
Plasma Membrane and Organellar Targets of STIM1 for Intracellular Calcium Handling in Health and Neurodegenerative Diseases

 , and
, and
Abstract
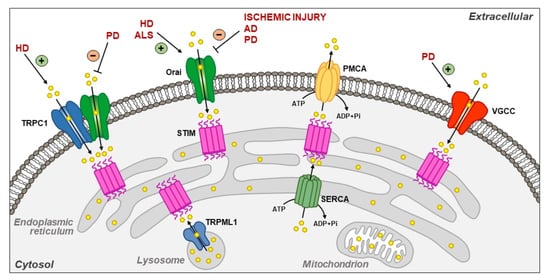
:
1. Introduction
2. Plasma Membrane Targets of STIM1
2.1. Orai and TRPC1
2.2. Plasma Membrane Ca2+-ATPase (PMCA)
2.3. Na+/K+ ATPase
2.4. L-Type Voltage-Gated Ca2+ Channels (VGCCs)
3. Intracellular Targets of STIM1
3.1. SERCA2A and SERCA3
3.2. TRPML1
3.3. Nuclear Proteins

{kind=link}
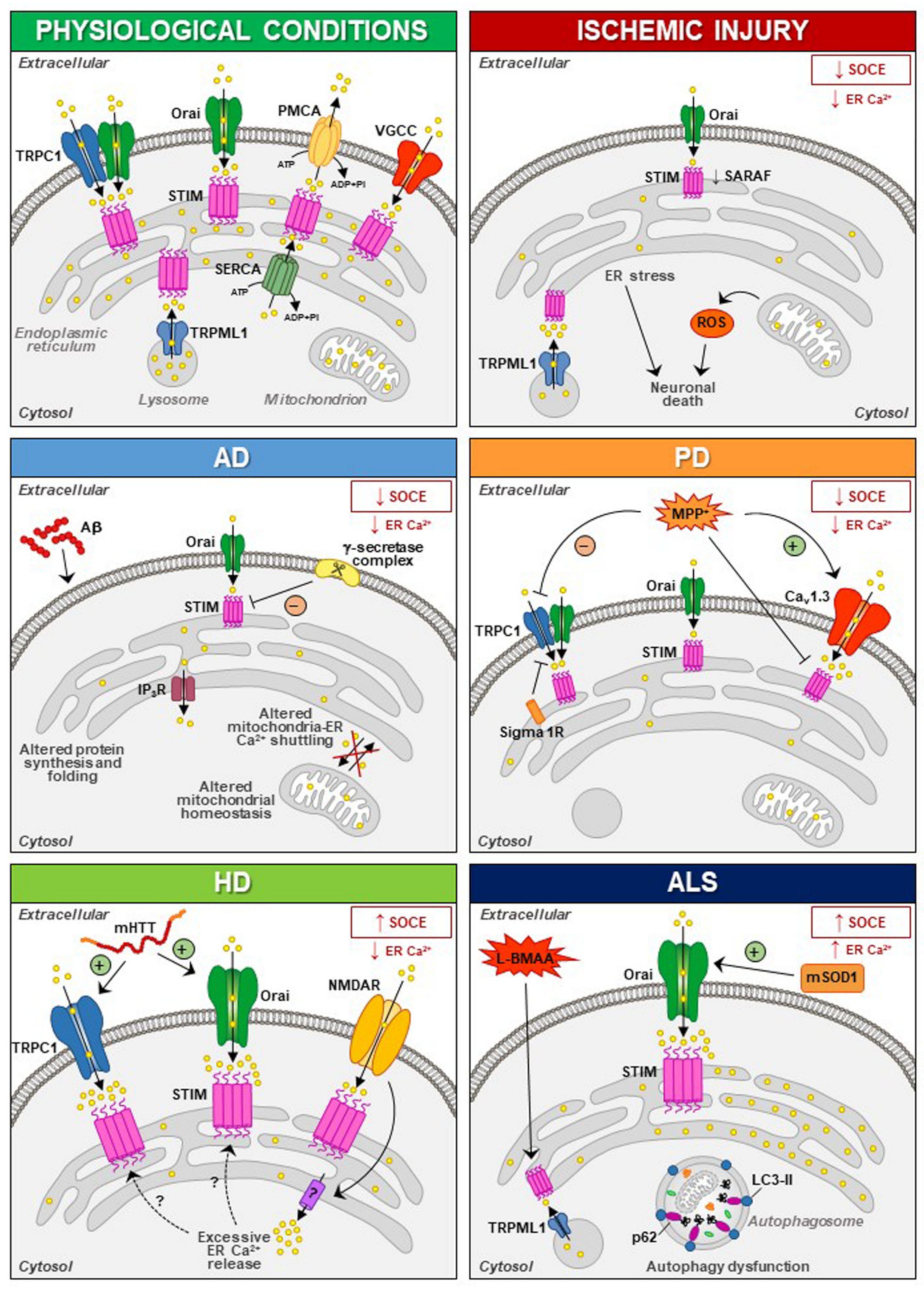{kind=link}
| STIM1 Target | Localization | Effect of the Interaction | References | |
|---|---|---|---|---|
| Orai | Plasma membrane | ER Ca2+ refilling through SOCE | [9,21,47,48] | PLASMA MEMBRANE TARGETS |
| TRPC1 | Plasma membrane | ER Ca2+ refilling through SOCE in an Orai1-dependent way | [28,29,32,33,34,35,49,50,57] | |
| PMCA | Plasma membrane | Regulation of intracellular Ca2+ homeostasis in T cells; regulation of NFAT-dependent cytokine production in Jurkat T cells | [45,46,53] | |
| Na+/K+ ATPase | Plasma membrane | Na+/K+ ATPase downregulation during hypoxia in alveolar epithelial cells | [54] | |
| L-type VGCC | Plasma membrane | Inhibition of L-type VGCC | [51,52] | |
| SERCA2A | Endoplasmic reticulum | ER Ca2+ refilling through Orai1 recruitment | [56] | INTRACELLULAR TARGETS |
| SERCA3 | Endoplasmic reticulum | Acidic Ca2+ store refilling in platelets in a SOC-independent way | [57] | |
| TRPML1 | Lysosome | Regulation of lysosomal and ER Ca2+ homeostasis | [68,69] | |
| Exportin 1 and transportin 1 | Nuclear envelope | ? | [70] | |
| Importins and exportins | Nuclear envelope | Possible role in the modulation of nuclear import/export, through the scaffold protein TMEM20/POST | [71] |
4. STIM1 Scaffold Proteins
5. Molecular Modulators of STIM1
| STIM1 Modulator | Localization | Effect | References |
|---|---|---|---|
| TMEM20/POST | ER membrane; plasma membrane (minor fraction) | Scaffold protein involved in the binding of STIM1 to plasma membrane proteins (i.e., Orai1, PMCA, SERCAs, Na+/K+ ATPase) and nuclear membrane proteins (importins and exportins) | [71] |
| SARAF (SOCE-associated regulatory factor) | ER membrane | It protects cells from Ca2+ overfilling by promoting the Ca2+-dependent slow inactivation of CRAC channels after the interaction with STIM1 | [80,81] |
| Fragment P100 of polycystin-1 | ? | Reduction in SOCE via direct inhibition of STIM1 translocation | [83] |
| ERp57 | ER lumen | Negative modulation of SOCE via regulation of STIM1 oligomerization | [84] |
| Stanniocalcin 2 | Extracellular (secreted); ER lumen | Negative regulation of SOCE | [86] |
| EB1 | Microtubules | It restricts STIM1 to ER-PM junctions, thus preventing excessive SOCE and ER Ca2+ overload | [89] |
| Golli | Cell body, nucleus and processes | By interacting with the C-terminal domain of STIM1, it negatively regulates the activity of SOCCs | [90,91] |
6. STIM1 Partners in Neurodegenerative Diseases
6.1. Ischemic Injury
6.2. Alzheimer’s Disease (AD)
6.3. Huntington’s Disease (HD)
6.4. Parkinson’s Disease (PD)
6.5. Amyotrophic Lateral Sclerosis (ALS)
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Csutora, P.; Peter, K.; Kilic, H.; Park, K.M.; Zarayskiy, V.; Gwozdz, T.; Bolotina, V.M. Novel role for STIM1 as a trigger for calcium influx factor production. J. Biol. Chem. 2008, 283, 14524–14531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Stathopulos, P.B.; Li, G.Y.; Plevin, M.J.; Ames, J.B.; Ikura, M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 2006, 281, 35855–35862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 2011, 194, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Dyrda, A.; Koenig, S.; Frieden, M. STIM1 long and STIM1 gate differently TRPC1 during store-operated calcium entry. Cell Calcium 2020, 86, 102134. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Jarzembowski, L.; Schwarz, Y.; Poth, V.; Konrad, M.; Knapp, M.L.; Schwär, G.; Lauer, A.A.; Grimm, M.O.W.; Alansary, D.; et al. A short isoform of STIM1 confers frequency-dependent synaptic enhancement. Cell Rep. 2021, 34, 108844. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, G.Y.; Subedi, K.P.; Ong, H.L.; Noyer, L.; Saadi, H.; Zheng, C.; Bhardwaj, R.; Feske, S.; Ambudkar, I.S. STIM2 targets Orai1/STIM1 to the AKAP79 signaling complex and confers coupling of Ca2+ entry with NFAT1 activation. Proc. Natl. Acad. Sci. USA 2020, 117, 16638–16648. [Google Scholar] [CrossRef]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Cahalan, M.D.; Lewis, R.S. Functional roles of ion channels in lymphocytes. Semin. Immunol. 1990, 2, 107–117. [Google Scholar]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef]
- Hoth, M.; Penner, R. Calcium release-activated calcium current in rat mast cells. J. Physiol. 1993, 465, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Parekh, B.; Fleig, A.; Penner, R. The store-operated calcium current ICRAC: Nonlinear activation by InsP3 and dissociation from calcium release. Cell 1997, 89, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Penner, R.; Matthews, G.; Neher, E. Regulation of calcium influx by second messengers in rat mast cells. Nature 1988, 334, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.; Neher, E.; Penner, R. Second messenger-activated calcium influx in rat peritoneal mast cells. J. Physiol. 1989, 418, 105–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, R.S.; Cahalan, M.D. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1989, 1, 99–112. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B.; Penner, R. Store depletion and calcium influx. Physiol. Rev. 1997, 77, 901–930. [Google Scholar] [CrossRef]
