
Intermittent High Glucose Elevates Nuclear Localization of EZH2 to Cause H3K27me3-Dependent Repression of KLF2 Leading to Endothelial Inflammation


 , and
, and
Abstract
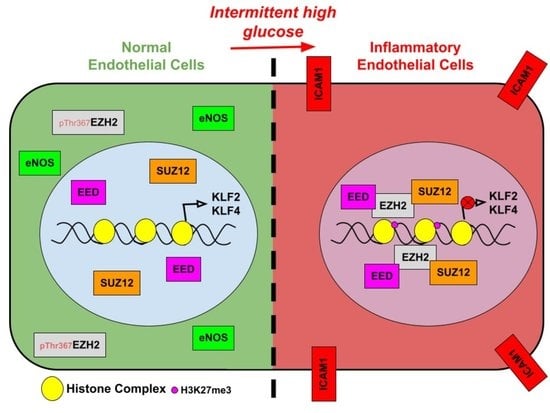
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and High Glucose Treatment Condition
2.2. Animal Dissection and Treatment Conditions
2.3. Inhibitor Treatment Condition and RNA Silencing in Cultured Endothelial Cells
2.4. Cell Viability Assay
2.5. RNA Isolation
2.6. cDNA Synthesis and Quantitative Analysis through Reverse Transcriptase-Quantitative Polymerase Chain Reaction
2.7. Subcellular Fractionation
2.8. Co-Immunoprecipitation
2.9. Immunoblotting
2.10. Immunofluorescence Imaging and Analysis
2.11. Chromatin Immunoprecipitation (ChIP) and Subsequent Quantitative PCR
2.12. Primer Sequences for Transcript and Promoter Primers
2.13. Statistics
3. Results
3.1. Intermittent High Glucose Treatment Maximally Induced Endothelial Inflammation In Vitro and Ex Vivo
3.2. Intermittent High Glucose Causes Nuclear Localization of EZH2 through Its Threonine 367 Phosphorylation, Thereby Elevating H3K27me3 Level
3.3. In Endothelial Cells Treated with Intermittent High Glucose, Nuclear EZH2 Assembles PRC2 That Endorses H3K27me3 Enrichment on KLF2 and KLF4 Promoters and Further Suppresses Their Expression
3.4. Inhibition of EZH2′s Methyltransferase Activity or siRNA-Mediated Knockdown of EZH2 Reverses Intermittent High Glucose-Dependent Endothelial Inflammation
3.5. Inhibition of EZH2 Blocks Intermittent High Glucose-Driven Endothelial Inflammation in Ex Vivo Rat Aortic Ring Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar] [PubMed]
- Tan, K.C.B.; Chow, W.-S.; Ai, V.H.G.; Lam, K.S.L. Effects of angiotensin II receptor antagonist on endothelial vasomotor function and urinary albumin excretion in type 2 diabetic patients with microalbuminuria. Diabetes Metab. Res. Rev. 2002, 18, 71–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Skidmore, J.C.; Porter-Turner, M.M.; Rea, C.A.; Khokher, M.A.; Singh, B.M. Relationship of glycation, antioxidant status and oxidativestress to vascular endothelial damage in diabetes. Diabetes Obes. Metab. 2002, 4, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Benton, M.C.; Johnstone, A.; Eccles, D.; Harmon, B.; Hayes, M.T.; Lea, R.A.; Griffiths, L.; Hoffman, E.P.; Stubbs, R.S.; Macartney-Coxson, D. An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 2015, 16, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Rönn, T.; Volkov, P.; Gillberg, L.; Kokosar, M.; Perfilyev, A.; Jacobsen, A.L.; Jørgensen, S.W.; Brøns, C.; Jansson, P.-A.; Eriksson, K.-F.; et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum. Mol. Genet. 2015, 24, 3792–3813. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, J.M.; Moreli, M.; Tewari, S.; Benite-Ribeiro, S.A. The effect of exercise on skeletal muscle glucose uptake in type 2 diabetes: An epigenetic perspective. Metabolism 2015, 64, 1619–1628. [Google Scholar] [CrossRef]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia Induces a Dynamic Cooperativity of Histone Methylase and Demethylase Enzymes Associated With Gene-Activating Epigenetic Marks That Coexist on the Lysine Tail. Diabetes 2009, 58, 1229–1236. [Google Scholar] [CrossRef] [Green Version]
