
The Role of Epithelial Damage in the Pulmonary Immune Response

Abstract
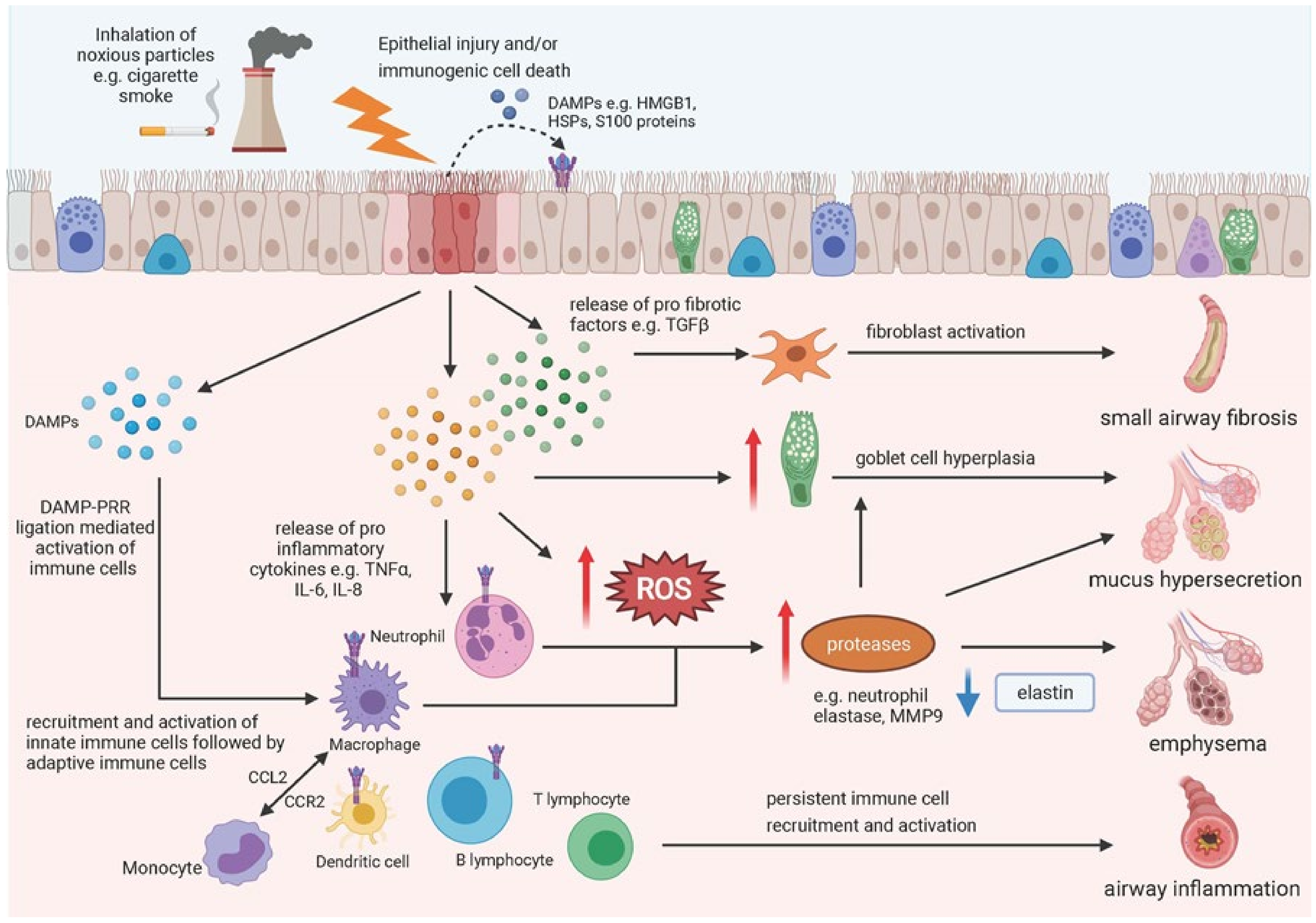
:1. Introduction
2. Sensing Danger: PAMP and DAMP Signalling
3. Epithelial–Immune Cell Cross Talk
4. Epithelial–Fibroblast Cross Talk
5. Epithelial Damage and COPD
6. Epithelial Damage and IPF
7. Epithelial Damage and COVID-19
8. Epithelial Damage and Senescence
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crystal, R.G.; Randell, S.H.; Engelhardt, J.; Voynow, J.; Sunday, M.E. Airway Epithelial Cells: Current Concepts and Challenges. Proc. Am. Thorac. Soc. 2008, 5, 772–777. [Google Scholar] [CrossRef]
- Davis, J.D.; Wypych, T.P. Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol. 2021, 14, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, Z.; Huang, H.; Li, J.; Wang, Z.; Yu, Y.; Zhang, C.; Li, J.; Dai, H.; Wang, F.; et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc. Natl. Acad. Sci. USA 2018, 115, 2407–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olajuyin, A.M.; Zhang, X.; Ji, H.-L. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discov. 2019, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, R.J.; Lloyd, C.M. Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 2021, 21, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Leiva-Juárez, M.M.; Kolls, J.K.; Evans, S.E. Lung epithelial cells: Therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. 2017, 11, 21–34. [Google Scholar] [CrossRef]
- Marchiando, A.M.; Graham, W.; Turner, J.R. Epithelial Barriers in Homeostasis and Disease. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 119–144. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Adivitiya; Kaushik, M.S.; Chakraborty, S.; Veleri, S.; Kateriya, S. Mucociliary Respiratory Epithelium Integrity in Molecular Defense and Susceptibility to Pulmonary Viral Infections. Biology 2021, 10, 95. [Google Scholar] [CrossRef]
- Bhat, A.A.; Uppada, S.; Achkar, I.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2019, 9, 1942. [Google Scholar] [CrossRef] [Green Version]
- Tam, A.; Wadsworth, S.; Dorscheid, D.; Man, S.P.; Sin, D.D. The airway epithelium: More than just a structural barrier. Ther. Adv. Respir. Dis. 2011, 5, 255–273. [Google Scholar] [CrossRef]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Cell. Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef] [Green Version]
- Pradeu, T.; Cooper, E.L. The danger theory: 20 years later. Front. Immunol. 2012, 3, 287. [Google Scholar] [CrossRef] [Green Version]
- Hiemstra, P.S.; McCray, P.B., Jr.; Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucchini, A.C.; Gachanja, N.N.; Rossi, A.G.; Dorward, D.A.; Lucas, C.D. Epithelial Cells and Inflammation in Pulmonary Wound Repair. Cells 2021, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadzic, S.; Wu, C.-Y.; Avdeev, S.; Weissmann, N.; Schermuly, R.T.; Kosanovic, D. Lung epithelium damage in COPD—An unstoppable pathological event? Cell. Signal. 2020, 68, 109540. [Google Scholar] [CrossRef]
- Bridges, J.P.; Vladar, E.K.; Huang, H.; Mason, R.J. Respiratory epithelial cell responses to SARS-CoV-2 in COVID-19. Thorax 2021. [Google Scholar] [CrossRef]
- Frey, A.; Lunding, L.P.; Ehlers, J.C.; Weckmann, M.; Zissler, U.M.; Wegmann, M. More than Just a Barrier: The Immune Functions of the Airway Epithelium in Asthma Pathogenesis. Front. Immunol. 2020, 11, 761. [Google Scholar] [CrossRef]
- Rohmann, K.; Tschernig, T.; Pabst, R.; Goldmann, T.; Drömann, D. Innate immunity in the human lung: Pathogen recognition and lung disease. Cell Tissue Res. 2010, 343, 167–174. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Allam, V.S.R.R.; Faiz, A.; Lam, M.; Rathnayake, S.N.H.; Ditz, B.; Pouwels, S.D.; Brandsma, C.; Timens, W.; Hiemstra, P.S.; Tew, G.W.; et al. RAGE and TLR4 differentially regulate airway hyperresponsiveness: Implications for COPD. Allergy 2020, 76, 1123–1135. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 1–18. [Google Scholar] [CrossRef]
- Crane, M.J.; Lee, K.M.; FitzGerald, E.S.; Jamieson, A.M. Surviving Deadly Lung Infections: Innate Host Tolerance Mechanisms in the Pulmonary System. Front. Immunol. 2018, 9, 1421. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.; Prince, A. Innate Immunity in the Respiratory Epithelium. Am. J. Respir. Cell Mol. Biol. 2011, 45, 189–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Botha, P.; Archer, L.; Anderson, R.L.; Lordan, J.; Dark, J.H.; Corris, P.A.; Gould, K.; Fisher, A.J. Pseudomonas aeruginosa Colonization of the Allograft After Lung Transplantation and the Risk of Bronchiolitis Obliterans Syndrome. Transplantation 2008, 85, 771–774. [Google Scholar] [CrossRef]
- Pragman, A.A.; Berger, J.P.; Williams, B.J. Understanding Persistent Bacterial Lung Infections. Clin. Pulm. Med. 2016, 23, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Matzinger, P. The Danger Model: A Renewed Sense of Self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 20, 95–112. [Google Scholar] [CrossRef]
- Piccinini, A.M.; Midwood, K.S. DAMPening Inflammation by Modulating TLR Signalling. Mediat. Inflamm. 2010, 2010, 672395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Shao, Y.; Tian, Y.; Ouyang, C.; Wang, X. Nuclear Alarmin Cytokines in Inflammation. J. Immunol. Res. 2020, 2020, 7206451. [Google Scholar] [CrossRef]
- Land, W.G. The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I - Promoting inflammation and immunity. Sultan Qaboos Univ. Med. J. 2015, 15, e9–e21. [Google Scholar] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Ellson, C.D.; Dunmore, R.; Hogaboam, C.M.; Sleeman, M.A.; Murray, L.A. DAMPs and Danger Signals in IPF. Am. J. Respir. Cell Mol. Biol. 2014, 51. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, Z.; Xu, Z.; Liu, J.; Zhang, J. High mobility group box 1 (HMGB1): A pivotal regulator of hematopoietic malignancies. J. Hematol. Oncol. 2020, 13, 1–19. [Google Scholar] [CrossRef]
