
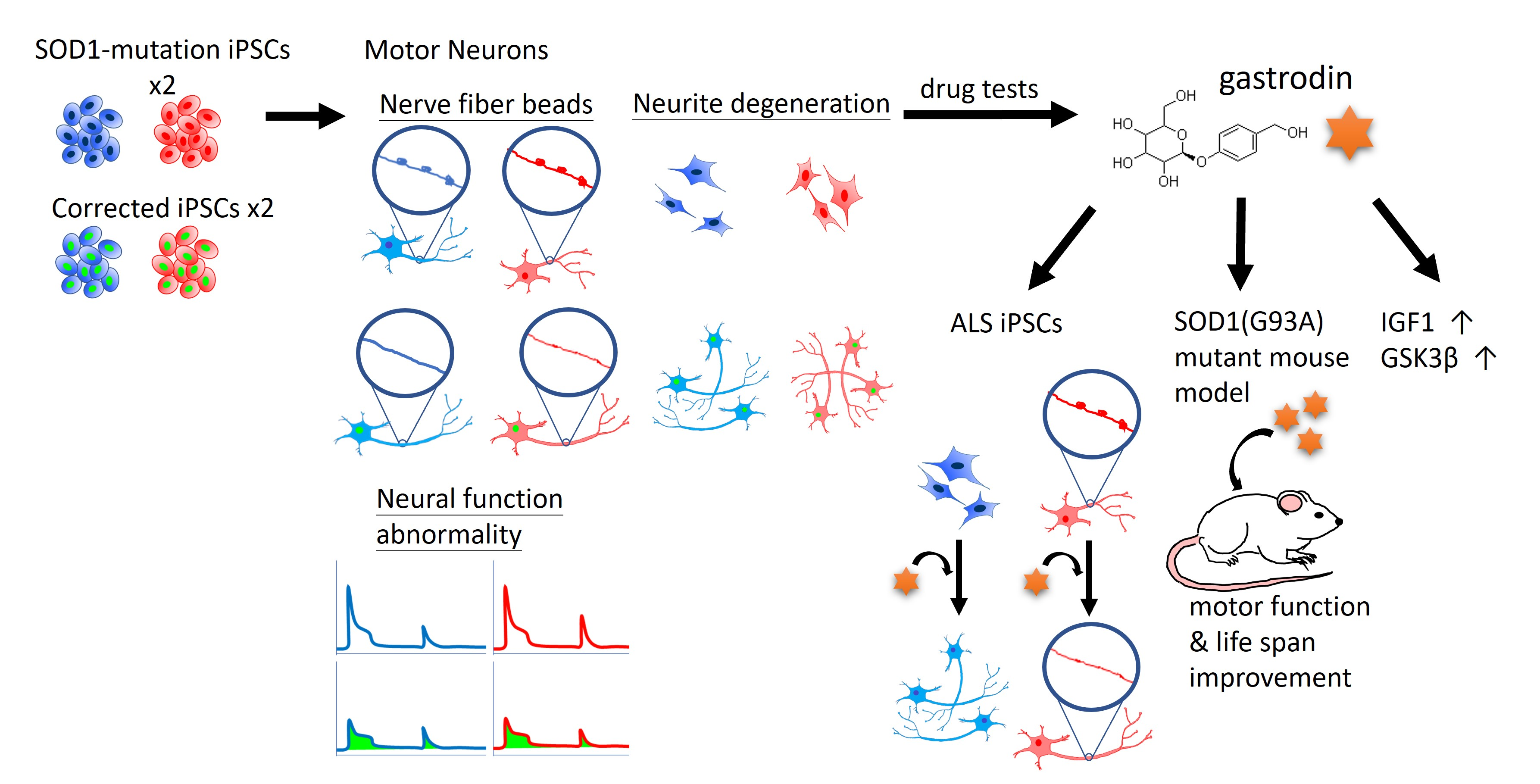
Coactivation of GSK3β and IGF-1 Attenuates Amyotrophic Lateral Sclerosis Nerve Fiber Cytopathies in SOD1 Mutant Patient-Derived Motor Neurons


 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
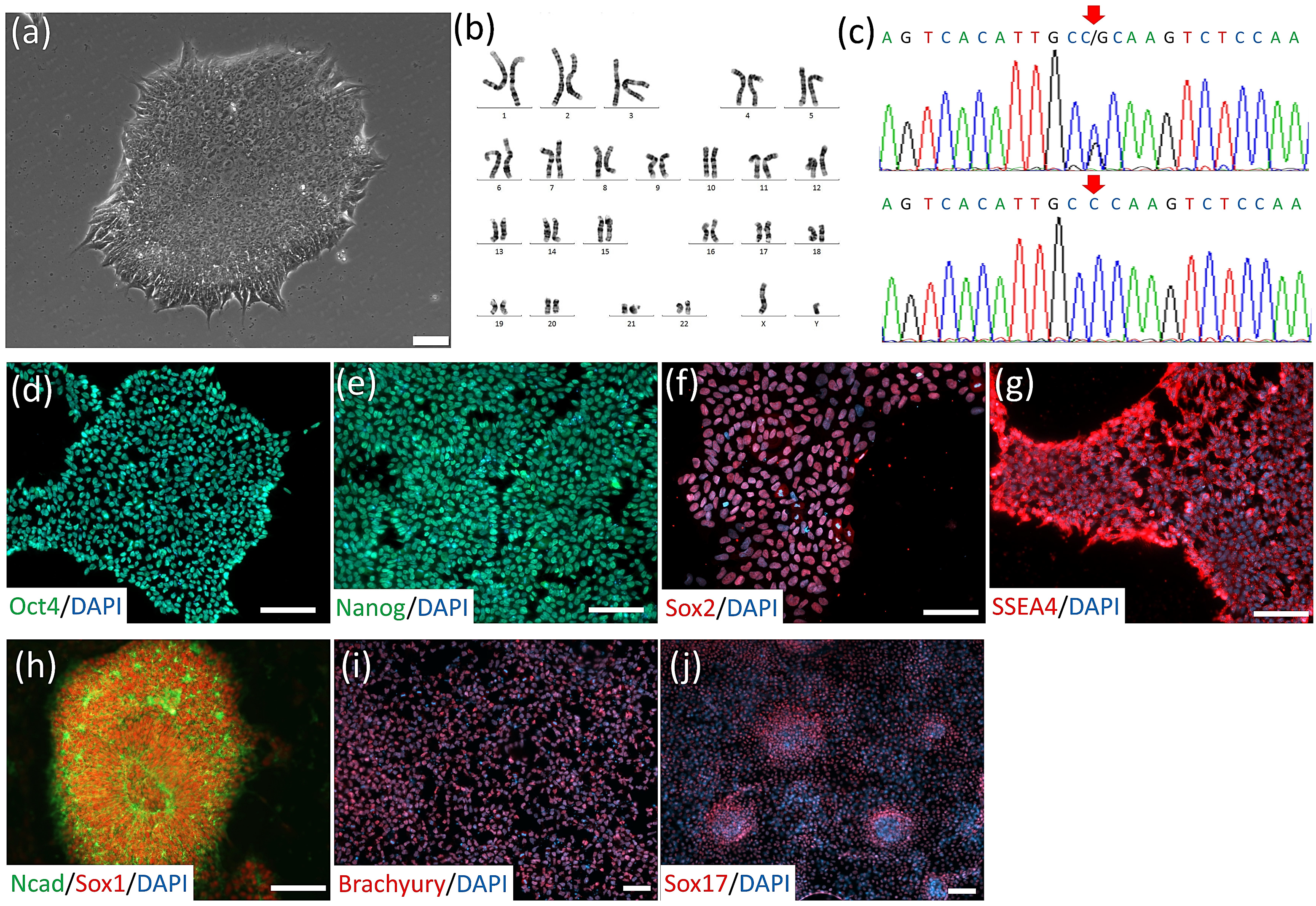
2.1. iPSC Line Establishment and Culture
2.2. SOD1G85R iPSC Site-Specific Correction by CRISPR/Cas9-Mediated Homologous Recombination
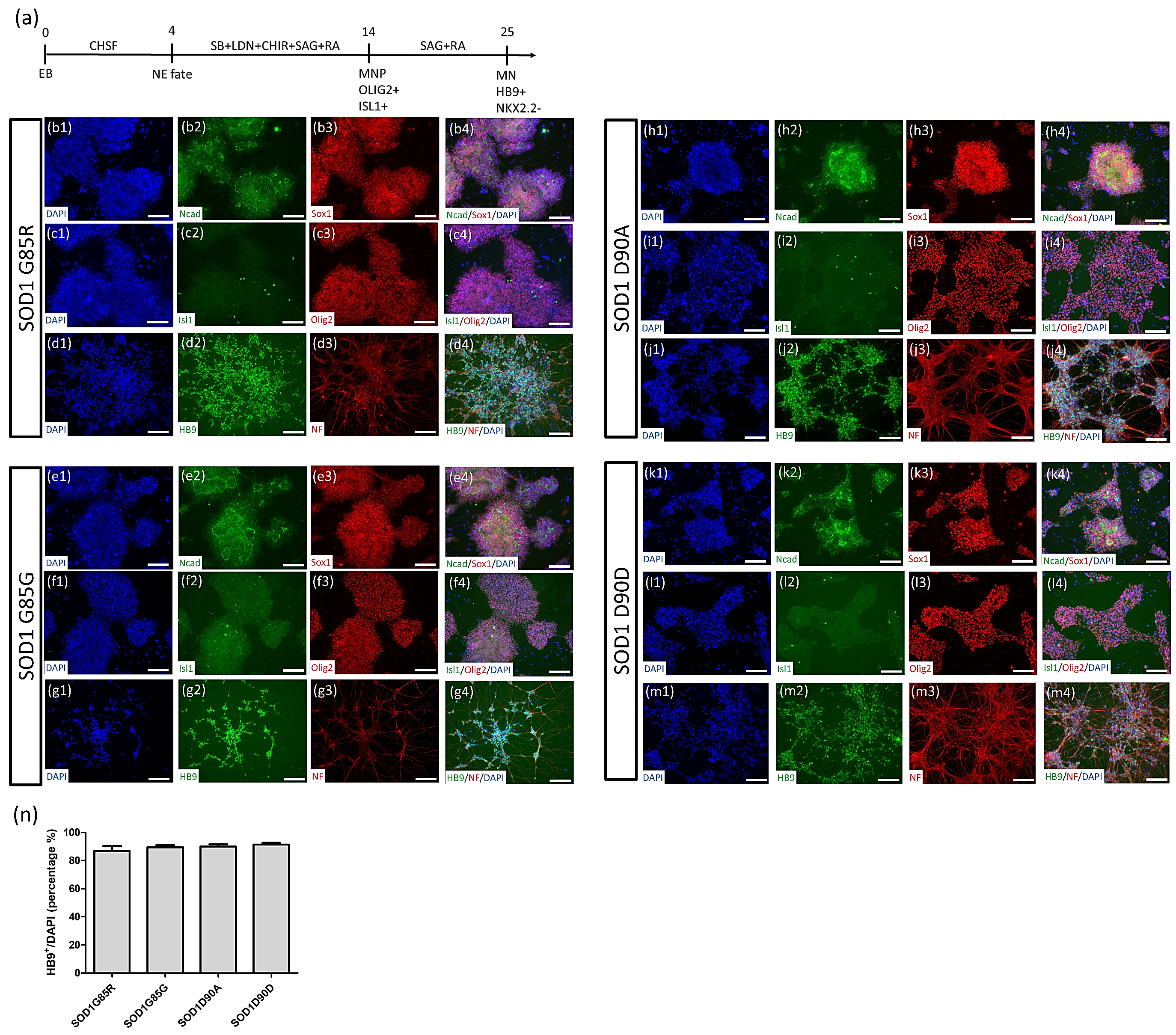
2.3. Robust MN Differentiation from iPSCs
2.4. Immunocytochemistry (ICC) Staining and Nerve Fiber Density/Beads Calculations
2.5. Calcium Imaging
2.6. ALS Animal Model and Small Compound Treatment
2.7. Animal Motor Behavior Test
2.8. Western Blotting
2.9. RNA-Microarray and Analysis
2.10. Statistical Analysis
3. Results
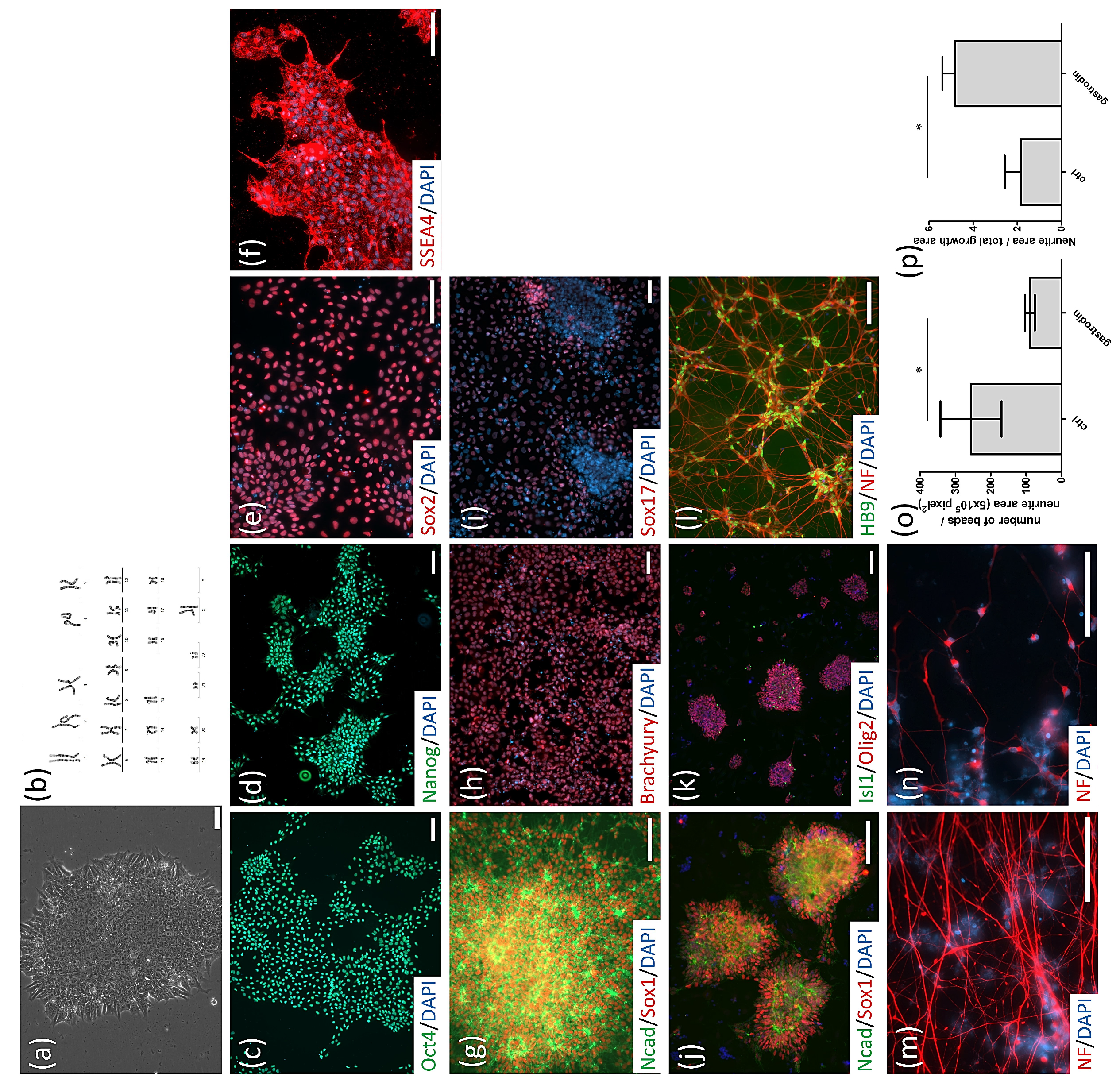
3.1. Generation of SOD1G85R ALS and SOD1G85G Isogenic iPSCs, and Robust MN Differentiation via the CHSF Method
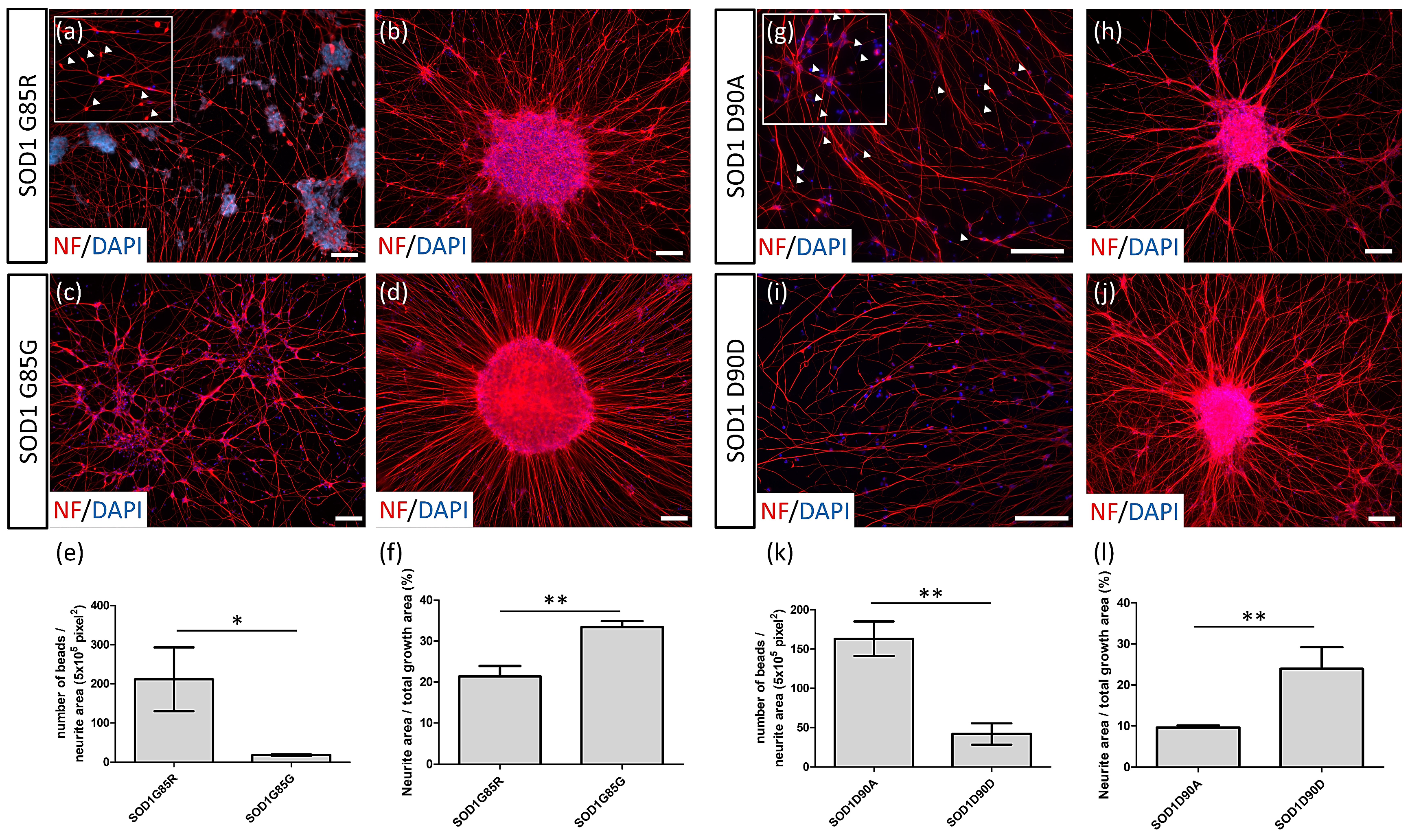
3.2. NF Aggregates and Nerve Fiber Degeneration in SOD1 ALS MNs
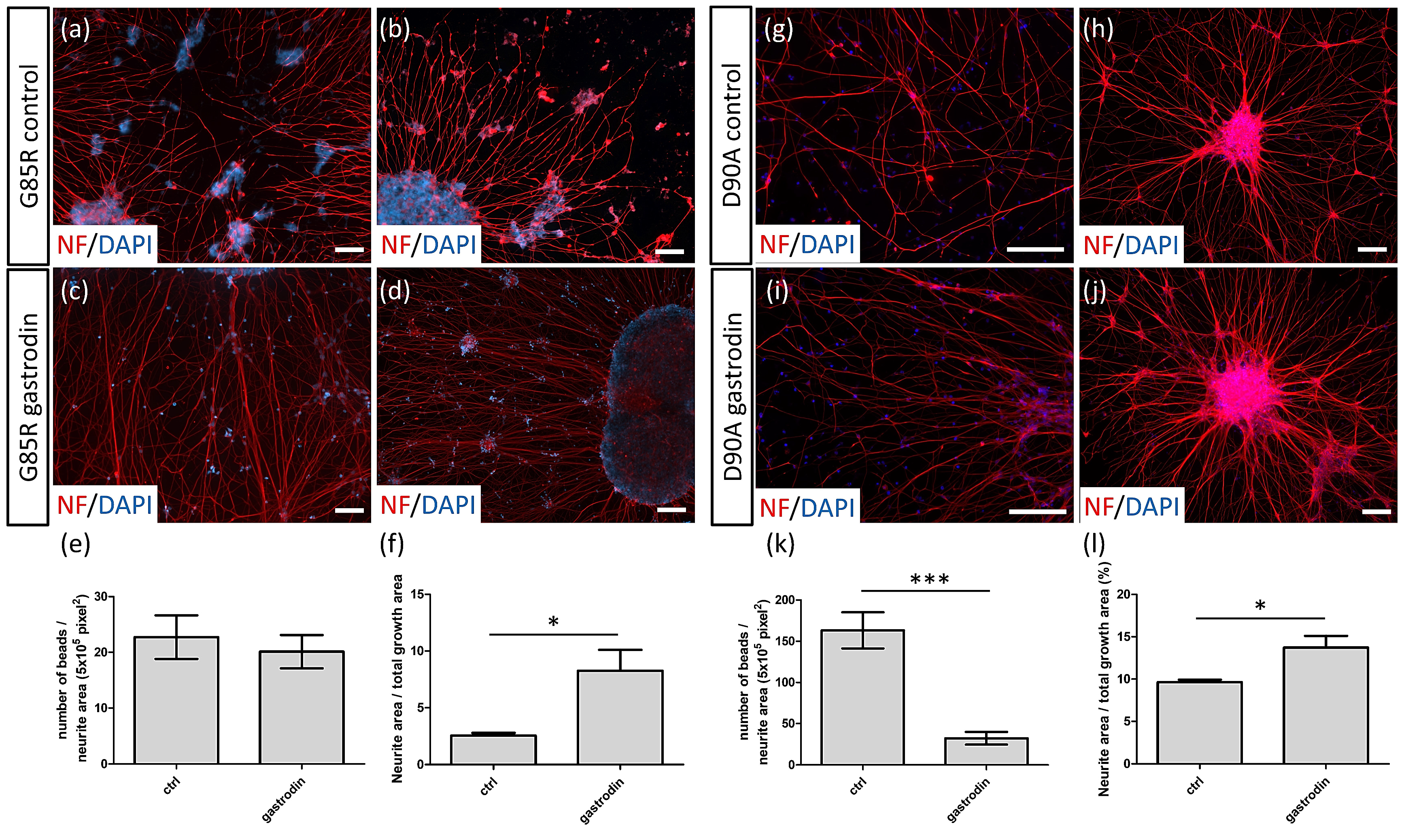
3.3. Gastrodin Reduces NF Aggregates and Promotes Nerve Fiber Extension in SOD1 ALS MNs
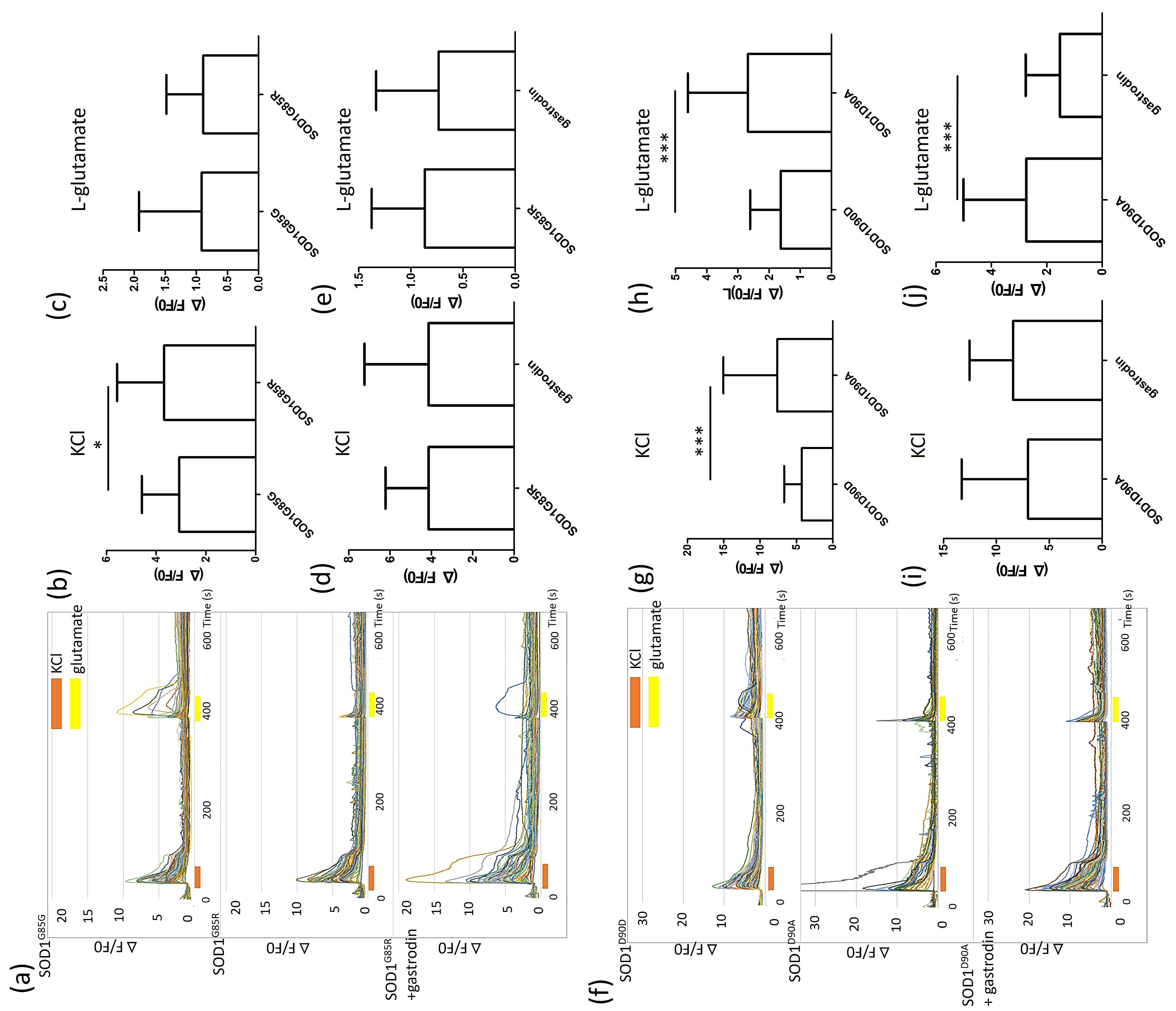
3.4. Gastrodin Reverses the Glutamate Induced Hyper-Calcium Flux in SOD1 ALS MNs
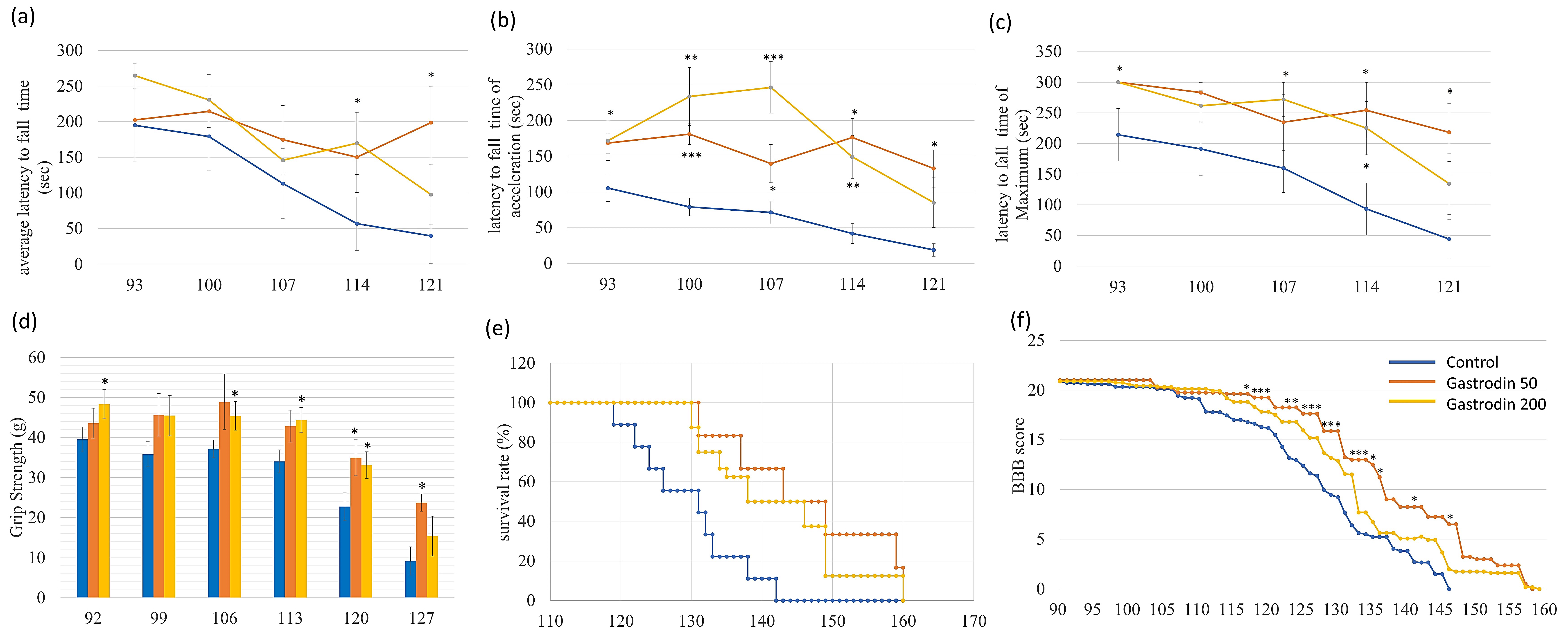
3.5. Gastrodin Rescues Nerve Fiber Cytopathies in Sporadic ALS MNs As Well As Restoring Motor Performance and Prolonging Survival in ALS Mice
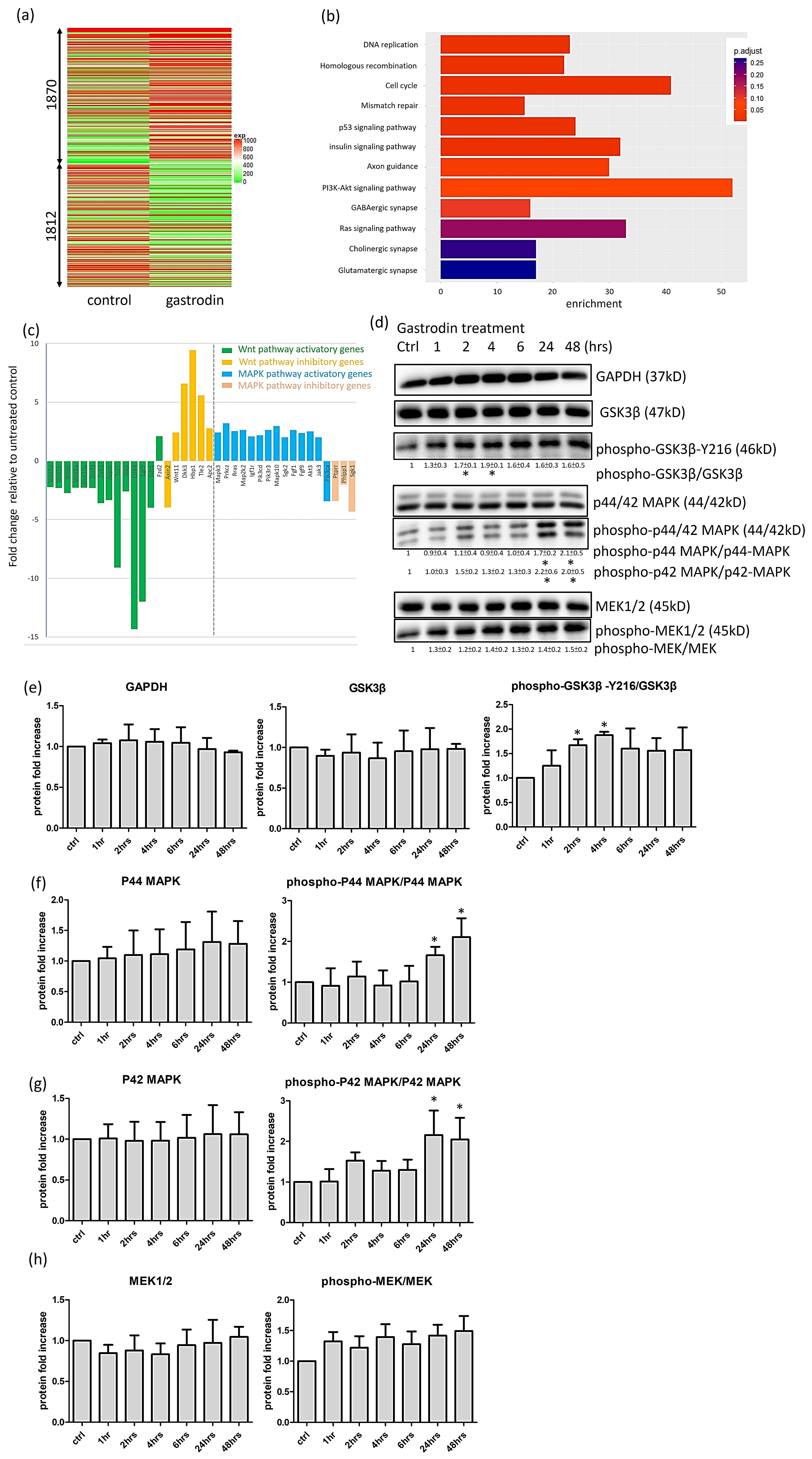
