, 2613–2625. [Google Scholar] [CrossRef]
- Ling, H.Y.; Hu, B.; Hu, X.B.; Zhong, J.; Feng, S.D.; Qin, L.; Liu, G.; Wen, G.B.; Liao, D.F. MiRNA-21 Reverses High Glucose and High Insulin Induced Insulin Resistance in 3T3-L1 Adipocytes through Targeting Phosphatase and Tensin Homologue. Exp. Clin. Endocrinol. Diabetes 2012, 120, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.Y.; Shen, B.R.; Zhang, Y.C.; Wan, X.J.; Yao, Q.P.; Wu, G.L.; Wang, J.Y.; Chen, S.G.; Yan, Z.Q.; Jiang, Z.L. Induction of Thoracic Aortic Remodeling by Endothelial-Specific Deletion of MicroRNA-21 in Mice. PLoS ONE 2013, 8, e59002. [Google Scholar]
- Esteves, J.V.; Enguita, F.J.; Machado, U.F. MicroRNAs-Mediated Regulation of Skeletal Muscle GLUT4 Expression and Translocation in Insulin Resistance. J. Diabetes Res. 2017, 2017, 7267910. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Lane, T.; Venugopal, R.; Parthasarathy, P.T.; Cho, Y.; Galam, L.; Lockey, R.; Kolliputi, N. MicroRNA-133a-1 Regulates Inflammasome Activation through Uncoupling Protein-2. Biochem. Biophys. Res. Commun. 2013, 439, 407e412. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Jiang, L.; Hu, Q.; Li, Y. MicroRNA-133b Ameliorates Allergic Inflammation and Symptom in Murine Model of Allergic Rhinitis by Targeting Nlrp3. Cell. Physiol. Biochem. 2017, 42, 901–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Gao, R.; Yan, B. Potential roles of MicroRNA-1 and MicroRNA-133 in Cardiovascular Disease. Rev. Cardiovasc. Med. 2020, 21, 57–64. [Google Scholar] [PubMed] [Green Version]
- Bhatt, K.; Lanting, L.L.; Jia, Y.; Yadav, S.; Reddy, M.A.; Magilnick, N.; Boldin, M.; Natarajan, N. Anti-inflammatory Role of MicroRNA-146a in the Pathogenesis of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2277–2288. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yue, Y.; Xu, W.; Xiong, S. MicroRNA-146a Represses Mycobacteriainduced Inflammatory Response and Facilitates Bacterial Replication via Targeting IRAK-1 and TRAF-6. PLoS ONE 2013, 8, e81438. [Google Scholar]
- Park, H.; Huang, X.; Lu, C.; Cairo, M.S.; Zhou, X. MicroRNA-146a and MicroRNA-146b Regulate Human Dendritic Cell Apoptosis and Cytokine Production by Targeting TRAF6 and IRAK1 Proteins. J. Biol. Chem. 2015, 290, 2831–2841. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.B.; Qing, Y.F.; Yin, C.C.; Zhou, L.; Liu, X.S.; Mi, Q.S.; Zhou, J.G. Mice with MiR-146a Deficiency Develop Severe Gouty Arthritis via Dysregulation of TRAF 6, IRAK 1 and NALP3 Inflammasome. Arthritis Res. Ther. 2018, 20, 45. [Google Scholar] [CrossRef] [Green Version]
- Cowan, C.; Muraleedharan, C.K.; O’Donnell, J.J. 3rd.; Singh, P.K.; Lum, H.; Kumar, A.; Xu, S. MicroRNA-146 Inhibits Thrombin-Induced NF-κB Activation and Subsequent Inflammatory Responses in Human Retinal Endothelial Cells. Invest. Ophthalmol. Vis. Sci. 2014, 55, 4944–4951. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.D.; Ohshiro, K.; Rayala, S.K.; Kumar, R. MicroRNA-7, A Homeobox D10 Target, Inhibits p21-Activated Kinase 1 and Regulates its Functions. Cancer Res. 2008, 68, 8195–8200. [Google Scholar] [CrossRef] [Green Version]
- Latreille, M.; Hausser, J.; Stützer, I.; Zhang, Q.; Hastoy, B.; Gargani, S.; Kerr-Conte, J.; Pattou, F.; Zavolan, M.; Esguerra, J.L.; et al. MicroRNA-7a Regulates Pancreatic β Cell Function. J. Clin. Investig. 2014, 124, 2722–2735. [Google Scholar] [CrossRef] [Green Version]
- Guay, C.; Regazzi, R. Role of Islet MicroRNAs in Diabetes: Which Model for Which Question? Diabetologia 2015, 58, 456–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliasson, L.; Regazzi, R. Micro (RNA) Management and Mismanagement of the Islet. J. Mol. Biol. 2020, 432, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Keller, M.P.; Rabaglia, M.E.; Oler, A.T.; Stapleton, D.S.; Schueler, K.L.; Neto, E.C.; Moon, J.Y.; Wang, P.; Wang, I.M.; et al. Obesity and Genetics Regulate MicroRNAs in Islets, Liver, and Adipose of Diabetic Mice. Mamm. Genome 2009, 20, 476–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijkerk, R.; Esguerra, J.L.S.; Ellenbroek, J.H.; Au, Y.W.; Hanegraaf, M.A.J.; de Koning, E.J.; Eliasson, L.; van Zonneveld, A.J. In Vivo Silencing of MicroRNA-132 Reduces Blood Glucose and Improves Insulin Decretion. Nucleic Acid Ther. 2019, 29, 67–72. [Google Scholar] [CrossRef]
