
Oxytocin Is a Positive Allosteric Modulator of κ-Opioid Receptors but Not δ-Opioid Receptors in the G Protein Signaling Pathway

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Establishment of HEK293 Cells Stably Expressing Human DOR or KOR
2.3. Cell Cultures
2.4. CellKeyTM Assay
2.5. GloSensorTM cAMP Assay
2.6. KOR Internalization Assay
2.7. Radioligand Competitive OR Binding Assay
2.8. Statistical Analysis
3. Results
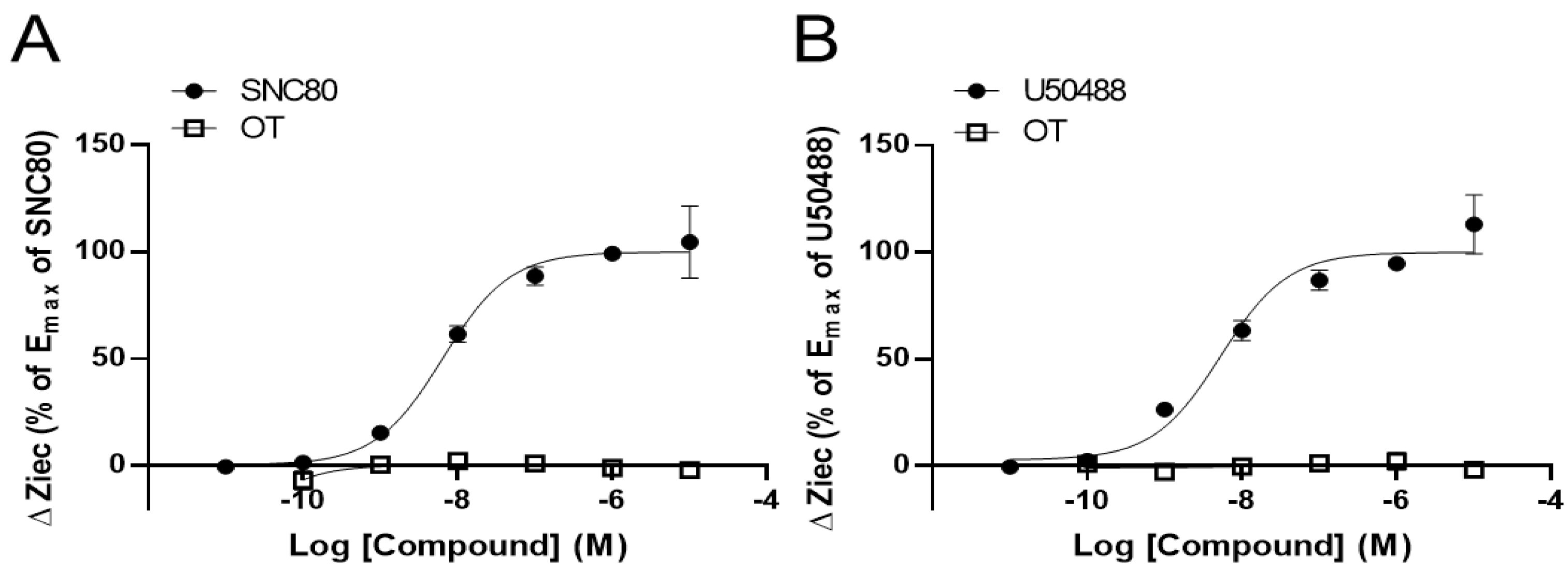
3.1. OT Alone Did Not Change Impedance (ΔZiec) in HEK293 Cells Stably Expressing Either Human DOR or KOR in the CellKeyTM Assay
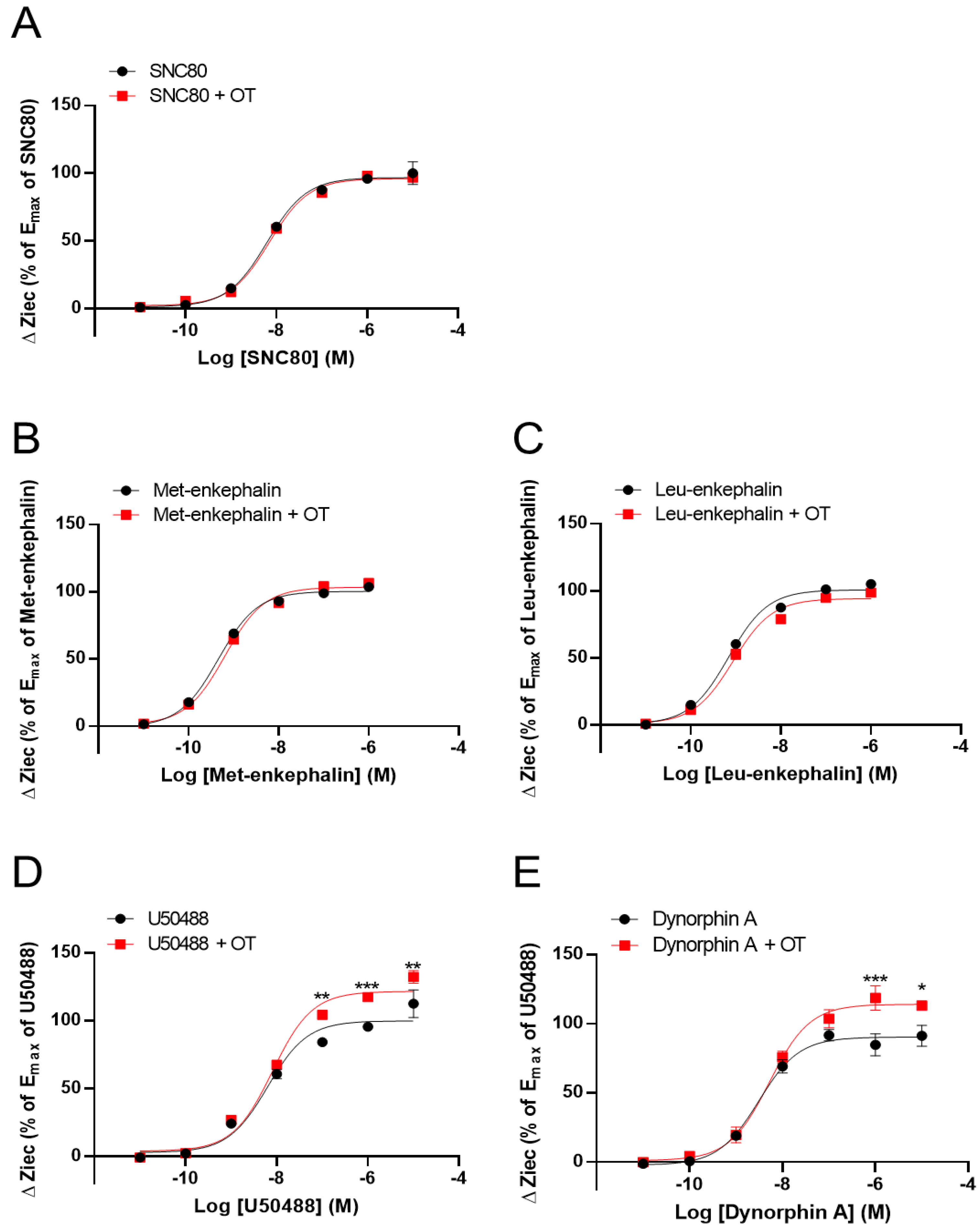
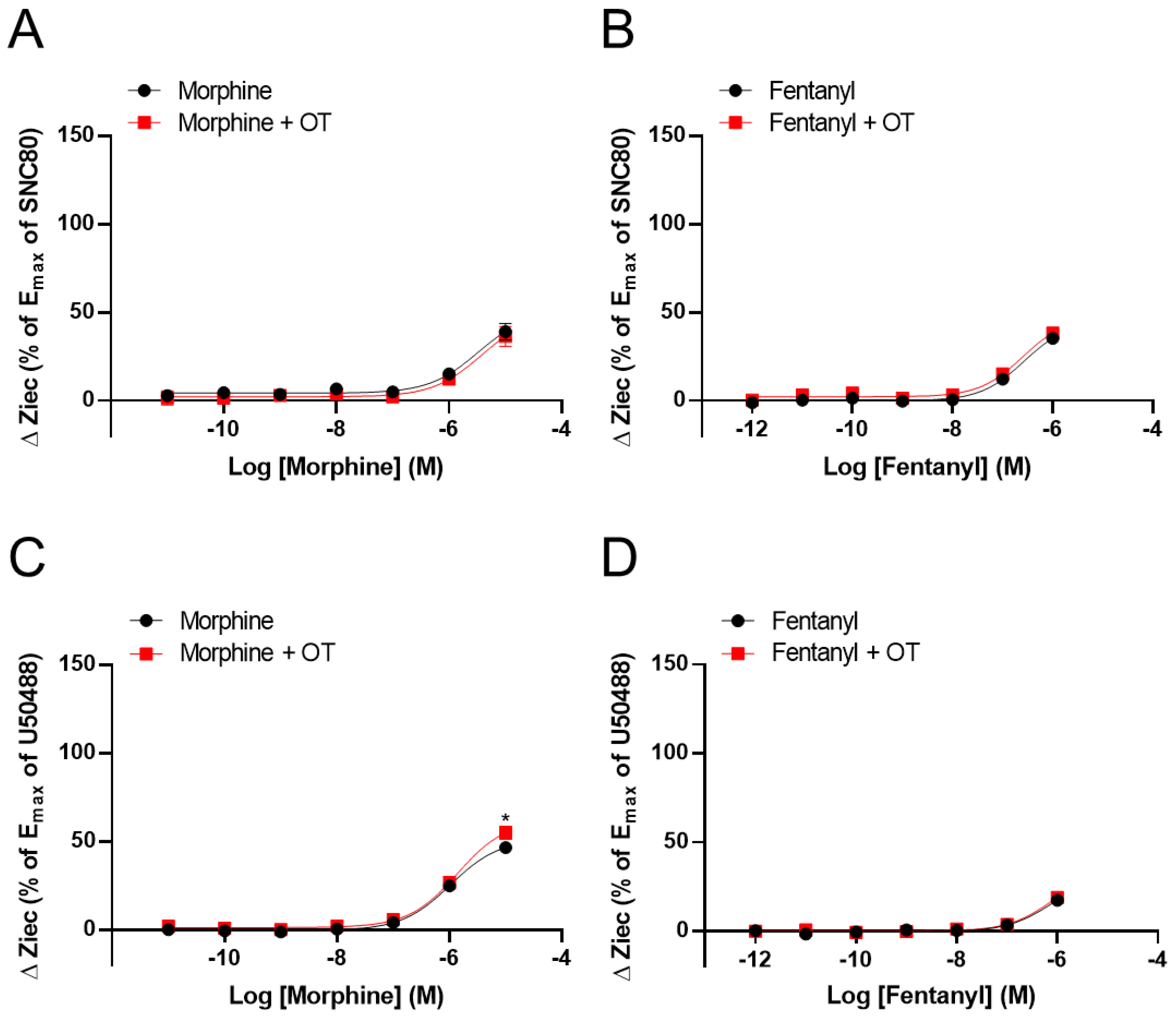
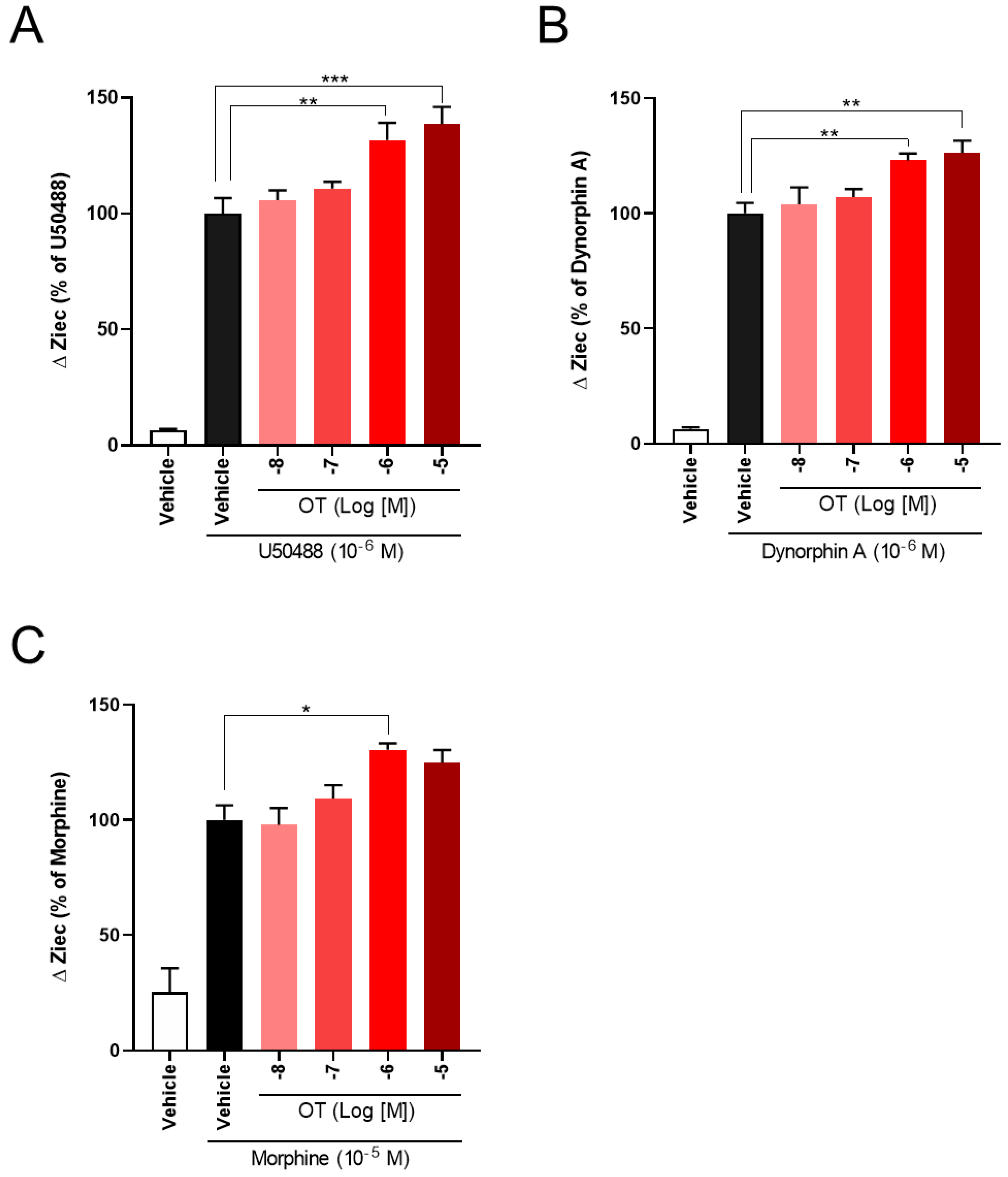
3.2. OT Enhanced ΔZiec Induced by KOR Agonists but Not DOR Agonists in HEK293 Cells Stably Expressing Human KOR or DOR, Respectively
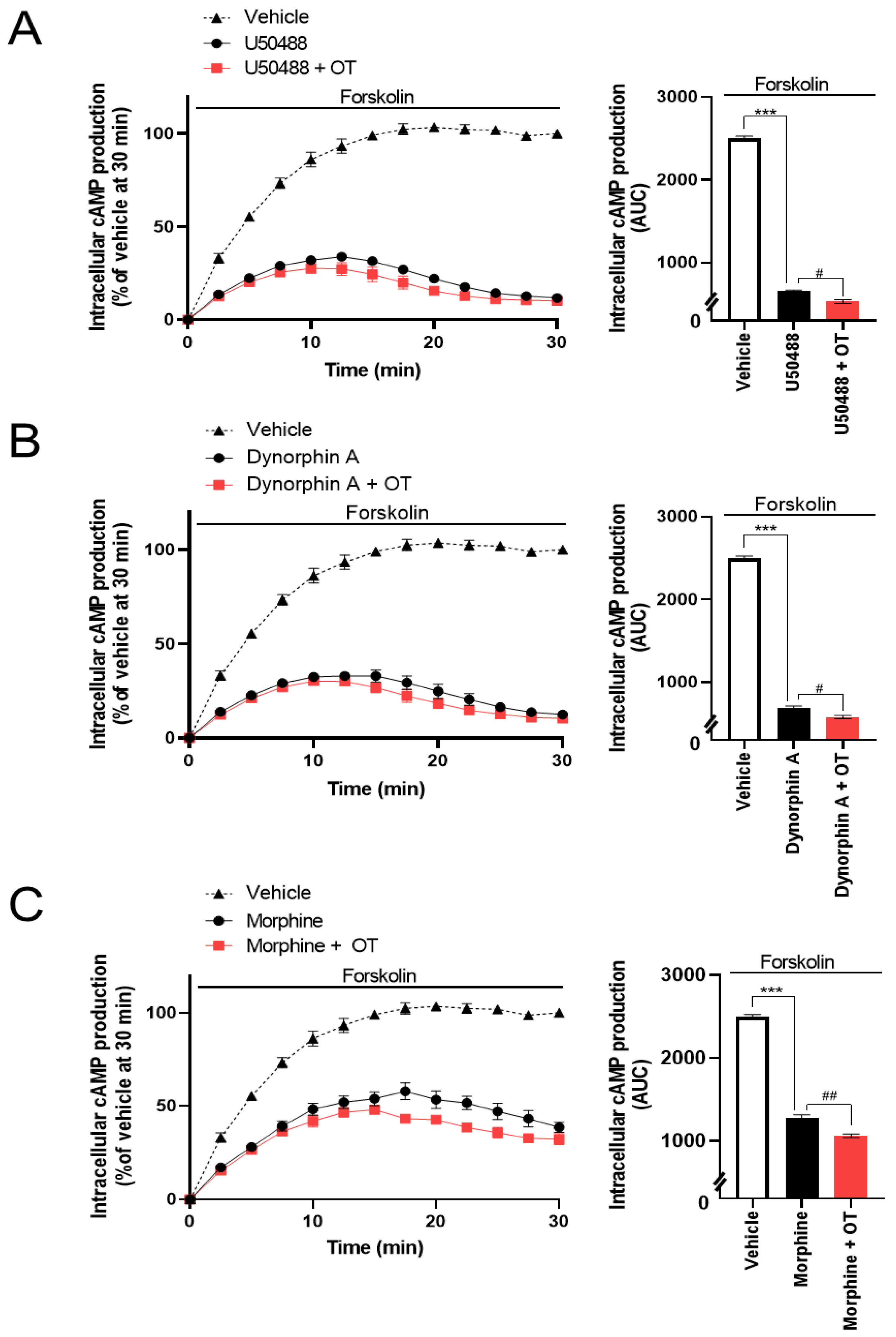
3.3. OT Enhanced Inhibition of Intracellular cAMP Induced by KOR Agonists in HEK293 Cells Stably Coexpressing Both Human KOR and Glosensor 22F Protein in the GlosensorTM cAMP Assay
3.4. OT Did Not Potentiate the KOR Internalization Induced by KOR Agonists in HEK293 Cells Stably Expressing the Human KOR Fused Halotag® in the Internalization Assay
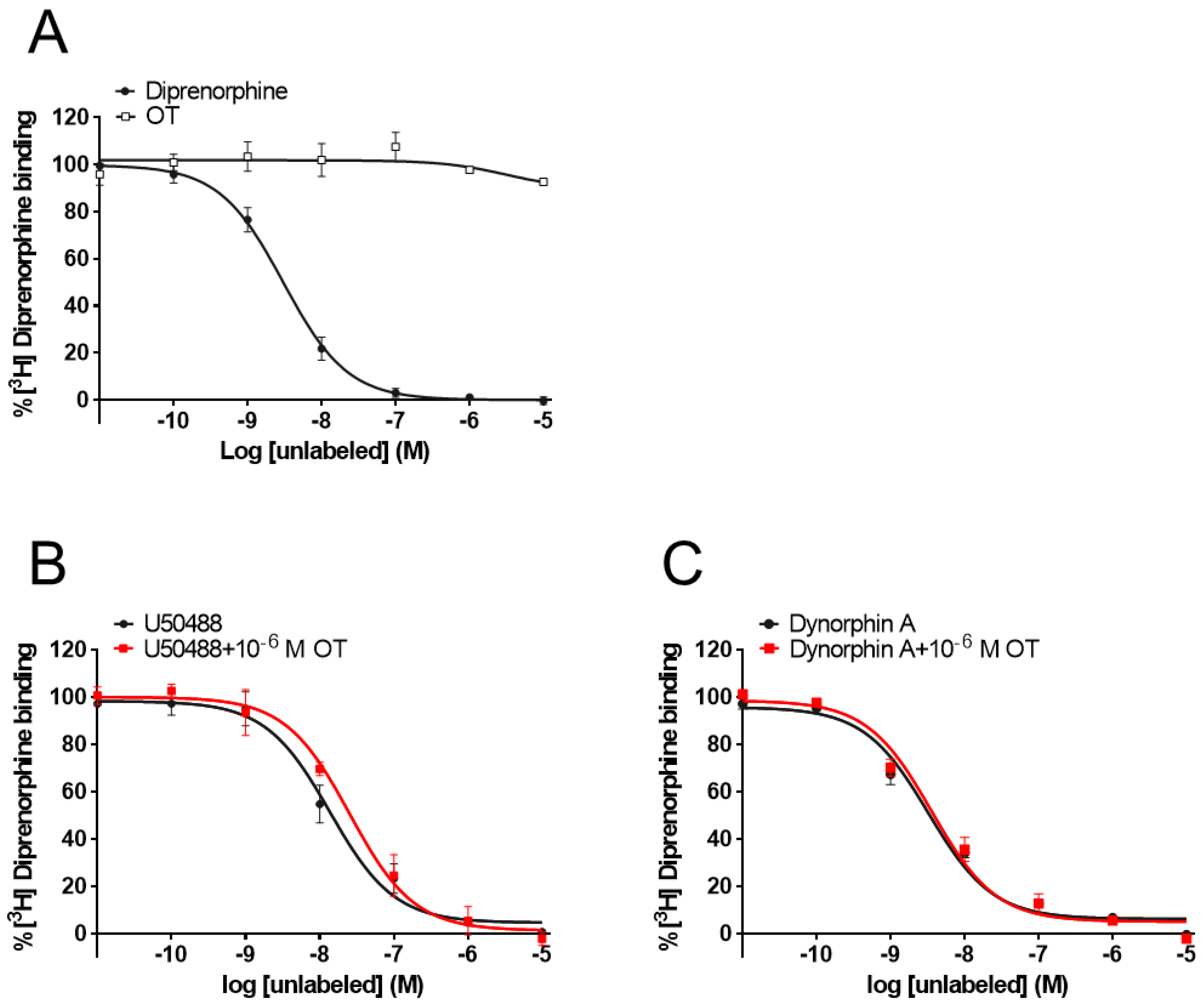
3.5. OT Did Not Affect [3H]Diprenorphine Binding to CHO Cells Stably Expressing Human KOR in the Radioligand Competitive OR Binding Assay
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GPCR | G protein-coupled receptor |
| HEK293 | human embryonic kidney 293 |
| OR | opioid receptor |
| OT | oxytocin |
| PAM | positive allosteric modulator |
| TRPV1 | transient receptor potential ankyrin 1 |
| MOR | µ-opioid receptor |
| DOR | δ-opioid receptor |
| KOR | κ-opioid receptor |
| PAM | positive allosteric modulator |
| U50488H | (-)-U-50488 hydrochloride |
| PVN | paraventricular nucleus |
| IBMX | 3-isobutyl-1-methylxanthine |
| DMEM | Dulbecco’s modified Eagle’s medium |
| ΔZiec | impedance of an induced extracellular current |
| AUC | area under the time-luminescence intensity curve |
| CHO | Chinese hamster ovary |
| n.d. | not detected |
| SEM | mean ± standard error of mean |
References
- Chen, Y.; Mestek, A.; Liu, J.; Hurley, J.A.; Yu, L. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol. Pharmacol. 1993, 44, 8–12. [Google Scholar] [CrossRef]
