Correcting Pervasive Errors in Genotypic Datasets to Develop Genetic Maps



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
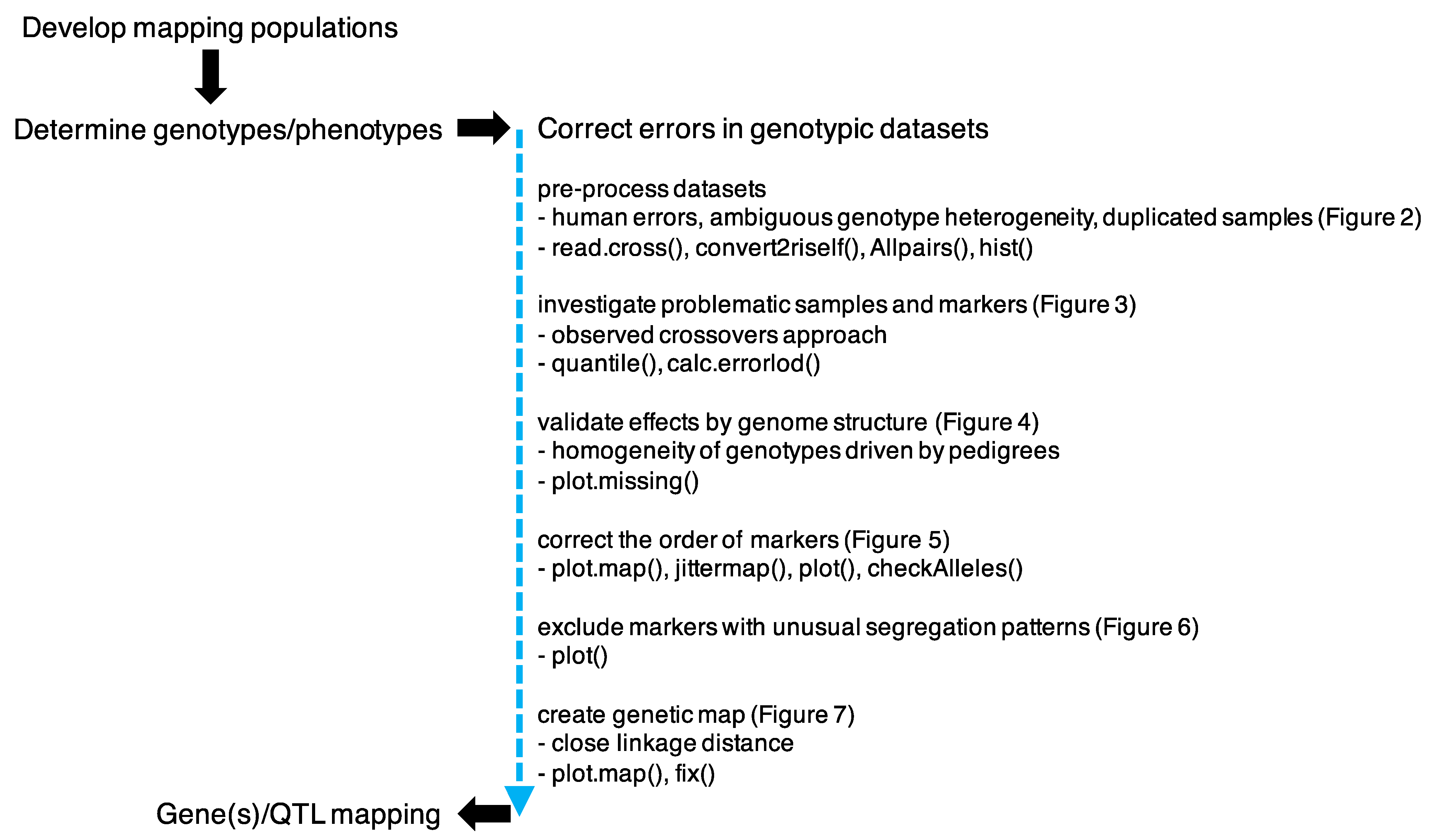
2. Materials and Methods
2.1. Preparation of the Plant Population