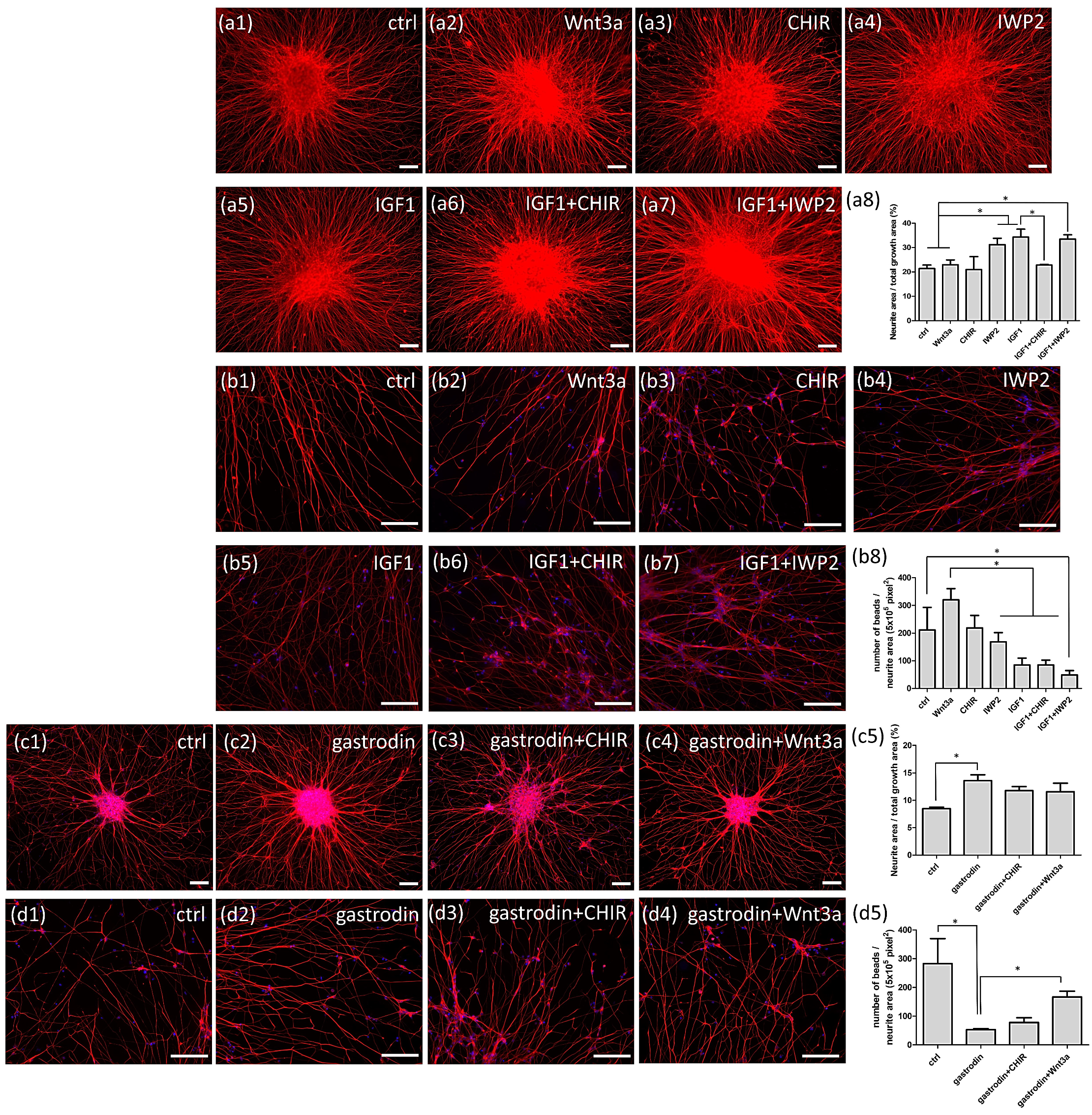
3.6. Gastrodin Activates the IGF-1 Signaling Pathway and GSK3β Activity to Rescue Nerve Fiber Cytopathies in SOD1 ALS MNs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef]
- Smith, R.; Pioro, E.; Myers, K.; Sirdofsky, M.; Goslin, K.; Meekins, G.; Yu, H.; Wymer, J.; Cudkowicz, M.; Macklin, E.A.; et al. Enhanced bulbar function in amyotrophic lateral sclerosis: The nuedexta treatment trial. Neurotherapeutics 2017, 14, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Sawada, H. Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin. Pharmacother. 2017, 18, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O'Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Tsuda, H.; Han, S.M.; Yang, Y.; Tong, C.; Lin, Y.Q.; Mohan, K.; Haueter, C.; Zoghbi, A.; Harati, Y.; Kwan, J.; et al. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell 2008, 133, 963–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, S. Proximal axonal enlargement in motor neuron disease. Neurology 1968, 18, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Hirano, A.; Nakano, I.; Kurland, L.T.; Mulder, D.W.; Holley, P.W.; Saccomanno, G. Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 1984, 43, 471–480. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Walker, A.K.; Atkin, J.D. Stress signaling from the endoplasmic reticulum: A central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life 2011, 63, 754–763. [Google Scholar] [CrossRef]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067. [Google Scholar] [CrossRef] [Green Version]