- Zweifach, A.; Lewis, R.S. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc. Natl. Acad. Sci. USA 1993, 90, 6295–6299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manjarrés, I.M.; Rodríguez-García, A.; Alonso, M.T.; García-Sancho, J. The sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) is the third element in capacitative calcium entry. Cell Calcium 2010, 47, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Nwokonko, R.M.; Cai, X.; Loktionova, N.A.; Abdulqadir, R.; Xin, P.; Niemeyer, B.A.; Wang, Y.; Trebak, M.; Gill, D.L. Cross-linking of Orai1 channels by STIM proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E3398–E3407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, J.C.; Dehaven, W.I.; Smyth, J.T.; Wedel, B.; Boyles, R.R.; Bird, G.S.; Putney, J.W., Jr. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J. Biol. Chem. 2006, 281, 24979–24990. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, M.; Tomita, T.; Janoshazi, A.; Putney, J.W. Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities. J. Cell Sci. 2012, 125, 4354–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feske, S. CRAC channels and disease—From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 2019, 80, 112–116. [Google Scholar] [CrossRef]
- McNally, B.A.; Somasundaram, A.; Yamashita, M.; Prakriya, M. Gated regulation of CRAC channel ion selectivity by STIM1. Nature 2012, 482, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.L.; Shuttleworth, T.J. Molecular basis of activation of the arachidonate-regulated Ca2+ (ARC) channel, a store-independent Orai channel, by plasma membrane STIM1. J. Physiol. 2013, 591, 3507–3523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, W.; Gonzalez-Cobos, J.C.; Jardin, I.; Romanin, C.; Matrougui, K.; Trebak, M. Complex role of STIM1 in the activation of store-independent Orai1/3 channels. J. Gen. Physiol. 2014, 143, 345–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, P.N.; Zhang, X.; Wu, S.; Janoshazi, A.; Bolimuntha, S.; Putney, J.W.; Trebak, M. Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message. Sci. Signal. 2015, 8, ra74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitt, C.; Zobel, A.; Obukhov, A.G.; Harteneck, C.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 1996, 16, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Jiang, M.; Peyton, M.; Boulay, G.; Hurst, R.; Stefani, E.; Birnbaumer, L. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 1996, 85, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, W.; Singh, B.B.; Lockwich, T.; Jadlowiec, J.; O’Connell, B.; Wellner, R.; Zhu, M.X.; Ambudkar, I.S. Trp1, a candidate protein for the store-operated Ca2+ influx mechanism in salivary gland cells. J. Biol. Chem. 2000, 275, 3403–3411. [Google Scholar] [CrossRef] [Green Version]
- Brough, G.H.; Wu, S.; Cioffi, D.; Moore, T.M.; Li, M.; Dean, N.; Stevens, T. Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway. FASEB J. 2001, 15, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC Channels in the SOCE Scenario. Cells 2020, 9, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Zeng, W.; Yuan, J.P.; Shin, D.M.; Worley, P.F.; Muallem, S. Native Store-operated Ca2+ Influx Requires the Channel Function of Orai1 and TRPC1. J. Biol. Chem. 2009, 284, 9733–9741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, H.L.; Cheng, K.T.; Liu, X.; Bandyopadhyay, B.C.; Paria, B.C.; Soboloff, J.; Pani, B.; Gwack, Y.; Srikanth, S.; Singh, B.B.; et al. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J. Biol. Chem. 2007, 282, 9105–9116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J. Biol. Chem. 2008, 283, 25296–25304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, A.P.; Large, W.A. A Ca2+-permeable non-selective cation channel activated by depletion of internal Ca2+ stores in single rabbit portal vein myocytes. J. Physiol. 2002, 538, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Swaim, W.; Ambudkar, I.S. Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol. 2011, 9, e1001025. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.; Vaca, L. SOC and now also SIC: Store-operated and store-inhibited channels. IUBMB Life 2011, 63, 856–863. [Google Scholar] [CrossRef]