- Ranzato, E.; Martinotti, S.; Patrone, M. Emerging roles for HMGB1 protein in immunity, inflammation, and cancer. ImmunoTargets Ther. 2015, 4, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y.; et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Suarez, J.S.; Minaai, M.; Li, S.; Gaudino, G.; Pass, H.I.; Carbone, M.; Yang, H. HMGB1 as a therapeutic target in disease. J. Cell. Physiol. 2020, 236, 3406–3419. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Tan, X.; Liang, Y.; Hou, C.; Qu, D.; Li, M.; Huang, Q. Differential DAMP release was observed in the sputum of COPD, asthma and asthma-COPD overlap (ACO) patients. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Liu, Z.; Zhang, W.; Cai, S. Ulinastatin protects the lungs of COPD rats through the HMGB1/TLR4 signaling pathway. Oncol. Lett. 2018, 16, 4057–4063. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Yang, D.; Tewary, P.; Li, Y.; Li, S.; Chen, X.; Howard, O.M.Z.; Bustin, M.; Oppenheim, J.J. The Alarmin HMGN1 Contributes to Antitumor Immunity and Is a Potent Immunoadjuvant. Cancer Res. 2014, 74, 5989–5998. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Postnikov, Y.V.; Li, Y.; Tewary, P.; De La Rosa, G.; Wei, F.; Klinman, D.; Gioannini, T.; Weiss, J.; Furusawa, T.; et al. High-mobility group nucleosome-binding protein 1 acts as an alarmin and is critical for lipopolysaccharide-induced immune responses. J. Exp. Med. 2011, 209, 157–171. [Google Scholar] [CrossRef] [Green Version]
- Postnikov, Y.V.; Furusawa, T.; Haines, D.C.; Factor, V.M.; Bustin, M. Loss of the nucleosome-binding protein HMGN1 affects the rate of N-nitrosodiethylamine-induced hepatocarcinogenesis in mice. Mol. Cancer Res. 2013, 12, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borthwick, L.A. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin. Immunopathol. 2016, 38, 517–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2016, 14, 43–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Y.; Yang, D.; Oppenheim, J.J. Alarmins and Antitumor Immunity. Clin. Ther. 2016, 38, 1042–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpa, M.; Kessler, S.; Sadler, T.; West, G.; Homer, C.; McDonald, C.; de la Motte, C.; Fiocchi, C.; Stylianou, E. The Epithelial Danger Signal IL-1α Is a Potent Activator of Fibroblasts and Reactivator of Intestinal Inflammation. Am. J. Pathol. 2015, 185, 1624–1637. [Google Scholar] [CrossRef]
- Altara, R.; Ghali, R.; Mallat, Z.; Cataliotti, A.; Booz, G.W.; A Zouein, F. Conflicting vascular and metabolic impact of the IL-33/sST2 axis. Cardiovasc. Res. 2018, 114, 1578–1594. [Google Scholar] [CrossRef] [Green Version]
- Du, X.X.; Shi, Y.; Yang, Y.; Yu, Y.; Lou, H.G.; Lv, F.F.; Chen, Z.; Yang, Q. DAMP molecular IL-33 augments monocytic inflammatory storm in hepatitis B-precipitated acute-on-chronic liver failure. Liver Int. 2017, 38, 229–238. [Google Scholar] [CrossRef]
- Kotsiou, O.S.; Gourgoulianis, K.I.; Zarogiannis, S. IL-33/ST2 Axis in Organ Fibrosis. Front. Immunol. 2018, 9, 2432. [Google Scholar] [CrossRef] [Green Version]
- Aziz, M.; Brenner, M.; Wang, P. Extracellular CIRP (eCIRP) and inflammation. J. Leukoc. Biol. 2019, 106, 133–146. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Tan, H.Y.; Yong, Y.K.; Shankar, E.M.; Paukovics, G.; Ellegård, R.; Larsson, M.; Kamarulzaman, A.; French, M.A.; Crowe, S.M. Aberrant Inflammasome Activation Characterizes Tuberculosis-Associated Immune Reconstitution Inflammatory Syndrome. J. Immunol. 2016, 196, 4052–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Xie, L.; Wang, X.; Ma, H.; Lv, L.; Liu, L.; Song, X. Circulating tumor DNA measurement provides reliable mutation detection in mice with human lung cancer xenografts. Lab. Investig. 2018, 98, 935–946. [Google Scholar] [CrossRef]
- Roers, A.; Hiller, B.; Hornung, V. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 2016, 44, 739–754. [Google Scholar] [CrossRef] [Green Version]
- Barrat, F.J.; Meeker, T.; Gregorio, J.; Chan, J.H.; Uematsu, S.; Akira, S.; Chang, B.; Duramad, O.; Coffman, R.L.; Ghiringhelli, F.; et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005, 202, 1131–1139. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Yang, J.; Guo, S.; Zhao, G.; Wu, H.; Deng, G. Peripheral Circulating Exosome-Mediated Delivery of miR-155 as a Novel Mechanism for Acute Lung Inflammation. Mol. Ther. 2019, 27, 1758–1771. [Google Scholar] [CrossRef]
- Bala, S.; Petrasek, J.; Mundkur, S.; Catalano, D.; Levin, I.; Ward, J.; Alao, H.; Kodys, K.; Szabo, G. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 2012, 56, 1946–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Liu, D.; Feng, Y.; Zhang, H.; Zeng, D.; Liu, Q.; Qu, J. Spliceosome-associated protein 130: A novel biomarker for idiopathic pulmonary fibrosis. Ann. Transl. Med. 2020, 8, 986. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Nakahara, M.; Masuda, Y.; Ono, S.; Yamada, S.; Ishikura, H.; Imaizumi, H.; Kamikokuryo, C.; Kakihana, Y.; Maruyama, I. Circulating histone H3 levels are increased in septic mice in a neutrophil-dependent manner: Preclinical evaluation of a novel sandwich ELISA for histone H3. J. Intensiv. Care 2018, 6, 79. [Google Scholar] [CrossRef]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating Histones Are Mediators of Trauma-associated Lung Injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Z.; Liu, Y.; Li, F.; Ren, F.; Chen, D.; Li, X.; Wen, T. Circulating histones exacerbate inflammation in mice with acute liver failure. J. Cell. Biochem. 2013, 114, 2384–2391. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Kumar, S.V.; Marschner, J.; Desai, J.; Holderied, A.; Rath, L.; Kraft, F.; Lei, Y.; Fukasawa, Y.; Moeckel, G.; et al. Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J. Am. Soc. Nephrol. 2017, 28, 1753–1768. [Google Scholar] [CrossRef] [Green Version]
- Bosmann, M.; Grailer, J.J.; Ruemmler, R.; Russkamp, N.F.; Zetoune, F.S.; Sarma, J.V.; Standiford, T.J.; Ward, P.A. Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J. 2013, 27, 5010–5021. [Google Scholar] [CrossRef] [Green Version]
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Fredholm, B.B. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007, 14, 1315–1323. [Google Scholar] [CrossRef] [Green Version]
- Bours, M.; Swennen, E.; Di Virgilio, F.; Cronstein, B.; Dagnelie, P. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 2006, 112, 358–404. [Google Scholar] [CrossRef]
- Dukay, B.; Csoboz, B.; Tóth, M.E. Heat-Shock Proteins in Neuroinflammation. Front. Pharmacol. 2019, 10, 920. [Google Scholar] [CrossRef] [Green Version]
- Singleton, K.D.; Wischmeyer, P.E. Effects of HSP70.1/3 gene knockout on acute respiratory distress syndrome and the inflammatory response following sepsis. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L956–L961. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Liu, Z.; Ding, H.; Zhou, Y.; Doan, H.A.; Sin, K.W.T.; Zhu, Z.J.; Flores, R.; Wen, Y.; Gong, X.; et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat. Commun. 2017, 8, 589. [Google Scholar] [CrossRef] [Green Version]