- Paneni, F.; Costantino, S.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Scavone, G.; Villano, A.; Pitocco, D.; Lanza, G.; et al. Adverse Epigenetic Signatures by Histone Methyltransferase Set7 Contribute to Vascular Dysfunction in Patients With Type 2 Diabetes Mellitus. Circ. Cardiovasc. Genet. 2015, 8, 150–158. [Google Scholar] [CrossRef]
- Inagaki, T.; Tachibana, M.; Magoori, K.; Kudo, H.; Tanaka, T.; Okamura, M.; Naito, M.; Kodama, T.; Shinkai, Y.; Sakai, J. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 2009, 14, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef]
- Pirola, L.; Balcerczyk, A.; Tothill, R.W.; Haviv, I.; Kaspi, A.; Lunke, S.; Ziemann, M.; Karagiannis, T.; Tonna, S.; Kowalczyk, A.; et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 2011, 21, 1601–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floris, I.; Descamps, B.; Vardeu, A.; Mitić, T.; Posadino, A.M.; Shantikumar, S.; Sala-Newby, G.; Capobianco, G.; Mangialardi, G.; Howard, L.; et al. Gestational Diabetes Mellitus Impairs Fetal Endothelial Cell Functions Through a Mechanism Involving MicroRNA-101 and Histone Methyltransferase Enhancer of Zester Homolog-2. Arter. Thromb. Vasc. Biol. 2015, 35, 664–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Martinelli, L.; Motz, E.; Ceriello, A. Intermittent High Glucose Enhances Apoptosis Related to Oxidative Stress in Human Umbilical Vein Endothelial Cells: The Role of Protein Kinase C and NAD(P)H-Oxidase Activation. Diabetes 2003, 52, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceriello, A.; Esposito, K.; Piconi, L.; Ihnat, M.A.; Thorpe, J.E.; Testa, R.; Boemi, M.; Giugliano, D. Oscillating Glucose Is More Deleterious to Endothelial Function and Oxidative Stress Than Mean Glucose in Normal and Type 2 Diabetic Patients. Diabetes 2008, 57, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Flynn, M.C.; Kraakman, M.J.; Tikellis, C.; Lee, M.K.; Hanssen, N.M.; Kammoun, H.L.; Pickering, R.J.; Dragoljevic, D.; Al-Sharea, A.; Barrett, T.; et al. Transient Intermittent Hyperglycemia Accelerates Atherosclerosis by Promoting Myelopoiesis. Circ. Res. 2020, 127, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Thieme, K.; Batchu, S.N.; Alghamdi, T.A.; Bowskill, B.B.; Kabir, M.G.; Liu, Y.; Advani, S.L.; White, K.E.; Geldenhuys, L.; et al. Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J. Clin. Investig. 2017, 128, 483–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, E.-F.; Yang, Y.; Cheng, L.; Deng, X.; Chen, S.-M.; Zhou, X.; Liu, S.-M. Hyperglycemia affects global 5-methylcytosine and 5-hydroxymethylcytosine in blood genomic DNA through upregulation of SIRT6 and TETs. Clin. Epigenetics 2019, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Su, I.H.; Dobenecker, M.W.; Dickinson, E.; Oser, M.; Basavaraj, A.; Marqueron, R.; Viale, A.; Reinberg, D.; Wülfing, C.; Tarakhovsky, A. Polycomb Group Protein Ezh2 Controls Actin Polymerization and Cell Signaling. Cell 2005, 121, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Anwar, T.; Arellano-Garcia, C.; Ropa, J.; Chen, Y.-C.; Kim, H.S.; Yoon, E.; Grigsby, S.; Basrur, V.; Nesvizhskii, A.I.; Muntean, A.; et al. p38-mediated phosphorylation at T367 induces EZH2 cytoplasmic localization to promote breast cancer metastasis. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Francis, N.J.; Kingston, R.E.; Woodcock, C.L. Chromatin Compaction by a Polycomb Group Protein Complex. Science 2004, 306, 1574–1577. [Google Scholar] [CrossRef] [Green Version]
