
Bile Acids Activate NLRP3 Inflammasome, Promoting Murine Liver Inflammation or Fibrosis in a Cell Type-Specific Manner

 , ,
, ,
Abstract
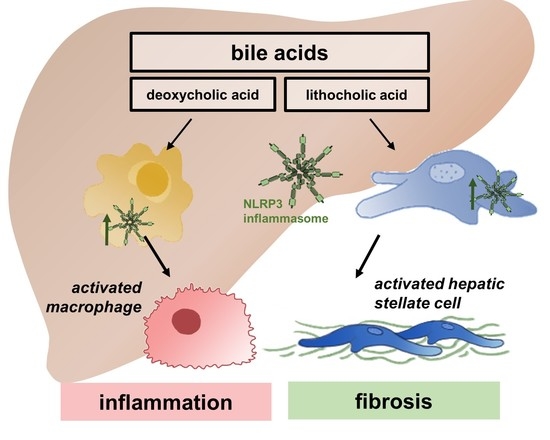
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Models
2.1.1. Bile Acid Feeding
2.1.2. Cell Isolation and Stimulation
2.2. Histology, Immunostaining and Sirius Red Staining
2.3. Real-Time PCR
2.4. Flow Cytometry Analysis
2.5. ELISA
2.6. Fluorescence in Situ Hybridization (FISH)
2.7. Statistics
3. Results
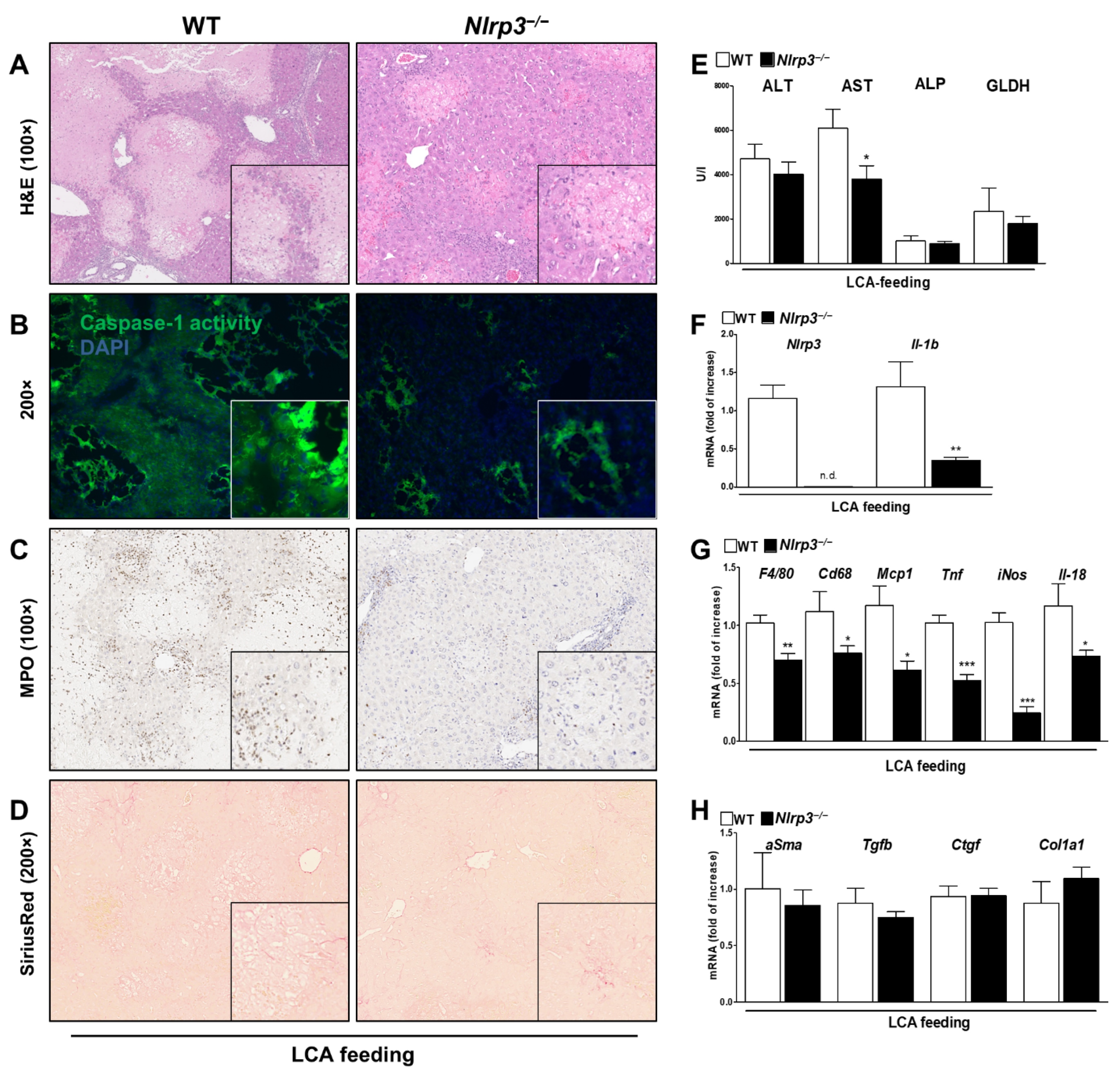
3.1. DCA and LCA Feeding Causes Liver Damage and Activation of NLRP3 Inflammasome
3.2. Bile Acid induced Liver Damage Was Ameliorated in Nlrp3−/− Mice
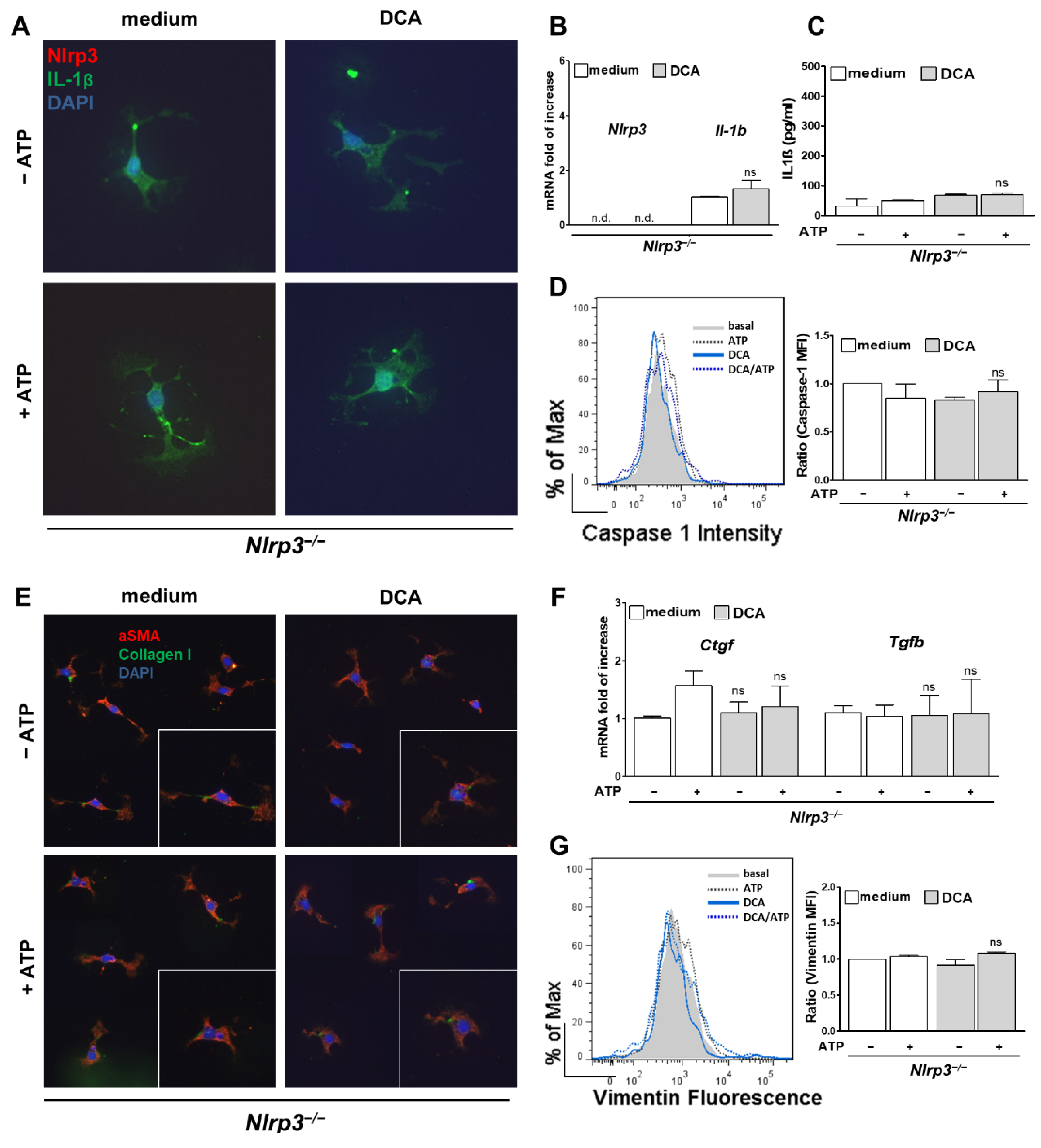
3.3. DCA, but Not LCA, Activates HSC to a Fibrotic Phenotype
3.4. Activation of HSC by DCA Was Abolished in Nlrp3−/−
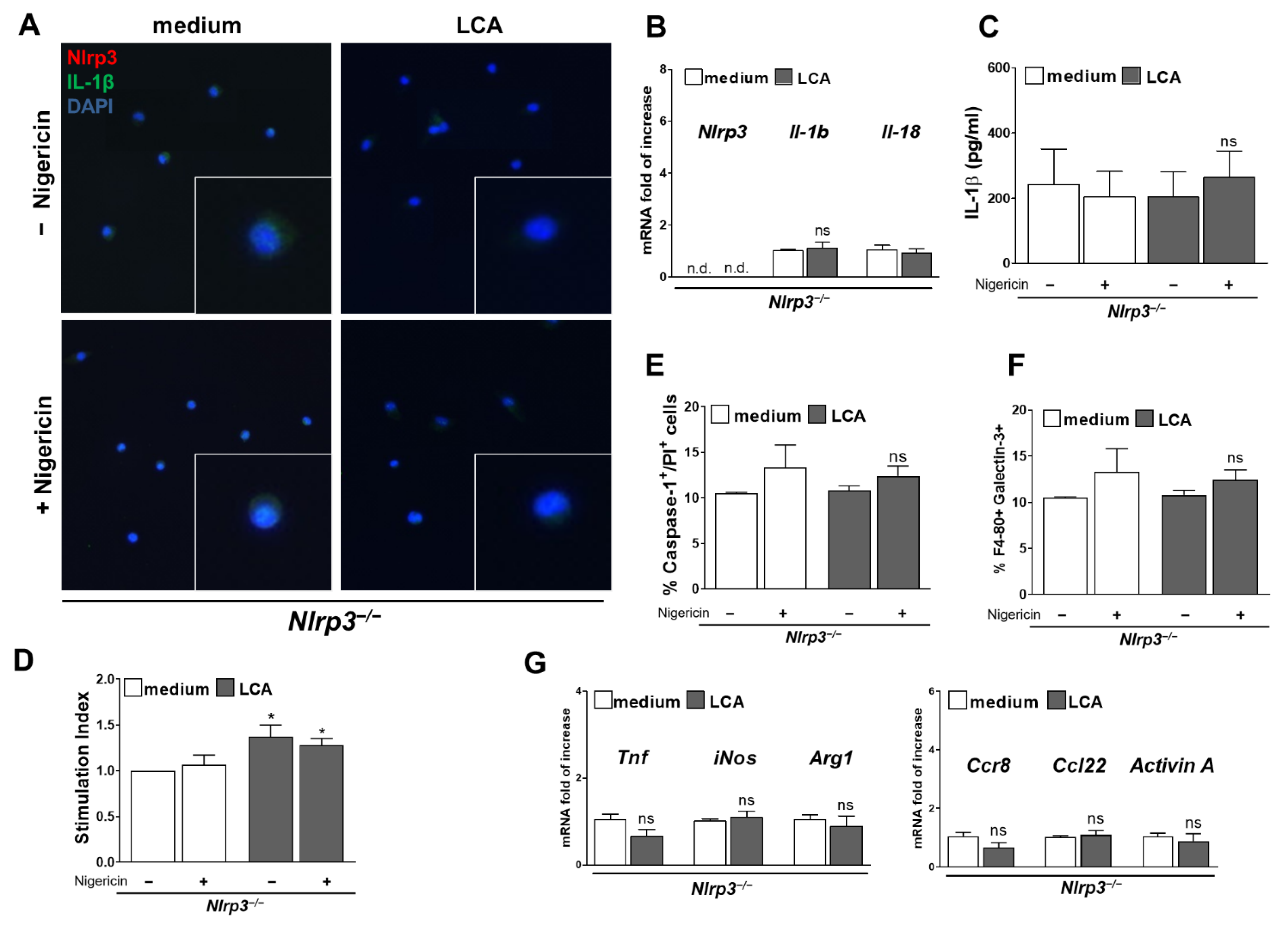
3.5. LCA Stimulates NLRP3 Activation in KC and Provokes a Pro-Inflammatory Phenotype
3.6. Activation of NLRP3 Inflammasome and Pro-Inflammatory Phenotype Was Blocked in Nlrp3−/− KC Despite LCA Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poupon, R.; Chazouilleres, O.; Poupon, R.E. Chronic cholestatic diseases. J. Hepatol. 2000, 32, 129–140. [Google Scholar] [CrossRef]
- Copple, B.L.; Jaeschke, H.; Klaassen, C.D. Oxidative stress and the pathogenesis of cholestasis. Semin. Live.r Dis 2010, 30, 195–204. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Bile acid-mediated hepatocyte apoptosis and cholestatic liver disease. Dig. Liver. Dis. 2002, 34, 387–392. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Novel insight into mechanisms of cholestatic liver injury. World. J. Gastroenterol. 2012, 18, 4985–4993. [Google Scholar] [CrossRef]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug. Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef] [PubMed]