- Kuipers, M.T.; Aslami, H.; Vlaar, A.P.J.; Juffermans, N.P.; Boer, A.M.T.-D.; Hegeman, M.A.; Jongsma, G.; Roelofs, J.; Van Der Poll, T.; Schultz, M.J.; et al. Pre-Treatment with Allopurinol or Uricase Attenuates Barrier Dysfunction but Not Inflammation during Murine Ventilator-Induced Lung Injury. PLoS ONE 2012, 7, e50559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.W.; Kim, S.-M.; Kim, Y.G.; Lee, S.-H.; Moon, J.-Y. Uric acid and inflammation in kidney disease. Am. J. Physiol. Physiol. 2020, 318, F1327–F1340. [Google Scholar] [CrossRef] [PubMed]
- Ehrchen, J.M.; Sunderkötter, C.; Foell, D.; Vogl, T.; Roth, J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J. Leukoc. Biol. 2009, 86, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Schelbergen, R.F.P.; De Munter, W.; van den Bosch, M.H.J.; Lafeber, F.P.J.G.; Sloetjes, A.; Vogl, T.; Roth, J.; van den Berg, W.B.; Van Der Kraan, P.M.; Blom, A.B.; et al. Alarmins S100A8/S100A9 aggravate osteophyte formation in experimental osteoarthritis and predict osteophyte progression in early human symptomatic osteoarthritis. Ann. Rheum. Dis. 2014, 75, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, L.; Dingemans, A.-M.C. Heat shock protein antagonists in early stage clinical trials for NSCLC. Expert Opin. Investig. Drugs 2017, 26, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; St-Pierre, C.; Bhaumik, P.; Nieminen, J. Galectins in innate immunity: Dual functions of host soluble β-galactoside-binding lectins as damage-associated molecular patterns (DAMPs) and as receptors for pathogen-associated molecular patterns (PAMPs). Immunol. Rev. 2009, 230, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Dapat, I.C.; Pascapurnama, D.N.; Iwasaki, H.; Labayo, H.K.; Chagan-Yasutan, H.; Egawa, S.; Hattori, T. Secretion of Galectin-9 as a DAMP during Dengue Virus Infection in THP-1 Cells. Int. J. Mol. Sci. 2017, 18, 1644. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.; Yang, W.-L.; Wu, R.; Zhou, M.; Jacob, A.; Dong, W.; Kuncewitch, M.; Ji, Y.; Yang, H.; Wang, H.; et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat. Med. 2013, 19, 1489–1495. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.A.; Vissel, B. Amyloid β: One of three danger-associated molecules that are secondary inducers of the proinflammatory cytokines that mediate Alzheimer’s disease. Br. J. Pharmacol. 2015, 172, 3714–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, N.; Schaffrath, A.; Welter, J.A.; Putzka, T.; Griep, A.; Ziegler, P.; Brandt, E.; Samer, S.; Heneka, M.T.; Kaddatz, H.; et al. Inhibition of formyl peptide receptors improves the outcome in a mouse model of Alzheimer disease. J. Neuroinflamm. 2020, 17, 131. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Wenceslau, C.F.; McCarthy, C.G.; Szasz, T.; Goulopoulou, S.; Webb, R.C. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am. J. Physiol. Circ. Physiol. 2015, 308, H768–H777. [Google Scholar] [CrossRef] [Green Version]
- Dorward, D.A.; Lucas, C.; Chapman, G.B.; Haslett, C.; Dhaliwal, K.; Rossi, A.G. The Role of Formylated Peptides and Formyl Peptide Receptor 1 in Governing Neutrophil Function during Acute Inflammation. Am. J. Pathol. 2015, 185, 1172–1184. [Google Scholar] [CrossRef] [Green Version]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. Cytochrome c as a Potentially Clinical Useful Marker of Mitochondrial and Cellular Damage. Front. Immunol. 2016, 7, 279. [Google Scholar] [CrossRef] [Green Version]
- Gouveia, A.; Bajwa, E.; Klegeris, A. Extracellular cytochrome c as an intercellular signaling molecule regulating microglial functions. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2017, 1861, 2274–2281. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Tumburu, L.; Ghosh-Choudhary, S.; Seifuddin, F.T.; Barbu, E.A.; Yang, S.; Ahmad, M.M.; Wilkins, L.H.W.; Tunc, I.; Sivakumar, I.; Nichols, J.S.; et al. Circulating mitochondrial DNA is a proinflammatory DAMP in sickle cell disease. Blood 2021, 137, 3116–3126. [Google Scholar] [CrossRef] [PubMed]
- Aswani, A.; Manson, J.; Itagaki, K.; Chiazza, F.; Collino, M.; Wupeng, W.L.; Chan, T.K.; Wong, W.S.F.; Hauser, C.; Thiemermann, C.; et al. Scavenging Circulating Mitochondrial DNA as a Potential Therapeutic Option for Multiple Organ Dysfunction in Trauma Hemorrhage. Front. Immunol. 2018, 9, 891. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Collins, L.E.; Troeberg, L. Heparan sulfate as a regulator of inflammation and immunity. J. Leukoc. Biol. 2018, 105, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Rajak, S.; Hussain, Y.; Singh, K.; Tiwari, S.; Ahmad, B.; Bharti, S.; Prakash, P. Cellular Fibronectin Containing Extra Domain A Causes Insulin Resistance via Toll-like Receptor 4. Sci. Rep. 2020, 10, 9102. [Google Scholar] [CrossRef]
- Kelsh-Lasher, R.M.; Ambesi, A.; Bertram, C.; McKeown-Longo, P.J. Integrin α4β1 and TLR4 Cooperate to Induce Fibrotic Gene Expression in Response to Fibronectin’s EDA Domain. J. Investig. Dermatol. 2017, 137, 2505–2512. [Google Scholar] [CrossRef] [Green Version]
- McKeown-Longo, P.J.; Higgins, P.J. Hyaluronan, Transforming Growth Factor β, and Extra Domain A-Fibronectin: A Fibrotic Triad. Adv. Wound Care 2021, 10, 137–152. [Google Scholar] [CrossRef]
- Rosin, D.L.; Okusa, M.D. Dangers Within: DAMP Responses to Damage and Cell Death in Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 416–425. [Google Scholar] [CrossRef] [Green Version]
- McQuitty, C.E.; Williams, R.; Chokshi, S.; Urbani, L. Immunomodulatory Role of the Extracellular Matrix within the Liver Disease Microenvironment. Front. Immunol. 2020, 11, 2903. [Google Scholar] [CrossRef]
- Docherty, N.G.; Godson, C. Fibrinogen as a damage-associated mitogenic signal for the renal fibroblast. Kidney Int. 2011, 80, 1014–1016. [Google Scholar] [CrossRef] [Green Version]
- Campo, G.M.; Avenoso, A.; Nastasi, G.; Micali, A.; Prestipino, V.; Vaccaro, M.; D’Ascola, A.; Calatroni, A.; Campo, S. Hyaluronan reduces inflammation in experimental arthritis by modulating TLR-2 and TLR-4 cartilage expression. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 1170–1181. [Google Scholar] [CrossRef] [Green Version]