- Kieffer, B.L.; Befort, K.; Gaveriaux-Ruff, C.; Hirth, C.G. The delta-opioid receptor: Isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. USA 1992, 89, 12048–12052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minami, M.; Toya, T.; Katao, Y.; Maekawa, K.; Nakamura, S.; Onogi, T.; Kaneko, S.; Satoh, M. Cloning and expression of a cDNA for the rat kappa-opioid receptor. FEBS Lett. 1993, 329, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Meunier, J.C. Utilizing functional genomics to identify new pain treatments: The example of nociceptin. Am. J. Pharm. Genom. 2003, 3, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Brott, N.R.; Peterson, E.; Cascella, M. Opioid, Risk Tool. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Inturrisi, C.E. Clinical pharmacology of opioids for pain. Clin. J. Pain 2002, 18, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Stein, C. Opioid receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [Green Version]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.T.; Pitis, P.M.; Gotchev, D.; Yuan, C.; et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Miyano, K.; Manabe, S.; Komatsu, A.; Fujii, Y.; Mizobuchi, Y.; Uezono, E.; Ohshima, K.; Nonaka, M.; Kuroda, Y.; Narita, M.; et al. The G protein signal-biased compound TRV130; Structures, its site of action and clinical studies. Curr. Top. Med. Chem. 2020, 20, 2822–2829. [Google Scholar] [CrossRef]
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased opioid ligands. Molecules 2020, 25, 4257. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conn, P.J.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug. Discov. 2009, 8, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentry, P.R.; Sexton, P.M.; Christopoulos, A. Novel allosteric modulators of G protein-coupled receptors. J. Biol. Chem. 2015, 290, 19478–19488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burford, N.T.; Clark, M.J.; Wehrman, T.S.; Gerritz, S.W.; Banks, M.; O’Connell, J.; Traynor, J.R.; Alt, A. Discovery of positive allosteric modulators and silent allosteric modulators of the μ-opioid receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 10830–10835. [Google Scholar] [CrossRef] [Green Version]