2.2. Genotypic Data Acquisition
2.3. Genetic Map Development
3. Results and Discussion
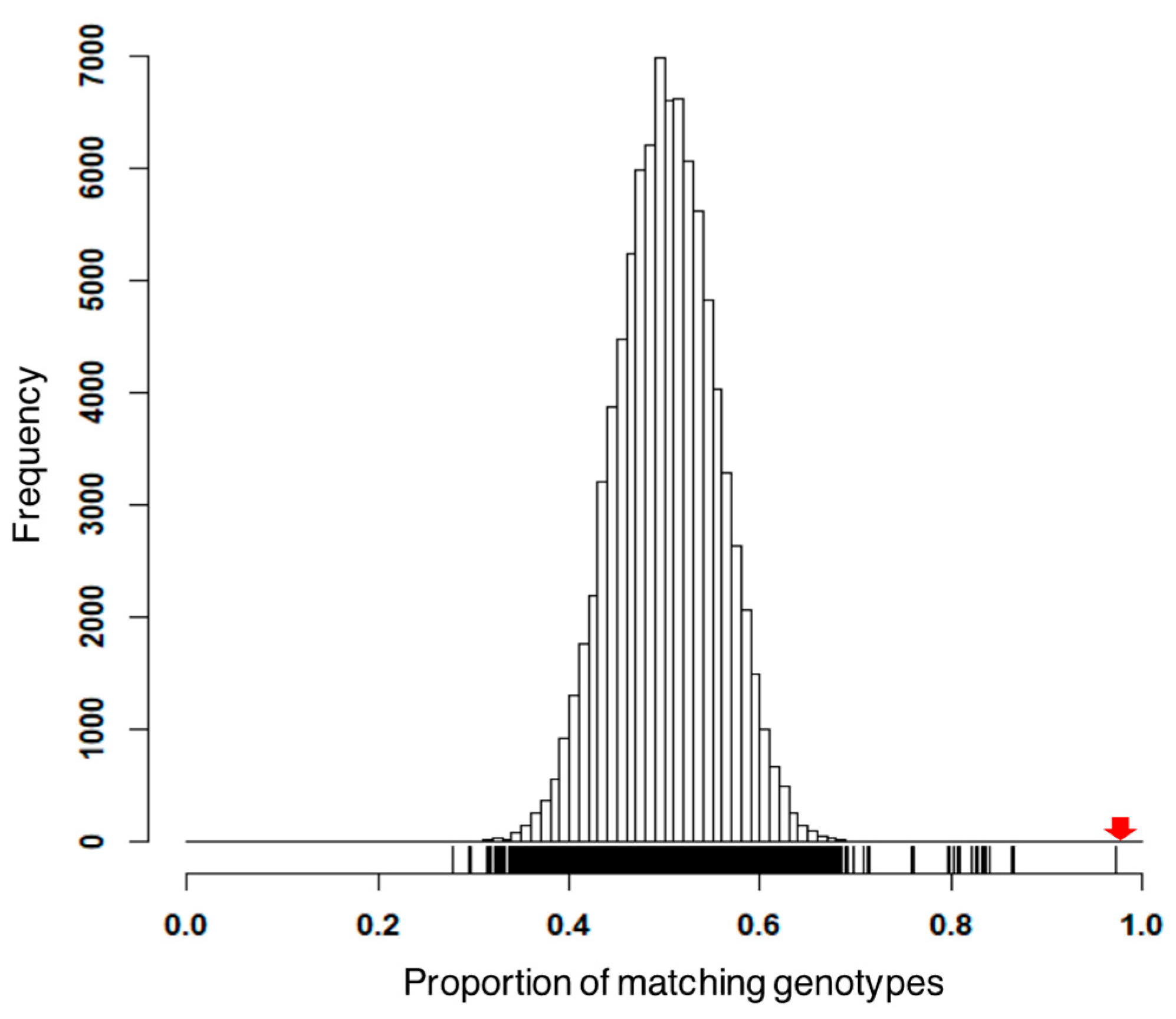