- Zarayskiy, V.; Monje, F.; Peter, K.; Csutora, P.; Khodorov, B.I.; Bolotina, V.M. Store-operated Orai1 and IP3 receptor-operated TRPC1 channel. Channels 2007, 1, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Ong, H.L.; Ambudkar, I.S. The dynamic complexity of the TRPC1 channelosome. Channels 2011, 5, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Cheng, X.; Tian, J.; Xiao, Y.; Tian, T.; Xu, F.; Hong, X.; Zhu, M.X. TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. 2020, 209, 107497. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.M.; Brough, G.H.; Babal, P.; Kelly, J.J.; Li, M.; Stevens, T. Store-operated calcium entry promotes shape change in pulmonary endothelial cells expressing Trp1. Am. J. Physiol. 1998, 275, L574–L582. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Ahmad, A.A.; Seidel, T.; Hunter, C.; Streiff, M.; Nikolova, L.; Spitzer, K.W.; Sachse, F.B. Location and function of transient receptor potential canonical channel 1 in ventricular myocytes. J. Mol. Cell. Cardiol. 2020, 139, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Elzamzamy, O.M.; Penner, R.; Hazlehurst, L.A. The Role of TRPC1 in Modulating Cancer Progression. Cells 2020, 9, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikanth, S.; Gwack, Y. Orai1-NFAT signalling pathway triggered by T cell receptor stimulation. Mol. Cells 2013, 35, 182–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.F.; Samakai, E.; Soboloff, J. STIM1 is required for attenuation of PMCA-mediated Ca2+ clearance during T-cell activation. EMBO J. 2012, 31, 1123–1133. [Google Scholar] [CrossRef] [Green Version]
- Peinelt, C.; Vig, M.; Koomoa, D.L.; Beck, A.; Nadler, M.J.; Koblan-Huberson, M.; Lis, A.; Fleig, A.; Penner, R.; Kinet, J.P. Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat. Cell Biol. 2006, 8, 771–773. [Google Scholar] [CrossRef] [Green Version]
- Soboloff, J.; Spassova, M.A.; Tang, X.D.; Hewavitharana, T.; Xu, W.; Gill, D.L. Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 2006, 281, 20661–20665. [Google Scholar] [CrossRef] [Green Version]
- Worley, P.F.; Zeng, W.; Huang, G.N.; Yuan, J.P.; Kim, J.Y.; Lee, M.G.; Muallem, S. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 2007, 42, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef]
- Park, C.Y.; Shcheglovitov, A.; Dolmetsch, R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 2010, 330, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, X.; Mancarella, S.; Hendron, E.; Eguchi, S.; Soboloff, J.; Tang, X.D.; Gill, D.L. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 2010, 330, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Go, C.K.; Hooper, R.; Aronson, M.R.; Schultz, B.; Cangoz, T.; Nemani, N.; Zhang, Y.; Madesh, M.; Soboloff, J. The Ca2+ export pump PMCA clears near-membrane Ca2+ to facilitate store-operated Ca2+ entry and NFAT activation. Sci. Signal 2019, 12, eaaw2627. [Google Scholar] [CrossRef] [PubMed]
- Gusarova, G.A.; Trejo, H.E.; Dada, L.A.; Briva, A.; Welch, L.C.; Hamanaka, R.B.; Mutlu, G.M.; Chandel, N.S.; Prakriya, M.; Sznajder, J.I. Hypoxia leads to Na,K-ATPase downregulation via Ca2+ release-activated Ca2+ channels and AMPK activation. Mol. Cell. Biol. 2011, 31, 3546–3556. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Erxleben, C.; Abramowitz, J.; Flockerzi, V.; Zhu, M.X.; Armstrong, D.L.; Birnbaumer, L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc. Natl. Acad. Sci. USA 2008, 105, 2895–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampieri, A.; Zepeda, A.; Asanov, A.; Vaca, L. Visualizing the store-operated channel complex assembly in real time: Identification of SERCA2 as a new member. Cell Calcium 2009, 45, 439–446. [Google Scholar] [CrossRef] [PubMed]
- López, J.J.; Jardín, I.; Bobe, R.; Pariente, J.A.; Enouf, J.; Salido, G.M.; Rosado, J.A. STIM1 regulates acidic Ca2+ store refilling by interaction with SERCA3 in human platelets. Biochem. Pharmacol. 2008, 75, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Godinho, L.F.; Costello, J.L.; Islinger, M. The different facets of organelle interplay—An overview of organelle interactions. Front. Cell Dev. Biol. 2015, 3, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Li, M.; Qiu, F.; Zhang, M.; Zhang, Y.H. Cell-permeable organic fluorescent probes for live-cell long-term super-resolution imaging reveal lysosome-mitochondrion interactions. Nat. Commun. 2017, 8, 1307. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.J.; Davis, L.C.; Wagner, S.K.; Lewis, A.M.; Parrington, J.; Churchill, G.C.; Galione, A. Bidirectional Ca2+ signaling occurs between the endoplasmic reticulum and acidic organelles. J. Cell Biol. 2013, 200, 789–805. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, B.S.; Magalhaes, J.; Beavan, M.S.; McNeill, A.; Gegg, M.E.; Cleeter, M.W.; Bloor-Young, D.; Churchill, G.C.; Duchen, M.R.; Schapira, A.H.; et al. Endoplasmic reticulum and lysosomal Ca2⁺ stores are remodelled in GBA1-linked Parkinson disease patient fibroblasts. Cell Calcium 2016, 59, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, V.; Petrozziello, T.; Sisalli, M.J.; Boscia, F.; Canzoniero, L.M.T.; Secondo, A. The activation of Mucolipin TRP channel 1 (TRPML1) protects motor neurons from L-BMAA neurotoxicity by promoting autophagic clearance. Sci. Rep. 2019, 9, 10743. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, V.; Sisalli, M.J.; Petrozziello, T.; Canzoniero, L.M.T.; Secondo, A. Lysosomal calcium is modulated by STIM1/TRPML1 interaction which participates to neuronal survival during ischemic preconditioning. FASEB J. 2021, 35, e21277. [Google Scholar] [CrossRef]
- Saitoh, N.; Oritani, K.; Saito, K.; Yokota, T.; Ichii, M.; Sudo, T.; Fujita, N.; Nakajima, K.; Okada, M.; Kanakura, Y. Identification of functional domains and novel binding partners of STIM proteins. J. Cell. Biochem. 2011, 112, 147–156. [Google Scholar] [CrossRef]