- Burford, N.T.; Livingston, K.E.; Canals, M.; Ryan, M.R.; Budenholzer, L.M.; Han, Y.; Shang, Y.; Herbst, J.J.; O’Connell, J.; Banks, M.; et al. Discovery, synthesis, and molecular pharmacology of selective positive allosteric modulators of the δ-opioid receptor. J. Med. Chem. 2015, 58, 4220–4229. [Google Scholar] [CrossRef]
- Rasmussen, N.A.; Farr, L.A. Beta-endorphin response to an acute pain stimulus. J. Neurosci. Methods 2009, 177, 285–288. [Google Scholar] [CrossRef]
- Gimpl, G.; Fahrenholz, F. The oxytocin receptor system: Structure, function, and regulation. Physiol. Rev. 2001, 81, 629–683. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Park, S.H.; Chung, C.; Kim, J.J.; Choi, S.Y.; Han, J.S. Oxytocin protects hippocampal memory and plasticity from uncontrollable stress. Sci. Rep. 2015, 5, 18540. [Google Scholar] [CrossRef] [PubMed]
- Yamasue, H.; Yee, J.R.; Hurlemann, R.; Rilling, J.K.; Chen, F.S.; Meyer-Lindenberg, A.; Tost, H. Integrative approaches utilizing oxytocin to enhance prosocial behavior: From animal and human social behavior to autistic social dysfunction. J. Neurosci. 2012, 32, 14109–14117. [Google Scholar] [CrossRef] [Green Version]
- Lundeberg, T.; Meister, B.; Björkstrand, E.; Uvnäs-Moberg, K. Oxytocin modulates the effects of galanin in carrageenan-induced hyperalgesia in rats. Brain Res. 1993, 608, 181–185. [Google Scholar] [CrossRef]
- Petersson, M.; Alster, P.; Lundeberg, T.; Uvnäs-Moberg, K. Oxytocin increases nociceptive thresholds in a long-term perspective in female and male rats. Neurosci. Lett. 1996, 212, 87–90. [Google Scholar] [CrossRef]
- Yang, J.; Yang, Y.; Chen, J.M.; Liu, W.Y.; Wang, C.H.; Lin, B.C. Central oxytocin enhances antinociception in the rat. Peptides 2007, 28, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Breton, J.D.; Veinante, P.; Uhl-Bronner, S.; Vergnano, A.M.; Freund-Mercier, M.J.; Schlichter, R.; Poisbeau, P. Oxytocin-induced antinociception in the spinal cord is mediated by a subpopulation of glutamatergic neurons in lamina I-II which amplify GABAergic inhibition. Mol. Pain 2008, 4, 19. [Google Scholar] [CrossRef]