- Ruppert, S.M.; Hawn, T.R.; Arrigoni, A.; Wight, T.N.; Bollyky, P.L. Tissue integrity signals communicated by high-molecular weight hyaluronan and the resolution of inflammation. Immunol. Res. 2014, 58, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, X.; Wu, J.-L.; Quan, W.-Q.; Ma, L.; Yang, F.; Wu, K.-Y.; Wan, H.-Y. Tumor-Produced Versican V1 Enhances hCAP18/LL-37 Expression in Macrophages through Activation of TLR2 and Vitamin D3 Signaling to Promote Ovarian Cancer Progression In Vitro. PLoS ONE 2013, 8, e56616. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J.; Schaefer, L. Beyond Tissue Injury—Damage-Associated Molecular Patterns, Toll-Like Receptors, and Inflammasomes Also Drive Regeneration and Fibrosis. J. Am. Soc. Nephrol. 2014, 25, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Babelova, A.; Moreth, K.; Tsalastra-Greul, W.; Zeng-Brouwers, J.; Eickelberg, O.; Young, M.F.; Bruckner, P.; Pfeilschifter, J.; Schaefer, R.M.; Gröne, H.-J.; et al. Biglycan, a Danger Signal That Activates the NLRP3 Inflammasome via Toll-like and P2X Receptors. J. Biol. Chem. 2009, 284, 24035–24048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreth, K.; Frey, H.; Hubo, M.; Zeng-Brouwers, J.; Nastase, M.-V.; Hsieh, L.T.-H.; Haceni, R.; Pfeilschifter, J.; Iozzo, R.; Schaefer, L. Biglycan-triggered TLR-2- and TLR-4-signaling exacerbates the pathophysiology of ischemic acute kidney injury. Matrix Biol. 2014, 35, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; Weiss, D.G.; Paronetto, F.; Veterans Affairs Cooperative Study 391 Group. Value of Fibrosis Markers for Staging Liver Fibrosis in Patients with Precirrhotic Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2008, 32, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Udalova, I.; Ruhmann, M.; Thomson, S.J.; Midwood, K.S. Expression and Immune Function of Tenascin-C. Crit. Rev. Immunol. 2011, 31, 115–145. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, A.M.; Midwood, K.S. Endogenous Control of Immunity against Infection: Tenascin-C Regulates TLR4-Mediated Inflammation via MicroRNA-155. Cell Rep. 2012, 2, 914–926. [Google Scholar] [CrossRef] [Green Version]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef] [Green Version]
- Ardain, A.; Marakalala, M.; Leslie, A. Tissue-resident innate immunity in the lung. Immunology 2019, 159, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Hartl, D.; Tirouvanziam, R.; Laval, J.; Greene, C.M.; Habiel, D.; Sharma, L.; Yildirim, A.; Cruz, C.S.D.; Hogaboam, C. Innate Immunity of the Lung: From Basic Mechanisms to Translational Medicine. J. Innate Immun. 2018, 10, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal. Transduct. Target. Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Girard, J.-P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Lloyd, C.M.; Snelgrove, R.J. Type 2 immunity: Expanding our view. Sci. Immunol. 2018, 3, eaat1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chen, X.; Cao, H.; Xiang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal. 2018, 16, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.-Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fanny, M.; Nascimento, M.; Baron, L.; Schricke, C.; Maillet, I.; Akbal, M.; Riteau, N.; Le Bert, M.; Quesniaux, V.; Ryffel, B.; et al. The IL-33 Receptor ST2 Regulates Pulmonary Inflammation and Fibrosis to Bleomycin. Front. Immunol. 2018, 9, 1476. [Google Scholar] [CrossRef]
- Xia, J.; Zhao, J.; Shang, J.; Li, M.; Zeng, Z.; Zhao, J.; Wang, J.; Xu, Y.; Xie, J. Increased IL-33 expression in chronic obstructive pulmonary disease. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L619–L627. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Li, Y.; Li, M.; Li, M.; Liu, X.; McSharry, C.; Xu, D. Anti-interleukin-33 inhibits cigarette smoke-induced lung inflammation in mice. Immunology 2012, 138, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Zhao, J.; Wu, X.; Xu, Y.; Xie, J.; Zhao, J. Interleukin-33 promotes inflammatory cytokine production in chronic airway inflammation. Biochem. Cell Biol. 2015, 93, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, S.; Wu, X.; Zhao, J.; Zhao, J.; Ning, Q.; Xu, Y.; Xie, J. Interleukin-33/ST2 signaling promotes production of interleukin-6 and interleukin-8 in systemic inflammation in cigarette smoke-induced chronic obstructive pulmonary disease mice. Biochem. Biophys. Res. Commun. 2014, 450, 110–116. [Google Scholar] [CrossRef]
- Kearley, J.; Silver, J.S.; Sanden, C.; Liu, Z.; Berlin, A.A.; White, N.; Mori, M.; Pham, T.-H.; Ward, C.K.; Criner, G.J.; et al. Cigarette Smoke Silences Innate Lymphoid Cell Function and Facilitates an Exacerbated Type I Interleukin-33-Dependent Response to Infection. Immunity 2015, 42, 566–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, C.; Hansbro, P. IL-33 in Chronic Respiratory Disease: From Preclinical to Clinical Studies. ACS Pharmacol. Transl. Sci. 2019, 3, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-U.; Chang, H.S.; Lee, H.J.; Jung, C.A.; Bae, D.J.; Song, H.J.; Park, J.S.; Uh, S.-T.; Kim, Y.H.; Seo, K.-H.; et al. Upregulation of interleukin-33 and thymic stromal lymphopoietin levels in the lungs of idiopathic pulmonary fibrosis. BMC Pulm. Med. 2017, 17, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Luzina, I.G.; Kopach, P.; Lockatell, V.; Kang, P.H.; Nagarsekar, A.; Burke, A.P.; Hasday, J.D.; Todd, N.W.; Atamas, S.P. Interleukin-33 Potentiates Bleomycin-Induced Lung Injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 999–1008. [Google Scholar] [CrossRef]
- Zhao, Y.; Santos, F.G.D.L.; Wu, Z.; Liu, T.; Phan, S.H. An ST2-dependent role of bone marrow-derived group 2 innate lymphoid cells in pulmonary fibrosis. J. Pathol. 2018, 245, 399–409. [Google Scholar] [CrossRef]
- Hamada, N.; Maeyama, T.; Kawaguchi, T.; Yoshimi, M.; Fukumoto, J.; Yamada, M.; Yamada, S.; Kuwano, K.; Nakanishi, Y. The Role of High Mobility Group Box1 in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 440–447. [Google Scholar] [CrossRef]
- Ferhani, N.; Letuve, S.; Kozhich, A.; Thibaudeau, O.; Grandsaigne, M.; Maret, M.; Dombret, M.-C.; Sims, G.P.; Kolbeck, R.; Coyle, A.J.; et al. Expression of High-Mobility Group Box 1 and of Receptor for Advanced Glycation End Products in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2010, 181, 917–927. [Google Scholar] [CrossRef]
- Chen, X.; Xu, H.; Hou, J.; Wang, H.; Zheng, Y.; Li, H.; Cai, H.; Han, X.; Dai, J. Epithelial cell senescence induces pulmonary fibrosis through Nanog-mediated fibroblast activation. Aging 2019, 12, 242–259. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Mi, Y.; Yang, H.; Hu, A.; Zhang, Q.; Shang, C. The activation of HMGB1 as a progression factor on inflammation response in normal human bronchial epithelial cells through RAGE/JNK/NF-κB pathway. Mol. Cell. Biochem. 2013, 380, 249–257. [Google Scholar] [CrossRef]