- Krapivinsky, G.; Krapivinsky, L.; Stotz, S.C.; Manasian, Y.; Clapham, D.E. POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters. Proc. Natl. Acad. Sci. USA 2011, 108, 19234–19239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.D.; Jung, H.R.; Seo, S.H.; Oh, S.C.; Ban, Y.; Tan, X.; Kim, J.M.; Lee, S.H.; Koh, D.S.; Jung, H.; et al. MicroRNA-150 modulates intracellular Ca2+ levels in naïve CD8+ T cells by targeting TMEM20. Sci. Rep. 2017, 7, 2623. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, S.; Gwack, Y. Orai1, STIM1, and their associating partners. J. Physiol. 2012, 590, 4169–4177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Quintana, A.; Findlay, G.M.; Mettlen, M.; Baust, B.; Jain, M.; Nilsson, R.; Rao, A.; Hogan, P.G. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 2013, 499, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Miner, C.; Zhang, L.; Hanson, P.I.; Dani, A.; Vig, M. An essential and NSF independent role for α-SNAP in store-operated calcium entry. eLife 2013, 2, e00802. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; He, L.; Sun, A.; Quintana, A.; Ding, Y.; Ma, G.; Tan, P.; Liang, X.; Zheng, X.; Chen, L.; et al. Proteomic mapping of ER-PM junctions identifies STIMATE as a regulator of Ca2⁺ influx. Nat. Cell Biol. 2015, 17, 1339–1347. [Google Scholar] [CrossRef] [Green Version]
- Hooper, R.; Soboloff, J. STIMATE reveals a STIM1 transitional state. Nat. Cell Biol. 2015, 17, 1232–1234. [Google Scholar] [CrossRef] [Green Version]
- López, J.J.; Albarrán, L.; Gómez, L.J.; Smani, T.; Salido, G.M.; Rosado, J.A. Molecular modulators of store-operated calcium entry. Biochim. Biophys. Acta 2016, 1863, 2037–2043. [Google Scholar] [CrossRef]
- Serwach, K.; Gruszczynska-Biegala, J. Target Molecules of STIM Proteins in the Central Nervous System. Front. Mol. Neurosci. 2020, 13, 617422. [Google Scholar] [CrossRef]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 2012, 49, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Albarrán, L.; López, J.J.; Gómez, L.J.; Salido, G.M.; Rosado, J.A. SARAF modulates TRPC1, but not TRPC6, channel function in a STIM1-independent manner. Biochem. J. 2016, 473, 3581–3595. [Google Scholar] [CrossRef] [PubMed]
- Albarrán, L.; López, J.J.; Ben, A.N.; Martin-Cano, F.E.; Berna-Erro, A.; Smani, T.; Salido, G.M.; Rosado, J.A. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci. Rep. 2016, 6, 24452. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Li, Y.; Yu, S.; Greenwell, P.; Wodarczyk, C.; Boletta, A.; Guggino, W.B.; Qian, F. Identification of a polycystin-1 cleavage product, P100, that regulates store operated Ca entry through interactions with STIM1. PLoS ONE 2010, 5, e12305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, D.; Groenendyk, J.; Touret, N.; Michalak, M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011, 12, 1182–1188. [Google Scholar] [CrossRef] [Green Version]
- Di Buduo, C.A.; Abbonante, V.; Marty, C.; Moccia, F.; Rumi, E.; Pietra, D.; Soprano, P.M.; Lim, D.; Cattaneo, D.; Iurlo, A.; et al. Defective interaction of mutant calreticulin and SOCE in megakaryocytes from patients with myeloproliferative neoplasms. Blood 2020, 135, 133–144. [Google Scholar] [CrossRef]
- Zeiger, W.; Ito, D.; Swetlik, C.; Oh-hora, M.; Villereal, M.L.; Thinakaran, G. Stanniocalcin 2 is a negative modulator of store-operated calcium entry. Mol. Cell. Biol. 2011, 31, 3710–3722. [Google Scholar] [CrossRef] [Green Version]
- López, E.; Gómez-Gordo, L.; Cantonero, C.; Bermejo, N.; Pérez-Gómez, J.; Granados, M.P.; Salido, G.N.; Dionisio, G.A.R.; Liberal, P.C.R. Stanniocalcin 2 Regulates Non-capacitative Ca2+ Entry and Aggregation in Mouse Platelets. Front. Physiol. 2018, 9, 266. [Google Scholar] [CrossRef] [Green Version]
- Grigoriev, I.; Gouveia, S.M.; van der Vaart, B.; Demmers, J.; Smyth, J.T.; Honnappa, S.; Splinter, D.; Steinmetz, M.O.; Putney, J.W., Jr.; Hoogenraad, C.C.; et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr. Biol. 2008, 18, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.L.; Chen, Y.J.; Quintanilla, C.G.; Hsieh, T.S.; Liou, J. EB1 binding restricts STIM1 translocation to ER-PM junctions and regulates store-operated Ca2+ entry. J. Cell Biol. 2018, 217, 2047–2058. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.M.; Doherty, M.K.; Tepikin, A.V.; Burgoyne, R.D. Evidence for an interaction between Golli and STIM1 in store-operated calcium entry. Biochem. J. 2010, 430, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Reyes, S.D.; Campagnoni, A.T. Two separate domains in the golli myelin basic proteins are responsible for nuclear targeting and process extension in transfected cells. J. Neurosci. Res. 2002, 69, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, J.; Liu, X.; Zhang, M.; An, J.; Sun, P.; Li, D.; Jin, T.; Wang, J. High expression of STIM1 in the early stages of diffuse axonal injury. Brain Res. 2013, 1495, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.F.; Liu, Z.H.; Li, N.; Cheng, W.J.; Guo, S.W. Knockdown of STIM1 improves neuronal survival after traumatic neuronal injury through regulating mGluR1-dependent Ca2+ signaling in mouse cortical neurons. Cell. Mol. Neurobiol. 