- Condés-Lara, M.; Rojas-Piloni, G.; Martínez-Lorenzana, G.; Rodríguez-Jiménez, J.; López Hidalgo, M.; Freund-Mercier, M.J. Paraventricular hypothalamic influences on spinal nociceptive processing. Brain Res. 2006, 1081, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Piloni, G.; Martínez-Lorenzana, G.; DelaTorre, S.; Condés-Lara, M. Nociceptive spinothalamic tract and postsynaptic dorsal column neurons are modulated by paraventricular hypothalamic activation. Eur. J. Neurosci. 2008, 28, 546–558. [Google Scholar] [CrossRef]
- Rojas-Piloni, G.; Mejía-Rodríguez, R.; Martínez-Lorenzana, G.; Condés-Lara, M. Oxytocin, but not vassopressin, modulates nociceptive responses in dorsal horn neurons. Neurosci. Lett. 2010, 476, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Schorscher-Petcu, A.; Sotocinal, S.; Ciura, S.; Dupré, A.; Ritchie, J.; Sorge, R.E.; Crawley, J.N.; Hu, S.B.; Nishimori, K.; Young, L.J.; et al. Oxytocin-induced analgesia and scratching are mediated by the vasopressin-1A receptor in the mouse. J. Neurosci. 2010, 30, 8274–8284. [Google Scholar] [CrossRef] [Green Version]
- Nersesyan, Y.; Demirkhanyan, L.; Cabezas-Bratesco, D.; Oakes, V.; Kusuda, R.; Dawson, T.; Sun, X.; Cao, C.; Cohen, A.M.; Chelluboina, B.; et al. Oxytocin modulates nociception as an agonist of pain-sensing TRPV1. Cell Rep. 2017, 21, 1681–1691. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liang, J.Y.; Li, P.; Pan, Y.J.; Qiu, P.Y.; Zhang, J.; Hao, F.; Wang, D.X. Oxytocin in the periaqueductal gray participates in pain modulation in the rat by influencing endogenous opiate peptides. Peptides 2011, 32, 1255–1261. [Google Scholar] [CrossRef]
- Taati, M.; Tamaddonfard, E. Ventrolateral orbital cortex oxytocin attenuates neuropathic pain through periaqueductal gray opioid receptor. Pharmacol. Rep. 2018, 70, 577–583. [Google Scholar] [CrossRef]
- Meguro, Y.; Miyano, K.; Hirayama, S.; Yoshida, Y.; Ishibashi, N.; Ogino, T.; Fujii, Y.; Manabe, S.; Eto, M.; Nonaka, M.; et al. Neuropeptide oxytocin enhances μ opioid receptor signaling as a positive allosteric modulator. J. Pharmacol. Sci. 2018, 137, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Q.; Lundeberg, T.; Yu, L.C. Involvement of oxytocin in spinal antinociception in rats with inflammation. Brain Res. 2003, 983, 13–22. [Google Scholar] [CrossRef]