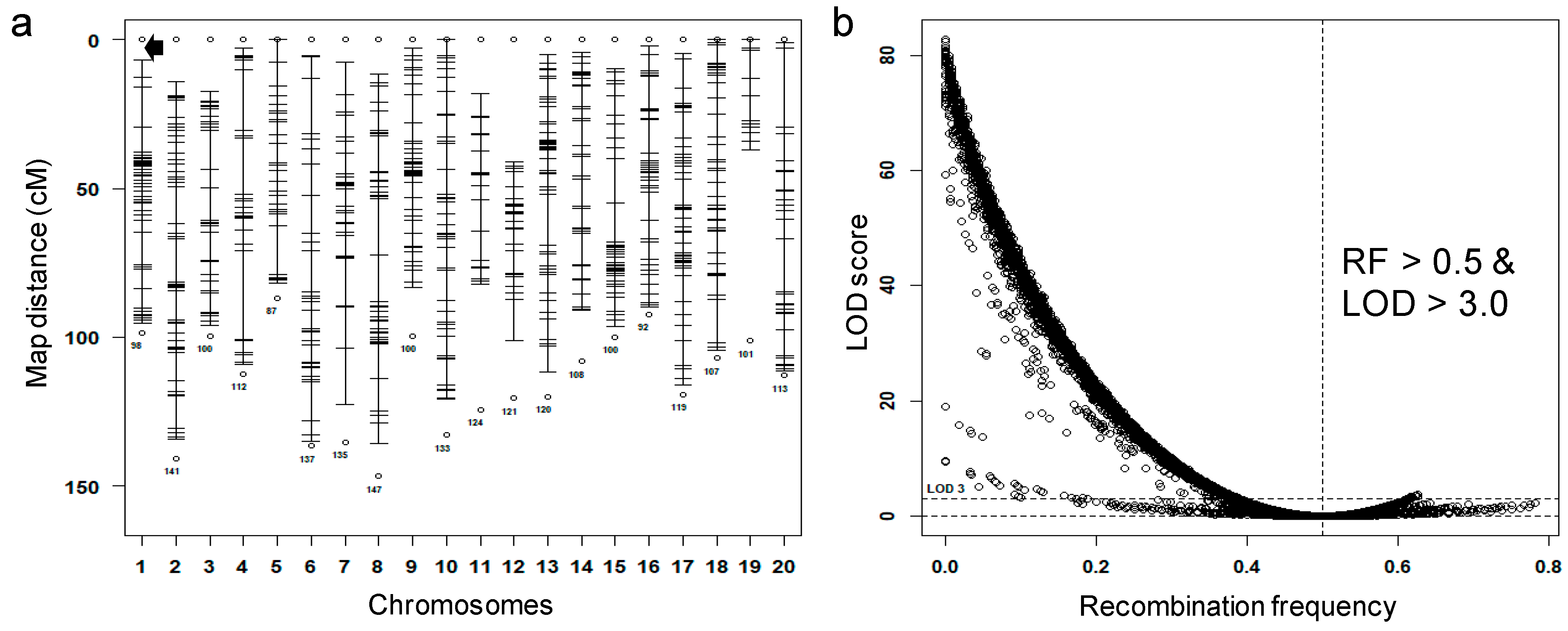
3.1. Preprocess Genotypic Datasets
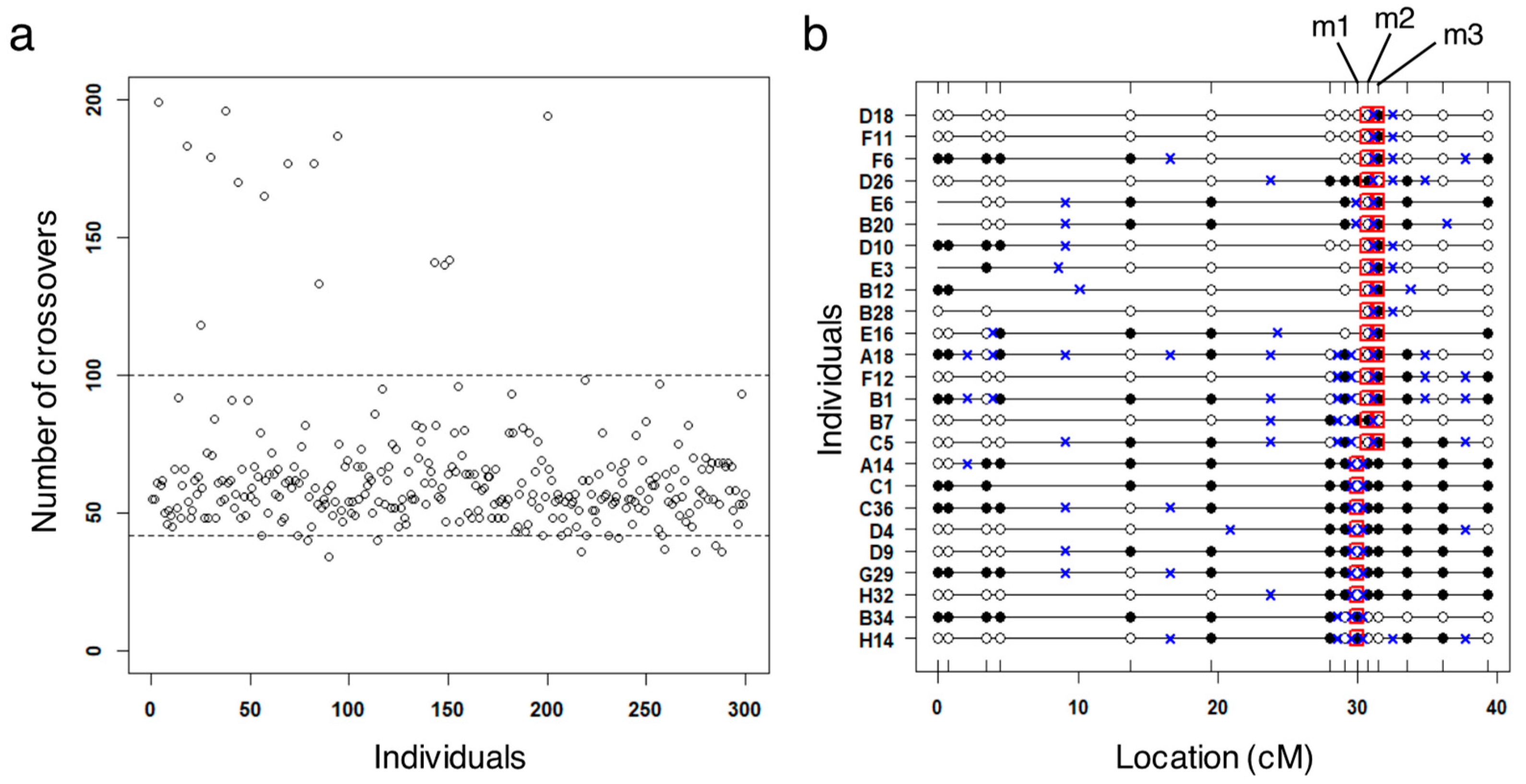