2015, 35, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Gruszczynska-Biegala, J.; Strucinska, K.; Maciag, F.; Majewski, L.; Sladowska, M.; Kuznicki, J. STIM protein-NMDA2 receptor interaction decreases NMDA-dependent calcium levels in cortical neurons. Cells 2020, 9, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmer, P.J.; Wild, A.R.; Dell’Acqua, M.L.; Sather, W.A. STIM1 Ca2+ sensor control of L-type Ca2+ channel-dependent dendritic spine structural plasticity and nuclear signaling. Cell Rep. 2017, 19, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Wegierski, T.; Kuznicki, J. Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 2018, 74, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Serwach, K.; Gruszczynska-Biegala, J. STIM Proteins and Glutamate Receptors in Neurons: Role in Neuronal Physiology and Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2289. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Gonzalez, J.; Jurado-Coronel, J.C.; Ávila, M.F.; Sabogal, A.; Capani, F.; Barreto, G.E. NMDARs in neurological diseases: A potential therapeutic target. Int. J. Neurosci. 2015, 125, 315–327. [Google Scholar] [CrossRef]
- Carvajal, F.J.; Mattison, H.A.; Cerpa, W. Role of NMDA Receptor-Mediated Glutamatergic Signaling in Chronic and Acute Neuropathologies. Neural Plast. 2016, 2016, 2701526. [Google Scholar] [CrossRef]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P.B.; Challiss, R.A.; Nahorski, S.R. Neuronal Ca2+ stores: Activation and function. Trends Neurosci. 1995, 18, 299–306. [Google Scholar] [CrossRef]
- Emptage, N.; Bliss, T.V.; Fine, A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 1999, 22, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Emptage, N.J.; Reid, C.A.; Fine, A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 2001, 29, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Bogeski, I.; Niemeyer, B.A. Redox regulation of ion channels. Antioxid. Redox Signal. 2014, 21, 859–862. [Google Scholar] [CrossRef] [Green Version]
- Nunes, P.; Demaurex, N. Redox regulation of store-operated Ca2+ entry. Antioxid. Redox Signal. 2014, 21, 915–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, R.; Hediger, M.A.; Demaurex, N. Redox modulation of STIM-ORAI signaling. Cell Calcium 2016, 60, 142–152. [Google Scholar] [CrossRef]
- Niemeyer, B.A. The STIM-Orai Pathway: Regulation of STIM and Orai by Thiol Modifications. Adv. Exp. Med. Biol. 2017, 993, 99–116. [Google Scholar] [CrossRef]
- Bogeski, I.; Kummerow, C.; Al-Ansary, D.; Schwarz, E.C.; Koehler, R.; Kozai, D.; Takahashi, N.; Peinelt, C.; Griesemer, D.; Bozem, M.; et al. Differential redox regulation of ORAI ion channels: A mechanism to tune cellular calcium signaling. Sci. Signal. 2010, 3, ra24. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, B.J.; Irrinki, K.M.; Mallilankaraman, K.; Lien, Y.C.; Wang, Y.; Bhanumathy, C.D.; Subbiah, R.; Ritchie, M.F.; Soboloff, J.; Baba, Y.; et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 2010, 190, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Gibhardt, C.S.; Cappello, S.; Bhardwaj, R.; Schober, R.; Kirsch, S.A.; Del Rio, Z.B.; Gahbauer, S.; Bochicchio, A.; Sumanska, M.; Ickes, C.; et al. Oxidative Stress-Induced STIM2 Cysteine Modifications Suppress Store-Operated Calcium Entry. Cell Rep. 2020, 33, 108292. [Google Scholar] [CrossRef] [PubMed]
- Gui, L.; Zhu, J.; Lu, X.; Sims, S.M.; Lu, W.Y.; Stathopulos, P.B.; Feng, Q. S-Nitrosylation of STIM1 by Neuronal Nitric Oxide Synthase Inhibits Store-Operated Ca2+ Entry. J. Mol. Biol. 2018, 430, 1773–1785. [Google Scholar] [CrossRef]
- Zhu, J.; Lu, X.; Feng, Q.; Stathopulos, P.B. A charge-sensing region in the stromal interaction molecule 1 luminal domain confers stabilization-mediated inhibition of SOCE in response to S-nitrosylation. J. Biol. Chem. 2018, 293, 8900–8911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secondo, A.; Petrozziello, T.; Tedeschi, V.; Boscia, F.; Vinciguerra, A.; Ciccone, R.; Pannaccione, A.; Molinaro, P.; Pignataro, G.; Annunziato, L. ORAI1/STIM1 Interaction Intervenes in Stroke and in Neuroprotection Induced by Ischemic Preconditioning Through Store-Operated Calcium Entry. Stroke 2019, 50, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Sirabella, R.; Secondo, A.; Pannaccione, A.; Scorziello, A.; Valsecchi, V.; Adornetto, A.; Bilo, L.; Di Renzo, G.; Annunziato, L. Anoxia-induced NF-kappaB-dependent upregulation of NCX1 contributes to Ca2+ refilling into endoplasmic reticulum in cortical neurons. Stroke 2009, 40, 922–929. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B. Store-operated CRAC channels: Function in health and disease. Nat. Rev. Drug Discov. 2010, 9, 399–410. [Google Scholar] [CrossRef]
- Lang, F.; Pelzl, L.; Hauser, S.; Hermann, A.; Stournaras, C.; Schöls, L. To die or not to die SGK1-sensitive ORAI/STIM in cell survival. Cell Calcium 2018, 74, 29–34. [Google Scholar] [CrossRef]