- Gu, X.L.; Yu, L.C. Involvement of opioid receptors in oxytocin-induced antinociception in the nucleus accumbens of rats. J. Pain 2007, 8, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Miyano, K.; Sudo, Y.; Yokoyama, A.; Hisaoka-Nakashima, K.; Morioka, N.; Takebayashi, M.; Nakata, Y.; Higami, Y.; Uezono, Y. History of the G protein-coupled receptor (GPCR) assays from traditional to a state-of-the-art biosensor assay. J. Pharmacol. Sci. 2014, 126, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manabe, S.; Miyano, K.; Fujii, Y.; Ohshima, K.; Yoshida, Y.; Nonaka, M.; Uzu, M.; Matsuoka, Y.; Sato, T.; Uezono, Y.; et al. Possible biased analgesic of hydromorphone through the G protein-over β-arrestin-mediated pathway: cAMP, CellKey™, and receptor internalization analyses. J. Pharmacol. Sci. 2019, 140, 171–177. [Google Scholar] [CrossRef]
- Uezono, Y.; Bradley, J.; Min, C.; McCarty, N.A.; Quick, M.; Riordan, J.R.; Chavkin, C.; Zinn, K.; Lester, H.A.; Davidson, N. Receptors that couple to 2 classes of G proteins increase cAMP and activate CFTR expressed in Xenopus oocytes. Recept. Channels 1993, 1, 233–241. [Google Scholar]
- Bisignano, P.; Burford, N.T.; Shang, Y.; Marlow, B.; Livingston, K.E.; Fenton, A.M.; Rockwell, K.; Budenholzer, L.; Traynor, J.R.; Gerritz, S.W.; et al. Ligand-Based discovery of a new scaffold for allosteric modulation of the μ-opioid receptor. J. Chem. Inf. Model. 2015, 55, 1836–1843. [Google Scholar] [CrossRef] [Green Version]
- Koole, C.; Wootten, D.; Simms, J.; Valant, C.; Sridhar, R.; Woodman, O.L.; Miller, L.J.; Summers, R.J.; Christopoulos, A.; Sexton, P.M. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: Implications for drug screening. Mol. Pharmacol. 2010, 78, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Leach, K.; Sexton, P.M.; Christopoulos, A. Allosteric GPCR modulators: Taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci. 2007, 28, 382–389. [Google Scholar] [CrossRef]
- Kenakin, T.P. Biased signalling and allosteric machines: New vistas and challenges for drug discovery. Br. J. Pharmacol. 2012, 165, 1659–1669. [Google Scholar] [CrossRef] [Green Version]