- Yang, J.; Agarwal, M.; Ling, S.; Teitz-Tennenbaum, S.; Zemans, R.L.; Osterholzer, J.J.; Sisson, T.H.; Kim, K.K. Diverse Injury Pathways Induce Alveolar Epithelial Cell CCL2/12, Which Promotes Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 622–632. [Google Scholar] [CrossRef]
- Foell, D.; Wittkowski, H.; Roth, J. Mechanisms of Disease: A ‘DAMP’ view of inflammatory arthritis. Nat. Clin. Pract. Rheumatol. 2007, 3, 382–390. [Google Scholar] [CrossRef]
- Ryckman, C.; Vandal, K.; Rouleau, P.; Talbot, M.; Tessier, P. Proinflammatory Activities of S100: Proteins S100A8, S100A9, and S100A8/A9 Induce Neutrophil Chemotaxis and Adhesion. J. Immunol. 2003, 170, 3233–3242. [Google Scholar] [CrossRef] [Green Version]
- Zemans, R.L.; Colgan, S.P.; Downey, G.P. Transepithelial Migration of Neutrophils. Am. J. Respir. Cell Mol. Biol. 2009, 40, 519–535. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Zenker, S.; Rossaint, J.; Hölscher, A.; Pohlen, M.; Zarbock, A.; Roth, J.; Vogl, T. Alarmin S100A8 Activates Alveolar Epithelial Cells in the Context of Acute Lung Injury in a TLR4-Dependent Manner. Front. Immunol. 2017, 8, 1493. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Wang, J.H.-C. Fibroblasts and myofibroblasts in wound healing: Force generation and measurement. J. Tissue Viability 2011, 20, 108–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Tang, L.; Kapoor, M. Fibroblasts and their responses to chronic injury in pulmonary fibrosis. Semin. Arthritis Rheum. 2020, 51, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Flavell, S.J.; Hou, T.Z.; Lax, S.; Filer, A.D.; Salmon, M.; Buckley, C.D. Fibroblasts as novel therapeutic targets in chronic inflammation. Br. J. Pharmacol. 2008, 153, S241–S246. [Google Scholar] [CrossRef] [Green Version]
- Suwara, M.; Green, N.; Borthwick, L.; Mann, J.R.; Mayerbarber, K.D.; Barron, L.D.; Corris, P.A.; Farrow, S.N.; Wynn, T.; Fisher, A.J.; et al. IL-1α released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol. 2013, 7, 684–693. [Google Scholar] [CrossRef]
- Osei, E.T.; Noordhoek, J.A.; Hackett, T.L.; Spanjer, A.I.; Postma, D.S.; Timens, W.; Brandsma, C.-A.; Heijink, I.H. Interleukin-1α drives the dysfunctional cross-talk of the airway epithelium and lung fibroblasts in COPD. Eur. Respir. J. 2016, 48, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Tsukui, T.; Sun, K.-H.; Wetter, J.B.; Wilson-Kanamori, J.; Hazelwood, L.A.; Henderson, N.C.; Adams, T.S.; Schupp, J.C.; Poli, S.; Rosas, I.O.; et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of inflammatory cells in airway remodeling in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 3341–3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brightling, C.; Greening, N. Airway inflammation in COPD: Progress to precision medicine. Eur. Respir. J. 2019, 54, 1900651. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Hu, S.; Li, C.; Ma, H.; Wang, Q.; Meng, G.; Guo, T.; Zhang, J. Cigarette Smoke Induced Lung Barrier Dysfunction, EMT, and Tissue Remodeling: A Possible Link between COPD and Lung Cancer. BioMed Res. Int. 2019, 2019, 2025636. [Google Scholar] [CrossRef]
- Demedts, I.K.; Brusselle, G.G.; Bracke, K.R.; Vermaelen, K.Y.; Pauwels, R.A. Matrix metalloproteinases in asthma and COPD. Curr. Opin. Pharmacol. 2005, 5, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Kosmider, B.; Messier, E.; Chu, H.W.; Mason, R.J. Human Alveolar Epithelial Cell Injury Induced by Cigarette Smoke. PLoS ONE 2011, 6, e26059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickenden, J.A.; Clarke, M.; Rossi, A.G.; Rahman, I.; Faux, S.P.; Donaldson, K.; MacNee, W. Cigarette Smoke Prevents Apoptosis through Inhibition of Caspase Activation and Induces Necrosis. Am. J. Respir. Cell Mol. Biol. 2003, 29, 562–570. [Google Scholar] [CrossRef] [Green Version]
- Faiz, A.; Heijink, I.H.; Vermeulen, C.; Guryev, V.; Berge, M.V.D.; Nawijn, M.; Pouwels, S.D. Cigarette smoke exposure decreases CFLAR expression in the bronchial epithelium, augmenting susceptibility for lung epithelial cell death and DAMP release. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Hulina-Tomašković, A.; Rajković, M.G.; Somborac-Bačura, A.; Čeri, A.; Dabelić, S.; Rumora, L. Extracellular Hsp70 modulates the inflammatory response of cigarette smoke extract in NCI-H292 cells. Exp. Physiol. 2018, 103, 1704–1716. [Google Scholar] [CrossRef] [Green Version]
- Hulina-Tomašković, A.; Heijink, I.H.; Jonker, M.R.; Bačura, A.S.; Rajković, M.G.; Rumora, L. Pro-inflammatory effects of extracellular Hsp70 and cigarette smoke in primary airway epithelial cells from COPD patients. Biochimie 2018, 156, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, S.D.; Faiz, A.; Den Boef, L.E.; Gras, R.; van den Berge, M.; Boezen, H.M.; Korstanje, R.; ten Hacken, N.H.T.; Van Oosterhout, A.J.M.; Heijink, I.H.; et al. Genetic variance is associated with susceptibility for cigarette smoke-induced DAMP release in mice. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L559–L580. [Google Scholar] [CrossRef] [Green Version]
- Pouwels, S.D.; Nawijn, M.; Bathoorn, E.; Riezebos-Brilman, A.; Van Oosterhout, A.J.M.; Kerstjens, H.; Heijink, I.H. Increased serum levels of LL37, HMGB1 and S100A9 during exacerbation in COPD patients. Eur. Respir. J. 2015, 45, 1482–1485. [Google Scholar] [CrossRef] [PubMed]
- Zabini, D.; Crnkovic, S.; Xu, H.; Tscherner, M.; Ghanim, B.; Klepetko, W.; Olschewski, A.; Kwapiszewska, G.; Marsh, L.M. High-mobility group box-1 induces vascular remodelling processes via c-Jun activation. J. Cell. Mol. Med. 2015, 19, 1151–1161. [Google Scholar] [CrossRef]
- Heijink, I.H.; Pouwels, S.D.; Leijendekker, C.; De Bruin, H.G.; Zijlstra, G.J.; Van Der Vaart, H.; ten Hacken, N.H.T.; Van Oosterhout, A.J.M.; Nawijn, M.C.; van der Toorn, M. Cigarette Smoke–Induced Damage-Associated Molecular Pattern Release from Necrotic Neutrophils Triggers Proinflammatory Mediator Release. Am. J. Respir. Cell Mol. Biol. 2015, 52, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 SIGNALS THROUGH TOLL-LIKE RECEPTOR (TLR) 4 AND TLR2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef]