- Lutz, C. Mouse models of ALS: Past, present and future. Brain Res. 2018, 1693, 1–10. [Google Scholar] [CrossRef]
- Morrice, J.R.; Gregory-Evans, C.Y.; Shaw, C.A. Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural Regen. Res. 2018, 13, 2050–2054. [Google Scholar] [CrossRef]
- Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; Asaka, I.; et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 2012, 4, 145ra104. [Google Scholar] [CrossRef]
- Mitne-Neto, M.; Machado-Costa, M.; Marchetto, M.C.; Bengtson, M.H.; Joazeiro, C.A.; Tsuda, H.; Bellen, H.J.; Silva, H.C.; Oliveira, A.S.; Lazar, M.; et al. Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum. Mol. Genet. 2011, 20, 3642–3652. [Google Scholar] [CrossRef] [Green Version]
- Alves, C.J.; Dariolli, R.; Jorge, F.M.; Monteiro, M.R.; Maximino, J.R.; Martins, R.S.; Strauss, B.E.; Krieger, J.E.; Callegaro, D.; Chadi, G. Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front. Cell. Neurosci. 2015, 9, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichiyanagi, N.; Fujimori, K.; Yano, M.; Ishihara-Fujisaki, C.; Sone, T.; Akiyama, T.; Okada, Y.; Akamatsu, W.; Matsumoto, T.; Ishikawa, M.; et al. Establishment of In Vitro FUS-Associated Familial Amyotrophic Lateral Sclerosis Model Using Human Induced Pluripotent Stem Cells. Stem Cell Reports 2016, 6, 496–510. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.t.; Huang, C.L.; Errigo, A.; Yin, Y.; et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar] [CrossRef] [Green Version]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkhardt, M.F.; Martinez, F.J.; Wright, S.; Ramos, C.; Volfson, D.; Mason, M.; Garnes, J.; Dang, V.; Lievers, J.; Shoukat-Mumtaz, U.; et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 2013, 56, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovas, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic correction of SOD1 mutant iPSCs reveals ERK and JNK activated AP1 as a driver of neurodegeneration in amyotrophic lateral sclerosis. Stem Cell Rep. 2017, 8, 856–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macarthur, C.C.; Fontes, A.; Ravinder, N.; Kuninger, D.; Kaur, J.; Bailey, M.; Taliana, A.; Vemuri, M.C.; Lieu, P.T. Generation of human-induced pluripotent stem cells by a nonintegrating RNA Sendai virus vector in feeder-free or xeno-free conditions. Stem Cells Int. 2012, 2012, 1–9. [Google Scholar] [CrossRef]
- Basso, D.M.; Beattie, M.S.; Bresnahan, J.C. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp. Neurol. 1996, 139, 244–256. [Google Scholar] [CrossRef] [Green Version]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Antonyuk, S.V.; Seetharaman, S.V.; Whitson, L.J.; Taylor, A.B.; Holloway, S.P.; Strange, R.W.; Doucette, P.A.; Valentine, J.S.; Tiwari, A.; et al. Structures of the G85R variant of SOD1 in familial amyotrophic lateral sclerosis. J. Biol. Chem. 2008, 283, 16169–16177. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-M.; Lee, M.-S.; Chang, C.-Y.; Lin, S.-Z.; Cheng, E.-H.; Liu, Y.-H.; Pan, H.-C.; Lee, H.-C.; Su, H.-L. Prerequisite OCT4 maintenance potentiates the neural induction of differentiating human embryonic stem cells and induced pluripotent stem cells. Cell Transplant. 2015, 24, 829–844. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Chen, S.-M.; Lu, H.-E.; Lai, S.-M.; Lai, P.-S.; Shen, P.-W.; Chen, P.-Y.; Shen, C.-I.; Harn, H.-J.; Lin, S.-Z.; et al. N-butylidenephthalide attenuates Alzheimer’s disease-like cytopathy in Down syndrome induced pluripotent stem cell-derived neurons. Sci Rep. 2015, 5, 8744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maury, Y.; Come, J.; Piskorowski, R.A.; Salah-Mohellibi, N.; Chevaleyre, V.; Peschanski, M.; Martinat, C.; Nedelec, S. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 2015, 33, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Wang, X.; Shang, B.; Fang, J.; Xi, Y.; Li, A.; Diao, Y. Gastrodin ameliorates spinal cord injury via antioxidant and anti-inflammatory effects. Acta Biochim. Pol. 2016, 63, 589–593. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.S.; Simon, N.G.; Menon, P.; Vucic, S.; Kiernan, M.C. The puzzling case of hyperexcitability in amyotrophic lateral sclerosis. J. Clin. Neurol. 2013, 9, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.L.; Zhong, X.; Fan, F.; Zhang, S.C. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, M.E.; Fleck, T.J.; Himes, C.S.; Hall, E.D. Riluzole preserves motor function in a transgenic model of familial amyotrophic lateral sclerosis. Neurology 1998, 50, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Hogg, M.C.; Halang, L.; Woods, I.; Coughlan, K.S.; Prehn, J.H.M. Riluzole does not improve lifespan or motor function in three ALS mouse models. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Warita, H.; Manabe, Y.; Murakami, T.; Shiro, Y.; Nagano, I.; Abe, K. Early decrease of survival signal-related proteins in spinal motor neurons of presymptomatic transgenic mice with a mutant SOD1 gene. Apoptosis 2001, 6, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Allodi, I.; Comley, L.; Nichterwitz, S.; Nizzardo, M.; Simone, C.; Benitez, J.A.; Cao, M.; Corti, S.; Hedlund, E. Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci. Rep. 2016, 6, 25960. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Cho, S.I.; Lim, H.R.; Lee, J.K.; Lee, Y.A.; Noh, J.S.; Joo, I.S.; Kim, K.W.; Gwag, B.J. Concurrent administration of Neu2000 and lithium produces marked improvement of motor neuron survival, motor function, and mortality in a mouse model of amyotrophic lateral sclerosis. Mol. Pharmacol. 2007, 71, 965–975. [Google Scholar] [CrossRef]
- Sugai, F.; Yamamoto, Y.; Miyaguchi, K.; Zhou, Z.; Sumi, H.; Hamasaki, T.; Goto, M.; Sakoda, S. Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur. J. Neurosci. 2004, 20, 3179–3183. [Google Scholar] [CrossRef]