- Livingston, K.E.; Stanczyk, M.A.; Burford, N.T.; Alt, A.; Canals, M.; Traynor, J.R. Pharmacologic evidence for a putative conserved allosteric site on opioid receptors. Mol. Pharmacol. 2018, 93, 157–167. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Interplay between two allosteric sites and their influence on agonist binding in human μ opioid receptor. J. Chem. Inf. Model. 2016, 56, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Livingston, K.E.; Traynor, J.R. Allostery at opioid receptors: Modulation with small molecule ligands. Br. J. Pharmacol. 2018, 175, 2846–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Y.; Yeatman, H.R.; Provasi, D.; Alt, A.; Christopoulos, A.; Canals, M.; Filizola, M. Proposed mode of binding and action of positive allosteric modulators at opioid receptors. ACS Chem. Boil. 2016, 11, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Provasi, D.; Filizola, M. The Dynamic Process of Drug-GPCR Binding at Either Orthosteric or Allosteric Sites Evaluated by Metadynamics. Methods Mol. Biol. 2015, 1335, 277–294. [Google Scholar] [CrossRef] [Green Version]
- Bruchas, M.R.; Land, B.B.; Aita, M.; Xu, M.; Barot, S.K.; Li, S.; Chavkin, C. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J. Neurosci. 2007, 27, 11614–11623. [Google Scholar] [CrossRef] [Green Version]
- Bruchas, M.R.; Chavkin, C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology 2010, 210, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Brust, T.F.; Morgenweck, J.; Kim, S.A.; Rose, J.H.; Locke, J.L.; Schmid, C.L.; Zhou, L.; Stahl, E.L.; Cameron, M.D.; Scarry, S.M.; et al. Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci. Signal. 2016, 9, ra117. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.H.; Stahl, E.L.; Schmid, C.L.; Scarry, S.M.; Aubé, J.; Bohn, L.M. G protein signaling-biased agonism at the κ-opioid receptor is maintained in striatal neurons. Sci. Signal. 2018, 11, eaar4309. [Google Scholar] [CrossRef] [Green Version]
- Remesic, M.; Hruby, V.J.; Porreca, F.; Lee, Y.S. Recent advances in the realm of allosteric modulators for opioid receptors for future therapeutics. ACS Chem. Neurosci. 2017, 8, 1147–1158. [Google Scholar] [CrossRef]
- Stanczyk, M.A.; Livingston, K.E.; Chang, L.; Weinberg, Z.Y.; Puthenveedu, M.A.; Traynor, J.R. The δ-opioid receptor positive allosteric modulator BMS 986187 is a G-protein-biased allosteric agonist. Br. J. Pharmacol. 2019, 176, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Lundeberg, T.; Yu, L.C. Blockade effect of mu and kappa opioid antagonists on the anti-nociception induced by intra-periaqueductal grey injection of oxytocin in rats. Brain Res. 2002, 927, 204–207. [Google Scholar] [CrossRef]