- Castaldi, P.J.; Cho, M.H.; Litonjua, A.A.; Bakke, P.; Gulsvik, A.; Lomas, D.A.; Anderson, W.; Beaty, T.H.; Hokanson, J.E.; Crapo, J.D.; et al. The Association of Genome-Wide Significant Spirometric Loci with Chronic Obstructive Pulmonary Disease Susceptibility. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1147–1153. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.; Angeli, J.P.F.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Chen, B.; Wang, Y.; Meng, C.; Huang, H.; Huang, X.-R.; Qin, J.; Mulay, S.R.; Anders, H.-J.; Qiu, A.; et al. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc. Natl. Acad. Sci. USA 2018, 115, E1475–E1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Duan, X.; Fu, X.; Jiang, Y.; Yang, P.; Cao, C.; Li, Q.; Zhang, J.; Hu, X.; Zhang, X.; et al. RIP1 Perturbation Induces Chondrocyte Necroptosis and Promotes Osteoarthritis Pathogenesis via Targeting BMP7. Front. Cell Dev. Biol. 2021, 9, 638382. [Google Scholar] [CrossRef] [PubMed]
- Luedde, M.; Lutz, M.; Carter, N.; Sosna, J.; Jacoby, C.; Vucur, M.; Gautheron, J.; Roderburg, C.; Borg, N.; Reisinger, F.; et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc. Res. 2014, 103, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gregory, A.D.; Li, X.; Wei, J.; Burton, C.L.; Gibson, G.; Scott, S.J.; Croix, C.M.S.; Zhang, Y.; Shapiro, S.D. RIP3-dependent necroptosis contributes to the pathogenesis of chronic obstructive pulmonary disease. JCI Insight 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Van Eeckhoutte, H.P.; Liu, G.; Nair, P.M.; Jones, B.; Gillis, C.M.; Nalkurthi, B.C.; Verhamme, F.; Buyle-Huybrecht, T.; Vandenabeele, P.; et al. Necroptosis Signaling Promotes Inflammation, Airway Remodeling, and Emphysema in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 667–681. [Google Scholar] [CrossRef]
- Yoshida, M.; Minagawa, S.; Araya, J.; Sakamoto, T.; Hara, H.; Tsubouchi, K.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 2019, 10, 3145. [Google Scholar] [CrossRef]
- Aghapour, M.; Remels, A.H.V.; Pouwels, S.D.; Bruder, D.; Hiemstra, P.S.; Cloonan, S.M.; Heijink, I.H. Mitochondria: At the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L149–L164. [Google Scholar] [CrossRef]
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R.; Bodas, M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am. J. Physiol. Physiol. 2018, 314, C73–C87. [Google Scholar] [CrossRef] [Green Version]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 157–179. [Google Scholar] [CrossRef] [Green Version]
- Heukels, P.; Moor, C.; von der Thusen, J.; Wijsenbeek, M.; Kool, M. Inflammation and immunity in IPF pathogenesis and treatment. Respir. Med. 2019, 147, 79–91. [Google Scholar] [CrossRef]
- Zhang, W.; Lavine, K.J.; Epelman, S.; Evans, S.A.; Weinheimer, C.J.; Barger, P.M.; Mann, D.L. Necrotic Myocardial Cells Release Damage-Associated Molecular Patterns That Provoke Fibroblast Activation In Vitro and Trigger Myocardial Inflammation and Fibrosis In Vivo. J. Am. Hear. Assoc. 2015, 4, e001993. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Guo, Y.; Fu, H.; Hu, S.; Pan, J.; Wang, Y.; Cheng, J.; Song, J.; Yu, Q.; Zhang, S.; et al. Chop deficiency prevents UUO-induced renal fibrosis by attenuating fibrotic signals originated from Hmgb1/TLR4/NFκB/IL-1β signaling. Cell Death Dis. 2015, 6, e1847. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Sakamoto, S.; Isshiki, T.; Furuya, K.; Kurosaki, A.; Homma, S. Association of serum high-mobility group box protein 1 level with outcomes of acute exacerbation of idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumonia. PLoS ONE 2018, 13, e0196558. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Iwamoto, H.; Sakamoto, S.; Horimasu, Y.; Masuda, T.; Miyamoto, S.; Nakashima, T.; Ohshimo, S.; Fujitaka, K.; Hamada, H.; et al. Serum high-mobility group box 1 is associated with the onset and severity of acute exacerbation of idiopathic pulmonary fibrosis. Respirology 2019, 25, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Ebina, M.; Taniguchi, H.; Miyasho, T.; Yamada, S.; Shibata, N.; Ohta, H.; Hisata, S.; Ohkouchi, S.; Tamada, T.; Nishimura, H.; et al. Gradual Increase of High Mobility Group Protein B1 in the Lungs after the Onset of Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Pulm. Med. 2011, 2011, 916486. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.; Fay, S.; Vieira, R.P.; Karmouty-Quintana, H.; Cicko, S.; Ayata, K.; Zissel, G.; Goldmann, T.; Lungarella, G.; Ferrari, D.; et al. The purinergic receptor subtype P2Y2 mediates chemotaxis of neutrophils and fibroblasts in fibrotic lung disease. Oncotarget 2017, 8, 35962–35972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, N.; Hozumi, H.; Isayama, T.; Okada, J.; Sugiura, K.; Yasui, H.; Suzuki, Y.; Kono, M.; Karayama, M.; Furuhashi, K.; et al. Clinical significance of serum S100 calcium-binding protein A4 in idiopathic pulmonary fibrosis. Respirology 2019, 25, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Janssen, L.J.; Farkas, L.; Rahman, T.; Kolb, M.R. ATP stimulates Ca2+-waves and gene expression in cultured human pulmonary fibroblasts. Int. J. Biochem. Cell Biol. 2009, 41, 2477–2484. [Google Scholar] [CrossRef] [PubMed]
- Hodono, S.; Shimokawa, A.; Stewart, N.J.; Yamauchi, Y.; Nishimori, R.; Yamane, M.; Imai, H.; Fujiwara, H.; Kimura, A. Ethyl Pyruvate Improves Pulmonary Function in Mice with Bleomycin-induced Lung Injury as Monitored with Hyperpolarized 129Xe MR Imaging. Magn. Reson. Med. Sci. 2018, 17, 331–337. [Google Scholar] [CrossRef]
- Pittet, J.-F.; Koh, H.; Fang, X.; Iles, K.; Christiaans, S.; Anjun, N.; Wagener, B.M.; Park, D.W.; Zmijewski, J.W.; Matthay, M.A.; et al. HMGB1 Accelerates Alveolar Epithelial Repair via an IL-1β- and αvβ6 Integrin-dependent Activation of TGF-β1. PLoS ONE 2013, 8, e63907. [Google Scholar] [CrossRef]
- Li, L.-C.; Li, D.-L.; Xu, L.; Mo, X.-T.; Cui, W.-H.; Zhao, P.; Zhou, W.-C.; Gao, J.; Li, J. High-Mobility Group Box 1 Mediates Epithelial-to-Mesenchymal Transition in Pulmonary Fibrosis Involving Transforming Growth Factor-β1/Smad2/3 Signaling. J. Pharmacol. Exp. Ther. 2015, 354, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Luo, R.; Zhang, M.; Wang, Y.; Song, T.; Tao, T.; Li, Z.; Jin, L.; Zheng, H.; Chen, W.; et al. A cross-talk between epithelium and endothelium mediates human alveolar–capillary injury during SARS-CoV-2 infection. Cell Death Dis. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Yoshida, M.; Kim, M.-S.; Lee, J.-H.; Baek, A.-R.; Jang, A.S.; Kim, D.J.; Minagawa, S.; Chin, S.S.; Park, C.-S.; et al. Involvement of Alveolar Epithelial Cell Necroptosis in Idiopathic Pulmonary Fibrosis Pathogenesis. Am. J. Respir. Cell Mol. Biol. 2018, 59, 215–224. [Google Scholar] [CrossRef]