- Nesca, V.; Guay, C.; Jacovetti, C.; Menoud, V.; Peyot, M.L.; Laybutt, D.R.; Prentki, M.; Regazzi, R. Identification of Particular Groups of MicroRNAs That Positively or Negatively Impact on Beta Cell Function in Obese Models of Type 2 Diabetes. Diabetologia 2013, 56, 2203–2212. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Zhang, M.; Tong, M.; Yang, L.; Pang, L.; Chen, L.; Xu, G.; Chi, X.; Hong, Q.; Ni, Y.; et al. miR-148a is Associated With Obesity and Modulates Adipocyte Differentiation of Mesenchymal Stem Cells through Wnt Signaling. Sci. Rep. 2015, 5, 9930. [Google Scholar] [CrossRef] [Green Version]
- Massart, J.; Sjögren, R.J.O.; Lundell, L.S.; Mudry, J.M.; Franck, N.; O’Gorman, D.J.; Egan, B.; Zierath, J.R.; Krook, A. Altered miR-29 Expression in Type 2 Diabetes Influences Glucose and Lipid Metabolism in Dkeletal Muscle. Diabetes 2017, 66, 1807–1818. [Google Scholar] [CrossRef] [Green Version]
- Pullen, T.J.; da Silva Xavier, G.; Kelsey, G.; Rutter, G.A. miR-29a and miR-29b Contribute to Pancreatic Beta-Cell-Specific Silencing of Monocarboxylate Transporter 1 (Mct1). Mol. Cell Biol. 2011, 31, 3182–3194. [Google Scholar] [CrossRef] [Green Version]
- Chamorro-Jorganes, A.; Araldi, E.; Suárez, Y. MicroRNAs as Pharmacological Targets in Endothelial Cell Function and Dysfunction. Pharmacol. Res. 2013, 75, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Willeit, P.; Skroblin, P.; Moschen, A.R.; Yin, X.; Kaudewitz, D.; Zampetaki, A.; Barwari, T.; Whitehead, M.; Ramírez, C.M.; Goedeke, L.; et al. Circulating MicroRNA-122 is Associated with the Risk of New-Onset Metabolic Syndrome and Type 2 Diabetes. Diabetes 2017, 66, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Simionescu, N.; Niculescu, L.S.; Carnuta, M.G.; Sanda, G.M.; Stancu, C.S.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; Simionescu, M.; et al. Hyperglycemia Determines Increased Specific MicroRNAs Levels in Sera and HDL of Acute Coronary Syndrome Patients and Stimulates MicroRNAs Production in Human Macrophages. PLoS ONE. 2016, 11, e0161201. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kaye, D.M. Mechanistic Insights into the Link Between a Polymorphism of the 3′UTR of the SLC7A1 Gene and Hypertension. Hum. Mutat. 2009, 30, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Du, X.; Yang, Y.; Liu, X.; Liu, X.; Zhang, N.; Li, Y.; Jiang, X.; Jiang, Y.; Yang, Z. Inhibition of mir-122 Reduced Atherosclerotic Lesion Formation by Regulating npas3-Mediated Endothelial to Mesenchymal Transition. Life Sci. 2021, 265, 118816. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, G.; Yang, C.; Zhou, K.; Shen, B.; Liang, H.; Jiang, X. The Role of Circulating Microrna-126 (mir-126): A Novel Biomarker for Screening Prediabetes and Newly Diagnosed Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2014, 15, 10567–10577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potus, F.; Ruffenach, G.; Dahou, A.; Thebault, C.; Breuils-Bonnet, S.; Tremblay, È.; Nadeau, V.; Paradis, R.; Graydon, C.; Wong, R.; et al. Downregulation of MicroRNA-126 Contributes to the Failing Right Ventricle in Pulmonary Srterial Hypertension. Circulation 2015, 132, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 Regulates Endothelial Expression of Vascular Cell Adhesion Molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.A.; Yamakuchi, M.; Kondo, M.; Oettgen, P.; Lowenstein, C.J. Ets-1 and Ets-2 Regulate the Expression of MicroRNA-126 in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1990–1997. [Google Scholar] [CrossRef]
- Xu, R.H.; Liu, B.; Wu, J.D.; Yan, Y.Y.; Wang, J.N. MiR-143 is involved In Endothelial Cell Dysfunction through Suppression of Glycolysis and Correlated with Atherosclerotic Plaques Formation. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4063–4071. [Google Scholar]
- Deng, L.; Blanco, F.J.; Stevens, H.; Lu, R.; Caudrillier, A.; McBride, M.; McClure, J.D.; Grant, J.; Thomas, M.; Frid, M.; et al. MicroRNA-143 Activation Regulates Smooth Muscle and Endothelial Cell Crosstalk in Pulmonary Srterial Hypertension. Circ. Res. 2015, 117, 870–883. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gora, I.M.; Ciechanowska, A.; Ladyzynski, P. NLRP3 Inflammasome at the Interface of Inflammation, Endothelial Dysfunction, and Type 2 Diabetes. Cells 2021, 10, 314. https://doi.org/10.3390/cells10020314
Gora IM, Ciechanowska A, Ladyzynski P. NLRP3 Inflammasome at the Interface of Inflammation, Endothelial Dysfunction, and Type 2 Diabetes. Cells. 2021; 10(2):314. https://doi.org/10.3390/cells10020314
Chicago/Turabian StyleGora, Ilona M., Anna Ciechanowska, and Piotr Ladyzynski. 2021. "NLRP3 Inflammasome at the Interface of Inflammation, Endothelial Dysfunction, and Type 2 Diabetes" Cells 10, no. 2: 314. https://doi.org/10.3390/cells10020314