- Lehotský, J.; Racay, P.; Pavlíková, M.; Tatarková, Z.; Urban, P.; Chomová, M.; Kovalská, M.; Kaplán, P. Cross-talk of intracellular calcium stores in the response to neuronal ischemia and ischemic tolerance. Gen. Physiol. Biophys. 2009, 28, F104–F114. [Google Scholar]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 Regulates Capacitive Ca2+ Entry in Neurons and Plays a Key Role in Hypoxic Neuronal Cell Death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Song, J.N.; Wu, Y.; Zhao, Y.L.; Pang, H.G.; Fu, Z.F.; Zhang, B.F.; Ma, X.D. Suppression of STIM1 in the early stage after global ischemia attenuates the injury of delayed neuronal death by inhibiting store-operated calcium entry-induced apoptosis in rats. Neuroreport 2014, 25, 507–513. [Google Scholar] [CrossRef]
- Soboloff, J.; Spassova, M.A.; Hewavitharana, T.; He, L.P.; Xu, W.; Johnstone, L.S.; Dziadek, M.A.; Gill, D.L. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr. Biol. 2006, 16, 1465–1470. [Google Scholar] [CrossRef] [Green Version]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Russa, D.; Frisina, M.; Secondo, A.; Bagetta, G.; Amantea, D. Modulation of Cerebral Store-operated Calcium Entry-regulatory Factor (SARAF) and Peripheral Orai1 Following Focal Cerebral Ischemia and Preconditioning in Mice. Neuroscience 2020, 441, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Ohara, H.; Nabika, T. A nonsense mutation of Stim1 identified in stroke-prone spontaneously hypertensive rats decreased the store-operated calcium entry in astrocytes. Biochem. Biophys. Res. Commun. 2016, 476, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Panza, F.; Solfrizzi, V.; Imbimbo, B.P.; Logroscino, G. Amyloid-directed monoclonal antibodies for the treatment of Alzheimer’s disease: The point of no return? Expert Opin. Biol. Ther. 2014, 14, 1465–1476. [Google Scholar] [CrossRef]
- Godyń, J.; Jończyk, J.; Panek, D.; Malawska, B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 2016, 68, 127–138. [Google Scholar] [CrossRef]
- Schon, E.A.; Area-Gomez, E. Mitochondria-associated ER membranes in Alzheimer disease. Mol. Cell. Neurosci. 2013, 55, 26–36. [Google Scholar] [CrossRef]
- Giacomello, M.; Barbiero, L.; Zatti, G.; Squitti, R.; Binetti, G.; Pozzan, T.; Fasolato, C.; Ghidoni, R.; Pizzo, P. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer’s disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol. Dis. 2005, 18, 638–648. [Google Scholar] [CrossRef]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G.J.D.S. The Interplay between Ca2+ Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, K.H.; Shineman, D.; Muller, M.; Cardenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.H.; Mei, L.; Mak, D.O.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 2010, 3, ra22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilling, D.; Muller, M.; Takano, H.; Mak, D.O.; Abel, T.; Coulter, D.A.; Foskett, J.K. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer’s disease pathogenesis. J. Neurosci. 2014, 34, 6910–6923. [Google Scholar] [CrossRef] [Green Version]
- Mak, D.O.; Cheung, K.H.; Toglia, P.; Foskett, J.K.; Ullah, G. Analyzing and quantifying the gain-of-function enhancement of IP3 receptor gating by familial Alzheimer’s disease-causing mutants in Presenilins. PLoS Comput. Biol. 2015, 11, e1004529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybalchenko, V.; Hwang, S.Y.; Rybalchenko, N.; Koulen, P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol. 2008, 40, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef] [Green Version]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef]
- Tong, B.C.; Lee, C.S.; Cheng, W.H.; Lai, K.O.; Kevin Foskett, J.; Cheung, K.H. Familial Alzheimer’s disease-associated presenilin 1 mutants promote gamma-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci. Signal. 2016, 9, ra89. [Google Scholar] [CrossRef] [Green Version]
- Zatti, G.; Ghidoni, R.; Barbiero, L.; Binetti, G.; Pozzan, T.; Fasolato, C.; Pizzo, P. The presenilin 2 M239I mutation associated with familial Alzheimer’s disease reduces Ca2+ release from intracellular stores. Neurobiol. Dis. 2004, 15, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Zatti, G.; Burgo, A.; Giacomello, M.; Barbiero, L.; Ghidoni, R.; Sinigaglia, G.; Florean, C.; Bagnoli, S.; Binetti, G.; Sorbi, S.; et al. Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 2006, 39, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J. Cell Biol. 2000, 149, 793–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, I.F.; Boyle, J.P.; Vaughan, P.F.; Pearson, H.A.; Cowburn, R.F.; Peers, C.S. Ca2+ stores and capacitative Ca2+ entry in human neuroblastoma (SH-SY5Y) cells expressing a familial Alzheimer’s disease presenilin-1 mutation. Brain Res. 2002, 949, 105–111. [Google Scholar] [CrossRef]
- Yoo, A.S.; Cheng, I.; Chung, S.; Grenfell, T.Z.; Lee, H.; Pack-Chung, E.; Handler, M.; Shen, J.; Xia, W.; Tesco, G.; et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron 2000, 27, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Herms, J.; Schneider, I.; Dewachter, I.; Caluwaerts, N.; Kretzschmar, H.; Van Leuven, F. Capacitive calcium entry is directly attenuated by mutant presenilin-1, independent of the expression of the amyloid precursor protein. J. Biol. Chem. 2003, 278, 2484–2489. [Google Scholar] [CrossRef] [Green Version]