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-H.; Cruz, S.A.; Qin, Z.; Stewart, A. Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regen. Res. 2018, 13, 252–256. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, Q.; Phan, N.; Ren, J.; Yang, H.; Feldman, C.C.; Feltenberger, J.B.; Ye, Z.; Wildman, S.; Tang, W.; et al. Identification of a novel class of RIP1/RIP3 dual inhibitors that impede cell death and inflammation in mouse abdominal aortic aneurysm models. Cell Death Dis. 2019, 10, 226. [Google Scholar] [CrossRef]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensiv. Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Sarzi-Puttini, P.; Giorgi, V.; Sirotti, S.; Marotto, D.; Ardizzone, S.; Rizzardini, G.; Antinori, S.; Galli, M. COVID-19, cytokines and immunosuppression: What can we learn from severe acute respiratory syndrome? Clin. Exp. Rheumatol. 2020, 38, 337–342. [Google Scholar]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Burke, H.; on behalf of the REACT COVID investigators; Freeman, A.; Cellura, D.C.; Stuart, B.L.; Brendish, N.J.; Poole, S.; Borca, F.; Phan, H.T.T.; Sheard, N.; et al. Inflammatory phenotyping predicts clinical outcome in COVID-19. Respir. Res. 2020, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Ge, Y.; Sun, J. IL-33 in COVID-19: Friend or foe? Cell. Mol. Immunol. 2021, 18, 1602–1604. [Google Scholar] [CrossRef]
- Filbin, M.R.; Mehta, A.; Schneider, A.M.; Kays, K.R.; Guess, J.R.; Gentili, M.; Fenyves, B.G.; Charland, N.C.; Gonye, A.L.; Gushterova, I.; et al. Longitudinal proteomic analysis of severe COVID-19 reveals survival-associated signatures, tissue-specific cell death, and cell-cell interactions. Cell Rep. Med. 2021, 2, 100287. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zuo, Y.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Woodward, W.; Lezak, S.P.; Lugogo, N.L.; et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J. Leukoc. Biol. 2020, 109, 67–72. [Google Scholar] [CrossRef]
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.-G.; Dubuisson, A.; Derosa, L.; Almire, C.; Hénon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418.e18. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef]
- Chen, R.; Huang, Y.; Quan, J.; Liu, J.; Wang, H.; Billiar, T.R.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. HMGB1 as a potential biomarker and therapeutic target for severe COVID-19. Heliyon 2020, 6, e05672. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.M.; Cheruiyot, I.; Benoit, S.W.; Sanchis-Gomar, F.; Lippi, G.; Benoit, J. Cytokeratin 18 cell death assays as biomarkers for quantification of apoptosis and necrosis in COVID-19: A prospective, observational study. J. Clin. Pathol. 2021. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.-L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal. Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.-C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nat. Cell Biol. 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Rall, G.F. Benefits and Perils of Necroptosis in Influenza Virus Infection. J. Virol. 2020, 94, e01101-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayflick, L.; Moorhead, P. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere Shortening Triggers Senescence of Human Cells through a Pathway Involving ATM, p53, and p21CIP1, but Not p16INK4a. Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wilson, C.; Passos, J.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 5, 4172. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Woldhuis, R.R.; De Vries, M.; Timens, W.; van den Berge, M.; Demaria, M.; Oliver, B.G.G.; Heijink, I.H.; Brandsma, C.-A. Link between increased cellular senescence and extracellular matrix changes in COPD. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L48–L60. [Google Scholar] [CrossRef]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Faner, R.; Rojas, M.; MacNee, W.; Agustí, A. Abnormal Lung Aging in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Nehme, J.; Borghesan, M.; Mackedenski, S.; Bird, T.G.; DeMaria, M. Cellular senescence as a potential mediator of COVID-19 severity in the elderly. Aging Cell 2020, 19, e13237. [Google Scholar] [CrossRef]
- Mohiuddin, M.; Kasahara, K. The emerging role of cellular senescence in complications of COVID-19. Cancer Treat. Res. Commun. 2021, 28, 100399. [Google Scholar] [CrossRef]
- Lomas, N.J.; Watts, K.L.; Akram, K.M.; Forsyth, N.R.; Spiteri, M.A. Idiopathic pulmonary fibrosis: Immunohistochemical analysis provides fresh insights into lung tissue remodelling with implications for novel prognostic markers. Int. J. Clin. Exp. Pathol. 2012, 5, 58–71. [Google Scholar] [PubMed]
- Kuwano, K.; Kunitake, R.; Kawasaki, M.; Nomoto, Y.; Hagimoto, N.; Nakanishi, Y.; Hara, N. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1996, 154, 477–483. [Google Scholar] [CrossRef]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef]
- DePianto, D.J.; Heiden, J.A.V.; Morshead, K.B.; Sun, K.-H.; Modrusan, Z.; Teng, G.; Wolters, P.J.; Arron, J.R. Molecular mapping of interstitial lung disease reveals a phenotypically distinct senescent basal epithelial cell population. JCI Insight 2021, 6, e143626. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; Lebrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, M.; Mutze, K.; Korfei, M.; Klee, S.; Wagner, D.; Costa, R.; Schiller, H.; Günther, A.; Königshoff, M. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J. 2017, 50, 1602367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calhoun, C.; Shivshankar, P.; Saker, M.; Sloane, L.B.; Livi, C.; Sharp, Z.D.; Orihuela, C.J.; Adnot, S.; White, E.S.; Richardson, A.; et al. Senescent Cells Contribute to the Physiological Remodeling of Aged Lungs. J. Gerontol. Ser. A: Boil. Sci. Med. Sci. 2015, 71, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaizen, Y.; Tachibana, Y.; Kashima, Y.; Bychkov, A.; Tabata, K.; Otani, K.; Kinoshita, Y.; Yamano, Y.; Kataoka, K.; Ichikado, K.; et al. Alveolar Epithelial Denudation Is a Major Factor in the Pathogenesis of Pleuroparenchymal Fibroelastosis. J. Clin. Med. 2021, 10, 895. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.-K.T.; Funari, V.A.; Gokey, J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.; Blumhagen, R.Z.; Yang, I.V.; Walts, A.; Powers, J.; Walker, T.; Bishop, M.; Russell, P.; Vestal, B.; Cardwell, J.; et al. Resequencing Study Confirms That Host Defense and Cell Senescence Gene Variants Contribute to the Risk of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Blokland, K.; Waters, D.; Schuliga, M.; Pouwels, S.; Grainge, C.; Mutsaers, S.; Prêle, C.; Westall, G.; Jaffar, J.; Knight, D.; et al. Alveolar epithelial wound repair is delayed by scenescent lung fibroblasts in IPF. Eur. Respir. J. 2019, 54, PA596. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1 is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef] [Green Version]