- El-Agamy, D.S.; Almaramhy, H.H.; Ahmed, N.; Bojan, B.; Alrohily, W.D.; Elkablawy, M.A. Anti-Inflammatory Effects of Vardenafil Against Cholestatic Liver Damage in Mice: A Mechanistic Study. Cell. Physiol. Biochem. 2018, 47, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Fickert, P.; Fuchsbichler, A.; Marschall, H.U.; Wagner, M.; Zollner, G.; Krause, R.; Zatloukal, K.; Jaeschke, H.; Denk, H.; Trauner, M. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am. J. Pathol. 2006, 168, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Benz, C.; Angermuller, S.; Tox, U.; Kloters-Plachky, P.; Riedel, H.D.; Sauer, P.; Stremmel, W.; Stiehl, A. Effect of tauroursodeoxycholic acid on bile-acid-induced apoptosis and cytolysis in rat hepatocytes. J. Hepatol. 1998, 28, 99–106. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; Fan, G.; Wong, P.Y.; Kren, B.T.; Steer, C.J. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol. Med. 1998, 4, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, K.; Jaeschke, H.; Copple, B.L. Bile acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis. Am. J. Pathol. 2011, 178, 175–186. [Google Scholar] [CrossRef]
- Zhou, X.; Cao, L.; Jiang, C.; Xie, Y.; Cheng, X.; Krausz, K.W.; Qi, Y.; Sun, L.; Shah, Y.M.; Gonzalez, F.J.; et al. PPARalpha-UGT axis activation represses intestinal FXR-FGF15 feedback signalling and exacerbates experimental colitis. Nat. Commun. 2014, 5, 4573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Gong, Z.; Zhou, J.; Tian, C.; Gao, Y.; Xu, C.; Chen, Y.; Cai, W.; Wu, J. Deoxycholic Acid Triggers NLRP3 Inflammasome Activation and Aggravates DSS-Induced Colitis in Mice. Front. Immunol. 2016, 7, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.; Zhou, J.; Zhao, S.; Tian, C.; Wang, P.; Xu, C.; Chen, Y.; Cai, W.; Wu, J. Chenodeoxycholic acid activates NLRP3 inflammasome and contributes to cholestatic liver fibrosis. Oncotarget 2016, 7, 83951–83963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroni, L.; Ninfole, E.; Pinto, C.; Benedetti, A.; Marzioni, M. Gut-Liver