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W.; et al. KLF2 Is a Novel Transcriptional Regulator of Endothelial Proinflammatory Activation. J. Exp. Med. 2004, 199, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Fledderus, J.; Boon, R.; Volger, O.L.; Hurttila, H.; Ylä-Herttuala, S.; Pannekoek, H.; Levonen, A.-L.; Horrevoets, A.J.G. KLF2 Primes the Antioxidant Transcription Factor Nrf2 for Activation in Endothelial Cells. Arter. Thromb. Vasc. Biol. 2008, 28, 1339–1346. [Google Scholar] [CrossRef] [Green Version]
- Hamik, A.; Lin, Z.; Kumar, A.; Balcells, M.; Sinha, S.; Katz, J.; Feinberg, M.W.; Gerszten, R.E.; Edelman, E.R.; Jain, M.K. Kruppel-like Factor 4 Regulates Endothelial Inflammation. J. Biol. Chem. 2007, 282, 13769–13779. [Google Scholar] [CrossRef] [Green Version]
- Sangwung, P.; Zhou, G.; Nayak, L.; Chan, E.R.; Kumar, S.; Kang, D.-W.; Zhang, R.; Liao, X.; Lu, Y.; Sugi, K.; et al. KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight 2017, 2, e91700. [Google Scholar] [CrossRef]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.C.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Bonora, E.; Muggeo, M. Postprandial blood glucose as a risk factor for cardiovascular disease in Type II diabetes: The epidemiological evidence. Diabetology 2001, 44, 2107–2114. [Google Scholar] [CrossRef]
- Hadi, H.A.R.; Al Suwaidi, J.A. Endothelial dysfunction in diabetes mellitus. Vasc. Health Risk Manag. 2007, 3, 853–876. [Google Scholar]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Da Ros, R.; Maier, A.; Zuodar, G.; Ceriello, A. Intermittent high glucose enhances ICAM-1, VCAM-1 and E-selectin expression in human umbilical vein endothelial cells in culture: The distinct role of protein kinase C and mitochondrial superoxide production. Atherosclerosis 2005, 183, 259–267. [Google Scholar] [CrossRef]
- Kim, S.-W.; Kim, C.E.; Kim, M.H. Flavonoids inhibit high glucose-induced up-regulation of ICAM-1 via the p38 MAPK pathway in human vein endothelial cells. Biochem. Biophys. Res. Commun. 2011, 415, 602–607. [Google Scholar] [CrossRef]
- Miao, F.; Wu, X.; Zhang, L.; Yuan, Y.-C.; Riggs, A.; Natarajan, R. Genome-wide Analysis of Histone Lysine Methylation Variations Caused by Diabetic Conditions in Human Monocytes. J. Biol. Chem. 2007, 282, 13854–13863. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Gao, D.; Zhang, W.; Liu, S.; Yang, S.; Li, X. Puerarin suppresses high glucose-induced MCP-1 expression via modulating histone methylation in cultured endothelial cells. Life Sci. 2015, 130, 103–107. [Google Scholar] [CrossRef]