- Akbari, Y.; Hitt, B.D.; Murphy, M.P.; Dagher, N.N.; Tseng, B.P.; Green, K.N.; Golde, T.E.; LaFerla, F.M. Presenilin regulates capacitative calcium entry dependently and independently of gamma-secretase activity. Biochem. Biophys. Res. Commun. 2004, 322, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Bojarski, L.; Pomorski, P.; Szybinska, A.; Drab, M.; Skibinska-Kijek, A.; Gruszczynska-Biegala, J.; Kuznicki, J. Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer’s disease. Biochim. Biophys. Acta 2009, 1793, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.Y.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.J.; Feske, S.; White, C.L.; Bezprozvanny, I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Sun, S.; Wu, L.; Pchitskaya, E.; Zakharova, O.; FonTacer, K.; Bezprozvanny, I. Store-operated calcium channel complex in postsynaptic spines: A new therapeutic target for Alzheimer’s disease treatment. J. Neurosci. 2016, 36, 11837–11850. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Caro, C.; Berrocal, M.; Lopez-Guerrero, A.M.; Alvarez-Barrientos, A.; Pozo-Guisado, E.; Gutierrez-Merino, C.; Mata, A.M.; Martin-Romero, F.J. STIM1 deficiency is linked to Alzheimer’s disease and triggers cell death in SH-SY5Y cells by upregulation of L-type voltage-operated Ca2+ entry. J. Mol. Med. 2018, 96, 1061–1079. [Google Scholar] [CrossRef] [Green Version]
- Gazda, K.; Kuznicki, J.; Wegierski, T. Knockdown of amyloid precursor protein increases calcium levels in the endoplasmic reticulum. Sci. Rep. 2017, 7, 14512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludewig, S.; Herrmann, U.; Michaelsen-Preusse, K.; Metzdorf, K.; Just, J.; Bold, C.; Müller, U.C.; Korte, M. APPsα rescues impaired Ca2+ homeostasis in APP- and APLP2-deficient hippocampal neurons. Proc. Natl. Acad. Sci. USA 2021, 118, e2011506118. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Karl, R.M.; Alexander, R.P.; Adelsberger, H.; Brill, M.S.; Ruhlmann, C.; Ansel, A.; Sakimura, K.; Baba, Y.; Kurosaki, T.; et al. STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 2014, 82, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.M.; Cheng, Y.C.; Xiao, Z.B.; Sun, M.Y.; Ren, P.C.; Sun, X.D. Down-regulation of Homer1b/c attenuates group I metabotropic glutamate receptors dependent Ca2+ signaling through regulating endoplasmic reticulum Ca2+ release in PC12 cells. Biochem. Biophys. Res. Commun. 2014, 450, 1568–1574. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alvarez, G.; Shetty, M.S.; Lu, B.; Yap, K.A.; Oh-Hora, M.; Sajikumar, S.; Bichler, Z.; Fivaz, M. Impaired spatial memory and enhanced long-term potentiation in mice with forebrain-specific ablation of the Stim genes. Front. Behav. Neurosci. 2015, 9, 180. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Caro, C.; Orantos-Aguilera, Y.; Sanchez-Lopez, I.; de Juan-Sanz, J.; Parys, J.B.; Area-Gomez, E.; Pozo-Guisado, E.; Martin-Romero, F.J. STIM1 Deficiency Leads to Specific Down-Regulation of ITPR3 in SH-SY5Y Cells. Int. J. Mol. Sci. 2020, 21, 6598. [Google Scholar] [CrossRef]
- Kumar, U.; Dunlop, D.M.; Richardson, J.S. Mitochondria from Alzheimer’s fibroblasts show decreased uptake of calcium and increased sensitivity to free radicals. Life Sci. 1994, 54, 1855–1860. [Google Scholar] [CrossRef]
- Pizzo, P.; Lissandron, V.; Capitanio, P.; Pozzan, T. Ca2+ signalling in the Golgi apparatus. Cell Calcium 2011, 50, 184–192. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Pihan, P.; Hetz, C. Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium 2018, 70, 24–31. [Google Scholar] [CrossRef]
- Martin, L.J. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals 2010, 3, 839–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell. Res. 2018, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Pierrot, N.; Ghisdal, P.; Caumont, A.S.; Octave, J.N. Intraneuronal amyloid-beta1-42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J. Neurochem. 2004, 88, 1140–1150. [Google Scholar] [CrossRef]
- Al-Mousa, F.; Michelangeli, F. Some commonly used brominated flame retardants cause Ca2+-ATPase inhibition, beta-amyloid peptide release and apoptosis in SH-SY5Y neuronal cells. PLoS ONE 2012, 7, e33059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linde, C.I.; Baryshnikov, S.G.; Mazzocco-Spezzia, A.; Golovina, V.A. Dysregulation of Ca2+ signaling in astrocytes from mice lacking amyloid precursor protein. Am. J. Physiol. Cell Physiol. 2011, 300, C1502–C1512. [Google Scholar] [CrossRef] [Green Version]
- Ronco, V.; Grolla, A.A.; Glasnov, T.N.; Canonico, P.L.; Verkhratsky, A.; Genazzani, A.A.; Lim, D. Differential deregulation of astrocytic calcium signalling by amyloid-β, TNFα, IL-1β and LPS. Cell Calcium 2014, 55, 219–229. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Gonzalez, A.; Andrade, V.; Cortes, N.; Tapia, J.P.; Guzman-Martinez, L. Alzheimer’s disease in the perspective of neuroimmunology. Open Neurol. J. 2018, 12, 50–56. [Google Scholar] [CrossRef]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Shih, H.P.; Vigont, V.; Hrdlicka, L.; Diggins, L.; Singh, C.; Mahoney, M.; Chesworth, R.; Shapiro, G.; Zimina, O.; et al. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington’s disease treatment. Chem. Biol. 2011, 18, 777–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czeredys, M.; Gruszczynska-Biegala, J.; Schacht, T.; Methner, A.; Kuznicki, J. Expression of genes encoding the calcium signalosome in cellular and transgenic models of Huntington’s disease. Front. Mol. Neurosci. 2013, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Ryskamp, D.A.; Liang, X.; Egorova, P.; Zakharova, O.; Hung, G.; Bezprozvanny, I. Enhanced store-operated calcium entry leads to striatal synaptic loss in a Huntington’s disease mouse model. J. Neurosci. 2016, 36, 125–141. [Google Scholar] [CrossRef]