Axis and Inflammasome Activation in Cholangiocyte Pathophysiology. Cells 2020, 9, 736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Cai, S.Y.; Ge, M.; Mennone, A.; Hoque, R.; Ouyang, X.; Boyer, J.L. Inflammasome Is Activated in the Liver of Cholestatic Patients and Aggravates Hepatic Injury in Bile Duct-Ligated Mouse. Cell. Mol. Gastroenterol. Hepatol. 2019, 9, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Yang, G.; Chen, H.Y.; Hsu, D.K.; Tomilov, A.; Olson, K.A.; Dehnad, A.; Fish, S.R.; Cortopassi, G.; Zhao, B.; et al. Galectin-3 regulates inflammasome activation in cholestatic liver injury. FASEB J. 2016, 30, 4202–4213. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Mizuochi, T.; Shivakumar, P.; Mourya, R.; Luo, Z.; Gutta, S.; Bezerra, J.A. Regulation of epithelial injury and bile duct obstruction by NLRP3, IL-1R1 in experimental biliary atresia. J. Hepatol. 2018, 69, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Dong, W.; Wang, S.; Zhang, Y.; Liu, T.; Xie, R.; Wang, B.; Cao, H. Deoxycholic acid disrupts the intestinal mucosal barrier and promotes intestinal tumorigenesis. Food Funct. 2018, 9, 5588–5597. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab. 2017, 25, 856–867. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Yuan, Z.; Wang, G.; Wang, X.; Li, K. The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice. Int. Immunopharmacol. 2019, 70, 147–155. [Google Scholar] [CrossRef]
- Yerushalmi, B.; Dahl, R.; Devereaux, M.W.; Gumpricht, E.; Sokol, R.J. Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology 2001, 33, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; Agostinelli, L.; Saccomanno, S.; Pinto, C.; Giordano, D.M.; Rychlicki, C.; De Minicis, S.; Trozzi, L.; Banales, J.M.; Melum, E.; et al. Nlrp3 Activation Induces Il-18 Synthesis and Affects the Epithelial Barrier Function in Reactive Cholangiocytes. Am. J. Pathol. 