- Mores, K.L.; Cummins, B.R.; Cassell, R.J.; van Rijn, R.M. A review of the therapeutic potential of recently developed G protein-biased kappa agonists. Front. Pharmacol. 2019, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Morgenweck, J.; Frankowski, K.J.; Prisinzano, T.E.; Aubé, J.; Bohn, L.M. Investigation of the role of βarrestin2 in kappa opioid receptor modulation in a mouse model of pruritus. Neuropharmacology 2015, 99, 600–609. [Google Scholar] [CrossRef] [Green Version]
- White, K.L.; Robinson, J.E.; Zhu, H.; DiBerto, J.F.; Polepally, P.R.; Zjawiony, J.K.; Nichols, D.E.; Malanga, C.J.; Roth, B.L. The G protein-biased κ-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J. Pharmacol. Exp. Ther. 2015, 352, 98–109. [Google Scholar] [CrossRef] [Green Version]
- De Los Monteros-Zúñiga, E.A.; Martínez-Lorenzana, G.; Condés-Lara, M.; González-Hernández, A. Intrathecal Oxytocin Improves Spontaneous Behavior and Reduces Mechanical Hypersensitivity in a Rat Model of Postoperative Pain. Front. Pharmacol. 2020, 11, 581544. [Google Scholar] [CrossRef]
- Loth, M.K.; Donaldson, Z.R. Oxytocin, Dopamine, and Opioid Interactions Underlying Pair Bonding: Highlighting a Potential Role for Microglia. Endocrinology 2021, 162, bqaa223. [Google Scholar] [CrossRef]
- Young, L.J.; Lim, M.M.; Gingrich, B.; Insel, T.R. Cellular mechanisms of social attachment. Horm. Behav. 2001, 40, 133–138. [Google Scholar] [CrossRef]
- Kubinyi, E.; Bence, M.; Koller, D.; Wan, M.; Pergel, E.; Ronai, Z.; Sasvari-Szekely, M.; Miklósi, Á. Oxytocin and Opioid Receptor Gene Polymorphisms Associated with Greeting Behavior in Dogs. Front. Psychol. 2017, 8, 1520. [Google Scholar] [CrossRef] [Green Version]
- Kutlu, S.; Yilmaz, B.; Canpolat, S.; Sandal, S.; Ozcan, M.; Kumru, S.; Kelestimur, H. Mu opioid modulation of oxytocin secretion in late pregnant and parturient rats. Involvement of noradrenergic neurotransmission. Neuroendocrinology 2004, 79, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Vuong, C.; Van Uum, S.H.; O’Dell, L.E.; Lutfy, K.; Friedman, T.C. The effects of opioids and opioid analogs on animal and human endocrine systems. Endocr. Rev. 2010, 31, 98–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Huang, Y.; Chen, L.; Yu, R. Lack of Evidence for the Effect of Oxytocin on Placebo Analgesia and Nocebo Hyperalgesia. Psychother. Psychosom. 2020, 89, 185–187. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OR | Compounds | LogEC50 (M) | Emax (%) | ||