- Okabe, J.; Orlowski, C.; Balcerczyk, A.; Tikellis, C.; Thomas, M.; Cooper, M.E.; El-Osta, A. Distinguishing Hyperglycemic Changes by Set7 in Vascular Endothelial Cells. Circ. Res. 2012, 110, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Yu, Q.; Shin, J.T.; Sebzda, E.; Bertozzi, C.; Chen, M.; Mericko, P.; Stadtfeld, M.; Zhou, D.; Cheng, L.; et al. Klf2 Is an Essential Regulator of Vascular Hemodynamic Forces In Vivo. Dev. Cell 2006, 11, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Atkins, G.B.; Wang, Y.; Mahabeleshwar, G.H.; Shi, H.; Gao, H.; Kawanami, D.; Natesan, V.; Lin, Z.; Simon, D.I.; Jain, M.K. Hemizygous Deficiency of Krüppel-Like Factor 2 Augments Experimental Atherosclerosis. Circ. Res. 2008, 103, 690–693. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, H.; Jacinto, F.V.; Villanueva, A.; Fernandez, A.; Yamamoto, H.; Carmona, F.J.; Puertas, S.; Marquez, V.E.; Shinomura, Y.; Imai, K.; et al. Silencing of Kruppel-like factor 2 by the histone methyltransferase EZH2 in human cancer. Oncogene 2011, 31, 1988–1994. [Google Scholar] [CrossRef]
- Jiang, Y.-Z.; Jiménez, J.M.; Ou, K.; McCormick, M.E.; Zhang, L.-D.; Davies, P.F. Hemodynamic Disturbed Flow Induces Differential DNA Methylation of Endothelial Kruppel-Like Factor 4 Promoter In Vitro and In Vivo. Circ. Res. 2014, 115, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Xiaoling, Y.; Li, Z.; Shuqiang, L.; Shengchao, M.; Anning, Y.; Ning, D.; Nan, L.; Yuexia, J.; Guizhong, L.; Yideng, J.; et al. Hyperhomocysteinemia in ApoE-/- Mice Leads to Overexpression of Enhancer of Zeste Homolog 2 via miR-92a Regulation. PLoS ONE 2016, 11, e0167744. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sequence of Forward Primer | Sequence of Reverse Primer |
|---|---|---|
| ICAM1 transcript | 5′TTCGTGTCCTGTATGGCCC3′ | 5′CACATTGGAGTCTGCTGGGA3′ |
| P-Selectin transcript | 5′CCAACCTGCAAAGGCATAGC3′ | 5′GCGTTGCAGCCAAAGTAACA3′ |
| VCAM1 transcript | 5′ACGAATGAGGGGACCACATC3′ | 5′TCCAGAGGGCCACTCAAATG3′ |
| UTX transcript | 5′GCAACAGTTAGGTTGGATGC3′ | 5′AGGCATCCTGAACTTTCCCA3′ |
| JMJD3 transcript | 5′GGTCTGTTGTACCCCACTGC3′ | 5′CCGCCTCAGTAACAGCCAGA3′ |
| KLF2 transcript | 5′CGGCAAGACCTACACCAAGA3′ | 5′TGGTAGGGCTTCTCACCTGT3′ |
| KLF4 transcript | 5′CCACCTTCTTCACCCCTAGA3′ | 5′AAGGTTTCTCACCTGTGTGG3′ |
| GAPDH transcript | 5′TCGGAGTCAACGGATTTGGT3′ | 5′TTCCCGTTCTCAGCCTTGAC3′ |
| KLF2 Promoter Primer | 5′TCCCATCCATCCAGGGTTCT3′ | 5′TCAGAGACTCTCAGGGGAGC3′ |
| KLF4 Promoter Primer | 5′TAGAGGGATTCCTGGCGTTG3′ | 5′GATTTTTCCACTCCTCGCCG3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thakar, S.; Katakia, Y.T.; Ramakrishnan, S.K.; Pandya Thakkar, N.; Majumder, S. Intermittent High Glucose Elevates Nuclear Localization of EZH2 to Cause H3K27me3-Dependent Repression of KLF2 Leading to Endothelial Inflammation. Cells 2021, 10, 2548. https://doi.org/10.3390/cells10102548
Thakar S, Katakia YT, Ramakrishnan SK, Pandya Thakkar N, Majumder S. Intermittent High Glucose Elevates Nuclear Localization of EZH2 to Cause H3K27me3-Dependent Repression of KLF2 Leading to Endothelial Inflammation. Cells. 2021; 10(10):2548. https://doi.org/10.3390/cells10102548
Chicago/Turabian StyleThakar, Sumukh, Yash T Katakia, Shyam Kumar Ramakrishnan, Niyati Pandya Thakkar, and Syamantak Majumder. 2021. "Intermittent High Glucose Elevates Nuclear Localization of EZH2 to Cause H3K27me3-Dependent Repression of KLF2 Leading to Endothelial Inflammation" Cells 10, no. 10: 2548. https://doi.org/10.3390/cells10102548