2017, 187, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Inzaugarat, M.E.; Johnson, C.D.; Holtmann, T.M.; McGeough, M.D.; Trautwein, C.; Papouchado, B.G.; Schwabe, R.; Hoffman, H.M.; Wree, A.; Feldstein, A.E. NLRP3 inflammasome activation in hepatic stellate cells induces murine liver fibrosis. Hepatology 2018. [Google Scholar] [CrossRef] [Green Version]
- Galle, P.R.; Theilmann, L.; Raedsch, R.; Otto, G.; Stiehl, A. Ursodeoxycholate reduces hepatotoxicity of bile salts in primary human hepatocytes. Hepatology 1990, 12, 486–491. [Google Scholar] [CrossRef]
- Attili, A.F.; Angelico, M.; Cantafora, A.; Alvaro, D.; Capocaccia, L. Bile acid-induced liver toxicity: Relation to the hydrophobic-hydrophilic balance of bile acids. Med. Hypotheses 1986, 19, 57–69. [Google Scholar] [CrossRef]
- Zhang, Y.; Hong, J.Y.; Rockwell, C.E.; Copple, B.L.; Jaeschke, H.; Klaassen, C.D. Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver. Int. 2012, 32, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolbright, B.L.; Jaeschke, H. Inflammation and Cell Death During Cholestasis: The Evolving Role of Bile Acids. Gene. Expr. 2019, 19, 215–228. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.M.; Allen, K.M.; Rockwell, C.E.; Towery, K.; Luyendyk, J.P.; Copple, B.L. IL-17A synergistically enhances bile acid-induced inflammation during obstructive cholestasis. Am. J. Pathol. 2013, 183, 1498–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trottier, J.; Bialek, A.; Caron, P.; Straka, R.J.; Milkiewicz, P.; Barbier, O. Profiling circulating and urinary bile acids in patients with biliary obstruction before and after biliary stenting. PLoS ONE 2011, 6, e22094. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Dorko, K.; Antoine, D.J.; Clarke, J.I.; Gholami, P.; Li, F.; Kumer, S.C.; Schmitt, T.M.; Forster, J.; Fan, F.; et al. Bile acid-induced necrosis in primary human hepatocytes and in patients with obstructive cholestasis. Toxicol. Appl. Pharm. 