|---|---|---|---|---|---|
| Agonist | Agonist + OT | Agonist | Agonist + OT | ||
| DOR | SNC-80 | −8.20 ± 0.11 | −8.15 ± 0.06 | 100 ± 3.23 | 99.13 ± 1.77 |
| Met-enkephalin | −9.38 ± 0.08 | −9.34 ± 0.11 | 107.8 ± 2.33 | 112.65 ± 3.43 | |
| Leu-enkephalin | −9.23 ± 0.09 | −9.04 ± 0.12 | 108.4 ± 2.71 | 103.1 ± 3.59 | |
| Morphine | n.d. | n.d. | n.d. | n.d. | |
| Fentanyl | n.d. | n.d. | n.d. | n.d. | |
| KOR | U50488 | −8.20 ± 0.12 | −8.09 ± 0.07 | 100.0 ± 3.47 | 121.7 ± 2.75 ** |
| Dynorphin A | −8.51 ± 0.11 | −8.28 ± 0.09 | 90.3 ± 2.71 | 114.1 ± 2.97 ** | |
| Morphine | n.d. | n.d. | n.d. | n.d. | |
| Fentanyl | n.d. | n.d. | n.d. | n.d. | |
| LogIC50 (M) | LogKi (M) | |||
|---|---|---|---|---|
| Agonist | Agonist + OT | Agonist | Agonist + OT | |
| Diprenorphine | −8.52 ± 0.06 | n.m. | −8.86 ± 0.06 | n.m. |
| U50488 | −7.86 ± 0.11 | −7.60 ± 0.12 n.s. | −8.20 ± 0.11 | −7.94 ± 0.12 n.s. |
| Dynorphin A | −8.49 ± 0.09 | −8.45 ± 0.09 n.s. | −8.83 ± 0.09 | −8.79 ± 0.09 n.s. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyano, K.; Yoshida, Y.; Hirayama, S.; Takahashi, H.; Ono, H.; Meguro, Y.; Manabe, S.; Komatsu, A.; Nonaka, M.; Mizuguchi, T.; et al. Oxytocin Is a Positive Allosteric Modulator of κ-Opioid Receptors but Not δ-Opioid Receptors in the G Protein Signaling Pathway. Cells 2021, 10, 2651. https://doi.org/10.3390/cells10102651
Miyano K, Yoshida Y, Hirayama S, Takahashi H, Ono H, Meguro Y, Manabe S, Komatsu A, Nonaka M, Mizuguchi T, et al. Oxytocin Is a Positive Allosteric Modulator of κ-Opioid Receptors but Not δ-Opioid Receptors in the G Protein Signaling Pathway. Cells. 2021; 10(10):2651. https://doi.org/10.3390/cells10102651
Chicago/Turabian StyleMiyano, Kanako, Yuki Yoshida, Shigeto Hirayama, Hideki Takahashi, Haruka Ono, Yoshiyuki Meguro, Sei Manabe, Akane Komatsu, Miki Nonaka, Takaaki Mizuguchi, and et al. 2021. "Oxytocin Is a Positive Allosteric Modulator of κ-Opioid Receptors but Not δ-Opioid Receptors in the G Protein Signaling Pathway" Cells 10, no. 10: 2651. https://doi.org/10.3390/cells10102651