- Feng, H.-L.; Leng, Y.; Ma, C.-H.; Zhang, J.; Ren, M.; Chuang, D.-M. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 2008, 155, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Koh, S.-H.; Kim, Y.; Kim, H.Y.; Hwang, S.; Lee, C.H.; Kim, S.H. Inhibition of glycogen synthase kinase-3 suppresses the onset of symptoms and disease progression of G93A-SOD1 mouse model of ALS. Exp. Neurol 2007, 205, 336–346. [Google Scholar] [CrossRef]
- Li, B.; Xu, W.; Luo, C.; Gozal, D.; Liu, R. VEGF-induced activation of the PI3-K/Akt pathway reduces mutant SOD1-mediated motor neuron cell death. Brain Res. Mol. Brain Res. 2003, 111, 155–164. [Google Scholar] [CrossRef]
- Group, U.K.-L.S.; Morrison, K.E.; Dhariwal, S.; Hornabrook, R.; Savage, L.; Burn, D.J.; Khoo, T.K.; Kelly, J.; Murphy, C.L.; Al-Chalabi, A.; et al. Lithium in patients with amyotrophic lateral sclerosis (LiCALS): A phase 3 multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2013, 12, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Fernandez, C.; Gonzalez, P.; Andres-Benito, P.; Ferrer, I.; Rodriguez, F.J. Wnt signaling alterations in the human spinal cord of amyotrophic lateral sclerosis cases: Spotlight on Fz2 and Wnt5a. Mol. Neurobiol. 2019, 56, 6777–6791. [Google Scholar] [CrossRef]
- Vohnoutka, R.B.; Boumil, E.F.; Liu, Y.; Uchida, A.; Pant, H.C.; Shea, T.B. Influence of a GSK3beta phosphorylation site within the proximal C-terminus of Neurofilament-H on neurofilament dynamics. Biol. Open 2017, 6, 1516–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Pant, H.C.; Shea, T.B. Divergent and convergent roles for kinases and phosphatases in neurofilament dynamics. J. Cell Sci. 2014, 127, 4064–4077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.