2015, 283, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billington, D.; Evans, C.E.; Godfrey, P.P.; Coleman, R. Effects of bile salts on the plasma membranes of isolated rat hepatocytes. Biochem. J. 1980, 188, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penman, S.L.; Sharma, P.; Aerts, H.; Park, B.K.; Weaver, R.J.; Chadwick, A.E. Differential toxic effects of bile acid mixtures in isolated mitochondria and physiologically relevant HepaRG cells. Toxicol. In Vitro 2019, 61, 104595. [Google Scholar] [CrossRef]
- Gujral, J.S.; Farhood, A.; Bajt, M.L.; Jaeschke, H. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology 2003, 38, 355–363. [Google Scholar] [CrossRef]
- Saito, J.M.; Maher, J.J. Bile duct ligation in rats induces biliary expression of cytokine-induced neutrophil chemoattractant. Gastroenterology 2000, 118, 1157–1168. [Google Scholar] [CrossRef]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef]
- Sokol, R.J.; Devereaux, M.; Khandwala, R.; O’Brien, K. Evidence for involvement of oxygen free radicals in bile acid toxicity to isolated rat hepatocytes. Hepatology 1993, 17, 869–881. [Google Scholar] [CrossRef]
- Sokol, R.J.; McKim, J.M., Jr.; Goff, M.C.; Ruyle, S.Z.; Devereaux, M.W.; Han, D.; Packer, L.; Everson, G. Vitamin E reduces oxidant injury to mitochondria and the hepatotoxicity of taurochenodeoxycholic acid in the rat. Gastroenterology 1998, 114, 164–174. [Google Scholar] [CrossRef]
- Aboutwerat, A.; Pemberton, P.W.; Smith, A.; Burrows, P.C.; McMahon, R.F.; Jain, S.K.; Warnes, T.W. Oxidant stress is a significant feature of primary biliary cirrhosis. Biochim. Biophys. Acta. 2003, 1637, 142–150. [Google Scholar] [CrossRef]
- Evangelakos, I.; Heeren, J.; Verkade, E.; Kuipers, F. Role of bile acids in inflammatory liver diseases. Semin. Immunopathol. 2021, 43, 577–590. [Google Scholar] [CrossRef]