- Wu, J.; Ryskamp, D.; Birnbaumer, L.; Bezprozvanny, I. Inhibition of TRPC1-dependent store-operated calcium entry improves synaptic stability and motor performance in a mouse model of Huntington’s disease. J. Huntingt. Dis. 2018, 7, 35–50. [Google Scholar] [CrossRef]
- Czeredys, M. Dysregulation of Neuronal Calcium Signaling via Store-Operated Channels in Huntington’s Disease. Front. Cell. Dev. Biol. 2020, 8, 611735. [Google Scholar] [CrossRef]
- Vigont, V.A.; Zimina, O.A.; Glushankova, L.N.; Kolobkova, J.A.; Ryazantseva, M.A.; Mozhayeva, G.N.; Kaznacheyeva, E.V. STIM1 protein activates store-operated calcium channels in cellular model of Huntington’s disease. Acta Nat. 2014, 6, 40–47. [Google Scholar] [CrossRef]
- Vigont, V.; Kolobkova, Y.; Skopin, A.; Zimina, O.; Zenin, V.; Glushankova, L.; Kaznacheyeva, E. Both Orai1 and TRPC1 are involved in excessive store-operated calcium entry in striatal neurons expressing mutant huntingtin exon 1. Front. Physiol. 2015, 6, 337. [Google Scholar] [CrossRef] [Green Version]
- Baba, A.; Yasui, T.; Fujisawa, S.; Yamada, R.X.; Yamada, M.K.; Nishiyama, N.; Matsuki, N.; Ikegaya, Y. Activity-evoked capacitative Ca2+ entry: Implications in synaptic plasticity. J. Neurosci. 2003, 23, 7737–7741. [Google Scholar] [CrossRef] [Green Version]
- Calì, T.; Ottolini, D.; Brini, M. Calcium signaling in Parkinson’s disease. Cell Tissue Res. 2014, 357, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Stefani, I.C.; Wright, D.; Polizzi, K.M.; Kontoravdi, C. The role of ER stress-induced apoptosis in neurodegeneration. Curr. Alzheimer Res. 2012, 9, 373–387. [Google Scholar] [CrossRef]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. “Rejuvenation” protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, H.; Selvaraj, S.; Sukumaran, P.; Lei, S.; Birnbaumer, L.; Singh, B.B. Inhibition of L-Type Ca2+ Channels by TRPC1-STIM1 Complex Is Essential for the Protection of Dopaminergic Neurons. J. Neurosci. 2017, 37, 3364–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Harding, M.; Pittman, A.; Dore, J.; Striessnig, J.; Rajadhyaksha, A.; Chen, X. Cav1.2 and Cav1.3 L-type calcium channels regulate dopaminergic firing activity in the mouse ventral tegmental area. J. Neurophysiol. 2014, 112, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, W.; Zhang, L.; Liu, W.B.; Fei, Z. Inhibition of store-operated calcium entry attenuates MPP(+)-induced oxidative stress via preservation of mitochondrial function in PC12 cells: Involvement of Homer1a. PLoS ONE 2013, 8, e83638. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sukumaran, P.; Singh, B.B. Sigma1 Receptor Inhibits TRPC1-Mediated Ca2+ Entry That Promotes Dopaminergic Cell Death. Cell. Mol. Neurobiol. 2021, 41, 1245–1255. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Watt, J.A.; Wang, S.; Lei, S.; Birnbaumer, L.; Singh, B.B. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Investig. 2012, 122, 1354–1367. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, V.; Petrozziello, T.; Secondo, A. Calcium Dyshomeostasis and Lysosomal Ca2+ Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2019, 8, 1216. [Google Scholar] [CrossRef] [Green Version]
- Kawamata, H.; Ng, S.K.; Diaz, N.; Burstein, S.; Morel, L.; Osgood, A.; Sider, B.; Higashimori, H.; Haydon, P.G.; Manfredi, G.; et al. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 2331–2348. [Google Scholar] [CrossRef] [Green Version]
- Goswami, A.; Jesse, C.M.; Chandrasekar, A.; Bushuven, E.; Vollrath, J.T.; Dreser, A.; Katona, I.; Beyer, C.; Johann, S.; Feller, A.C.; et al. Accumulation of STIM1 is associated with the degenerative muscle fibre phenotype in ALS and other neurogenic atrophies. Neuropathol. Appl. Neurobiol. 2015, 41, 304–318. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tedeschi, V.; La Russa, D.; Franco, C.; Vinciguerra, A.; Amantea, D.; Secondo, A. Plasma Membrane and Organellar Targets of STIM1 for Intracellular Calcium Handling in Health and Neurodegenerative Diseases. Cells 2021, 10, 2518. https://doi.org/10.3390/cells10102518
Tedeschi V, La Russa D, Franco C, Vinciguerra A, Amantea D, Secondo A. Plasma Membrane and Organellar Targets of STIM1 for Intracellular Calcium Handling in Health and Neurodegenerative Diseases. Cells. 2021; 10(10):2518. https://doi.org/10.3390/cells10102518
Chicago/Turabian StyleTedeschi, Valentina, Daniele La Russa, Cristina Franco, Antonio Vinciguerra, Diana Amantea, and Agnese Secondo. 2021. "Plasma Membrane and Organellar Targets of STIM1 for Intracellular Calcium Handling in Health and Neurodegenerative Diseases" Cells 10, no. 10: 2518. https://doi.org/10.3390/cells10102518