- Chilosi, M.; Carloni, A.; Rossi, A.; Poletti, V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl. Res. 2013, 162, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Kheradmand, F.; You, R.; Hee Gu, B.; Corry, D.B. Cigarette Smoke and DNA Cleavage Promote Lung Inflammation and Emphysema. Trans. Am. Clin. Climatol. Assoc. 2017, 128, 222–233. [Google Scholar] [PubMed]
- Cottage, C.T.; Peterson, N.; Kearley, J.; Berlin, A.; Xiong, X.; Huntley, A.; Zhao, W.; Brown, C.; Migneault, A.; Zerrouki, K.; et al. Targeting p16-induced senescence prevents cigarette smoke-induced emphysema by promoting IGF1/Akt1 signaling in mice. Commun. Biol. 2019, 2, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.-Y.; Li, Y.-Y.; Huang, C.; Li, J.; Yao, H.-W. AMP-activated protein kinase reduces inflammatory responses and cellular senescence in pulmonary emphysema. Oncotarget 2017, 8, 22513–22523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, X.; He, B.; Ning, Q.; Liu, Y.; Guo, J.; Niu, G.; Chen, M. Alantolactone suppresses inflammation, apoptosis and oxidative stress in cigarette smoke-induced human bronchial epithelial cells through activation of Nrf2/HO-1 and inhibition of the NF-κB pathways. Respir. Res. 2020, 21, 95. [Google Scholar] [CrossRef]
- Sundar, I.K.; Rashid, K.; Gerloff, J.; Li, D.; Rahman, I. Genetic Ablation of p16INK4a Does Not Protect against Cellular Senescence in Mouse Models of Chronic Obstructive Pulmonary Disease/Emphysema. Am. J. Respir. Cell Mol. Biol. 2018, 59, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.; Topisirovic, I.; Hulea, L. mTOR as a central regulator of lifespan and aging. F1000Research 2019, 8, 998. [Google Scholar] [CrossRef] [PubMed]
- Houssaini, A.; Breau, M.; Kebe, K.; Abid, S.; Marcos, E.; Lipskaia, L.; Rideau, D.; Parpaleix, A.; Huang, J.; Amsellem, V.; et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight 2018, 3, e93203. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2020, 65, 101205. [Google Scholar] [CrossRef]
- Cox, L.S.; Bellantuono, I.; Lord, J.M.; Sapey, E.; Mannick, J.B.; Partridge, L.; Gordon, A.L.; Steves, C.J.; Witham, M.D. Tackling immunosenescence to improve COVID-19 outcomes and vaccine response in older adults. Lancet Heal. Longev. 2020, 1, e55–e57. [Google Scholar] [CrossRef]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.G.P.L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics reduce coronavirus-related mortality in old mice. Science 2021, 373. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DAMP Ligand | Known Receptors | Evidence in Disease | Reference |
|---|---|---|---|
| Intracellular-Nuclear DAMPs | |||
| HMGB1 | TLR2, TLR4, TLR9, RAGE, CXCR4 | Ischaemia reperfusion injury, sepsis, acute lung injury, chronic inflammatory and autoimmune diseases, viral infection, cancer | [34,36,37,38,39,40,41,42] |
| HMGN1 | TLR4 | Cancer | [43,44,45] |
| IL-1α | TLR2, TLR4, TLR7, TLR8, TLR9, IL-1R | Chronic inflammatory disease, sepsis, cancer | [46,47,48,49] |
| IL-33 | ST2 | Asthma, atopic dermatitis, anaphylaxis, allergic inflammation, fibrosis | [46,47,48,50,51,52] |
| DNA | TLR9, AIM2 | Kidney disease, inflammation, cancer | [53,54,55,56,57,58] |
| RNA | TLR3, TLR7, TLR8, MDA5 | Autoimmune disease, sepsis, acute lung injury, liver injury | [34,56,59,60,61,62] |
| SAP130 | MINCLE | Inflammatory disease and fibrosis | [30,63] |
| Histones | TLR2, TLR4 | Sepsis, trauma, acute liver failure, kidney injury, acute lung injury | [64,65,66,67,68] |
| Intracellular-Cytosolic DAMPs | |||
| ATP | P2X7, P2Y2 | Sepsis, hypoxia, mechanical ventilation, migraine, stroke, cancer | [34,69,70,71] |
| Heat shock proteins | TLR2, TLR4, CD91, CD14, CD40 | Sepsis, acute lung injury, cancer, rheumatoid arthritis, neuroinflammation | [34,72,73,74] |
| Uric acid | NLRP3, P2X7 | Acute inflammation, ventilator-induced lung injury, kidney disease, gout | [34,75,76] |
| S100 proteins | TLR2, TLR4, RAGE | Endotoxin-induced shock, cancer, autoimmune disease, kidney disease, osteoarthritis | [34,77,78,79] |
| Galectins | CD2 | Cancer, fibrosis, chronic inflammation, infection | [80,81] |
| CIRP | TLR-MD2 | Haemorrhagic shock, sepsis | [53,82] |
| Amyloid-β | TLR2, NLRP1, NLRP3, RAGE | Alzheimer’s disease | [83,84] |
| Intracellular-Mitochondrial DAMPs | |||
| mROS | NLRP3 | Neurodegenerative disorders, cancer, pulmonary disease, cardiovascular disease, gastrointestinal disorders | [85,86] |
| Formyl peptides | FPR1 | systemic inflammatory response syndrome, acute inflammation | [87,88] |
| Cytochrome C | TLR4 | Neuroinflammation | [89,90] |
| mtDNA | TLR9 | Trauma haemorrhage, sickle cell disease, arthritis, inflammation | [85,91,92,93,94] |
| Extracellular DAMPs | |||
| Heparan sulfate | TLR4 | Alzheimer’s disease, autoimmune disease, inflammatory disease | [95] |
| Fibronectin | TLR4 | Diabetes, inflammation, fibrosis, liver disease, kidney disease | [96,97,98,99,100] |
| Fibrinogen | TLR4 | Cancer, fibrosis, kidney disease | [99,101] |
| Hyaluronan | TLR2, TLR4, NRLP3 | Fibrosis, arthritis, inflammation, kidney disease | [98,102,103] |
| Versican | TLR2, TLR6, CD14 | Inflammatory lung diseases, cancer, kidney disease | [104] |
| Decorin | TLR2, TLR4 | Chronic inflammation, fibrosis | [105] |
| Biglycan | TLR2, TLR4, NRLP3 | Acute kidney injury, chronic inflammation | [106,107] |
| Laminin | TLR4 | Autoimmune disease, fibrosis, chronic inflammatory disease, liver disease, cancer | [100,108] |
| Tenascin C | TLR4 | Injury, fibrosis, infection, cancer | [109,110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgoyne, R.A.; Fisher, A.J.; Borthwick, L.A. The Role of Epithelial Damage in the Pulmonary Immune Response. Cells 2021, 10, 2763. https://doi.org/10.3390/cells10102763
Burgoyne RA, Fisher AJ, Borthwick LA. The Role of Epithelial Damage in the Pulmonary Immune Response. Cells. 2021; 10(10):2763. https://doi.org/10.3390/cells10102763
Chicago/Turabian StyleBurgoyne, Rachel Ann, Andrew John Fisher, and Lee Anthony Borthwick. 2021. "The Role of Epithelial Damage in the Pulmonary Immune Response" Cells 10, no. 10: 2763. https://doi.org/10.3390/cells10102763