- Petrescu, A.D.; DeMorrow, S. Farnesoid X Receptor as Target for Therapies to Treat Cholestasis-Induced Liver Injury. Cells 2021, 10, 1846. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.Y.; Boyer, J.L. The role of bile acids in cholestatic liver injury. Ann. Transl. Med. 2021, 9, 737. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Ridolfi, F.; Hannivoort, R.; Saccomanno, S.; Homan, M.; De Minicis, S.; Jansen, P.L.; Candelaresi, C.; Benedetti, A.; Moshage, H. Bile acids induce hepatic stellate cell proliferation via activation of the epidermal growth factor receptor. Gastroenterology 2005, 128, 1042–1055. [Google Scholar] [CrossRef]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 944. [Google Scholar] [CrossRef] [Green Version]
- Frissen, M.; Liao, L.; Schneider, K.M.; Djudjaj, S.; Haybaeck, J.; Wree, A.; Rolle-Kampczyk, U.; Bergen, M.V.; Latz, E.; Boor, P.; et al. Bidirectional role of NLRP3 during acute and chronic cholestatic liver injury. Hepatology 2020, 73, 1836–1854. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holtmann, T.M.; Inzaugarat, M.E.; Knorr, J.; Geisler, L.; Schulz, M.; Bieghs, V.; Frissen, M.; Feldstein, A.E.; Tacke, F.; Trautwein, C.; et al. Bile Acids Activate NLRP3 Inflammasome, Promoting Murine Liver Inflammation or Fibrosis in a Cell Type-Specific Manner. Cells 2021, 10, 2618. https://doi.org/10.3390/cells10102618
Holtmann TM, Inzaugarat ME, Knorr J, Geisler L, Schulz M, Bieghs V, Frissen M, Feldstein AE, Tacke F, Trautwein C, et al. Bile Acids Activate NLRP3 Inflammasome, Promoting Murine Liver Inflammation or Fibrosis in a Cell Type-Specific Manner. Cells. 2021; 10(10):2618. https://doi.org/10.3390/cells10102618
Chicago/Turabian StyleHoltmann, Theresa Maria, Maria Eugenia Inzaugarat, Jana Knorr, Lukas Geisler, Marten Schulz, Veerle Bieghs, Mick Frissen, Ariel E. Feldstein, Frank Tacke, Christian Trautwein, and et al. 2021. "Bile Acids Activate NLRP3 Inflammasome, Promoting Murine Liver Inflammation or Fibrosis in a Cell Type-Specific Manner" Cells 10, no. 10: 2618. https://doi.org/10.3390/cells10102618