-C.; Chen, Y.-F.; Lee, W.-C.; Wu, Y.-T.; Tsai, T.-H. Pharmacokinetics of gastrodin and its metabolite p-hydroxybenzyl alcohol in rat blood, brain and bile by microdialysis coupled to LC-MS/MS. J. Pharm. Biomed. Anal. 2008, 48, 909–917. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ting, H.-C.; Yang, H.-I.; Harn, H.-J.; Chiu, I.-M.; Su, H.-L.; Li, X.; Chen, M.-F.; Ho, T.-J.; Liu, C.-A.; Tsai, Y.-J.; et al. Coactivation of GSK3β and IGF-1 Attenuates Amyotrophic Lateral Sclerosis Nerve Fiber Cytopathies in SOD1 Mutant Patient-Derived Motor Neurons. Cells 2021, 10, 2773. https://doi.org/10.3390/cells10102773
Ting H-C, Yang H-I, Harn H-J, Chiu I-M, Su H-L, Li X, Chen M-F, Ho T-J, Liu C-A, Tsai Y-J, et al. Coactivation of GSK3β and IGF-1 Attenuates Amyotrophic Lateral Sclerosis Nerve Fiber Cytopathies in SOD1 Mutant Patient-Derived Motor Neurons. Cells. 2021; 10(10):2773. https://doi.org/10.3390/cells10102773
Chicago/Turabian StyleTing, Hsiao-Chien, Hui-I Yang, Horng-Jyh Harn, Ing-Ming Chiu, Hong-Lin Su, Xiang Li, Mei-Fang Chen, Tsung-Jung Ho, Ching-Ann Liu, Yung-Jen Tsai, and et al. 2021. "Coactivation of GSK3β and IGF-1 Attenuates Amyotrophic Lateral Sclerosis Nerve Fiber Cytopathies in SOD1 Mutant Patient-Derived Motor Neurons" Cells 10, no. 10: 2773. https://doi.org/10.3390/cells10102773