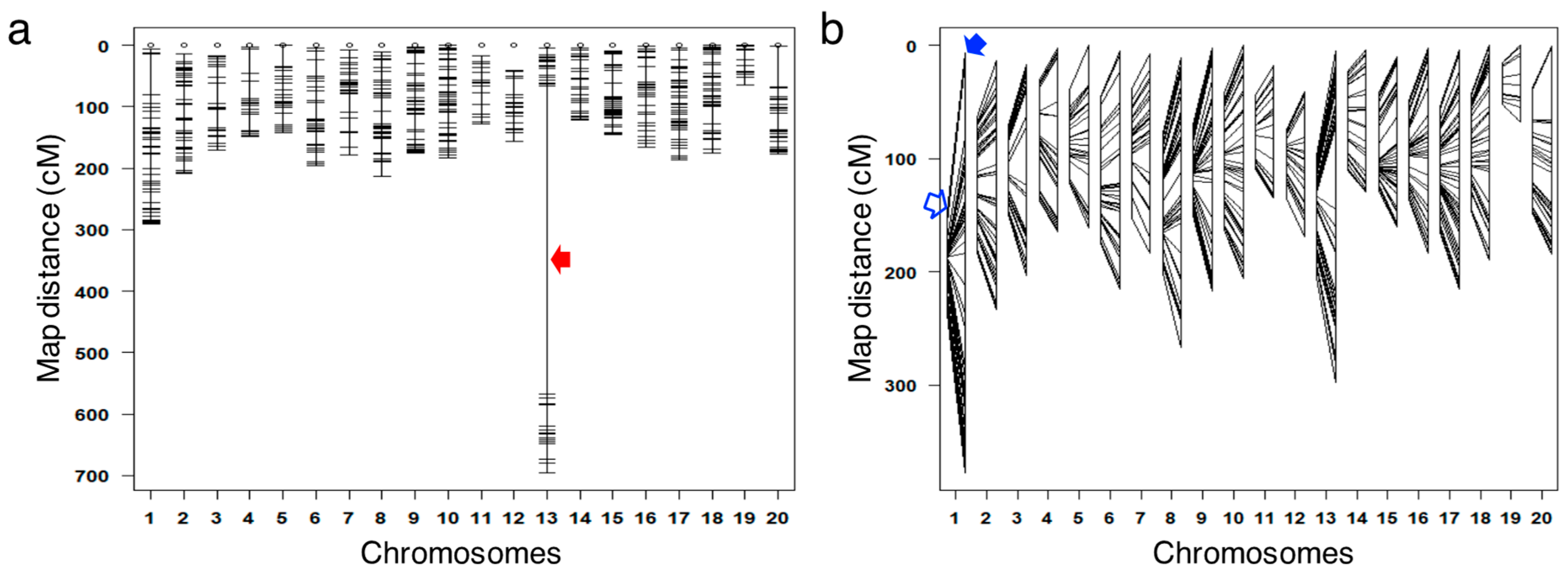
3.2. Identification of Potential False Crossovers
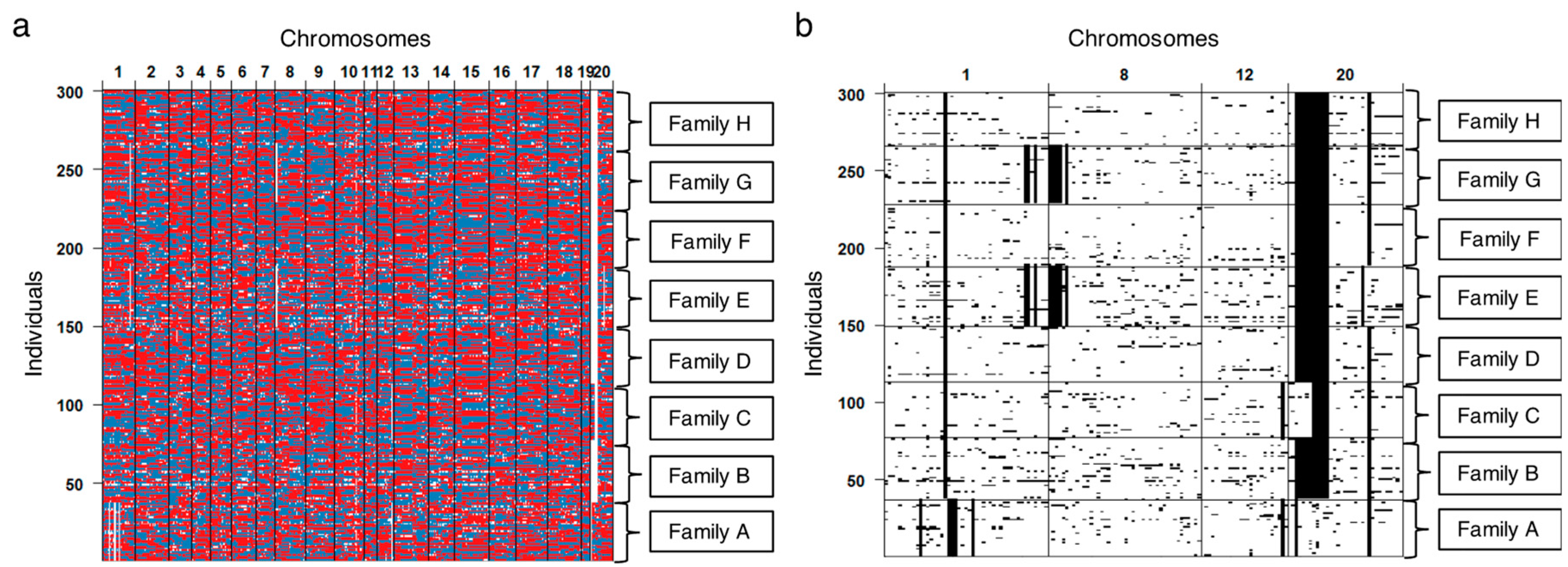
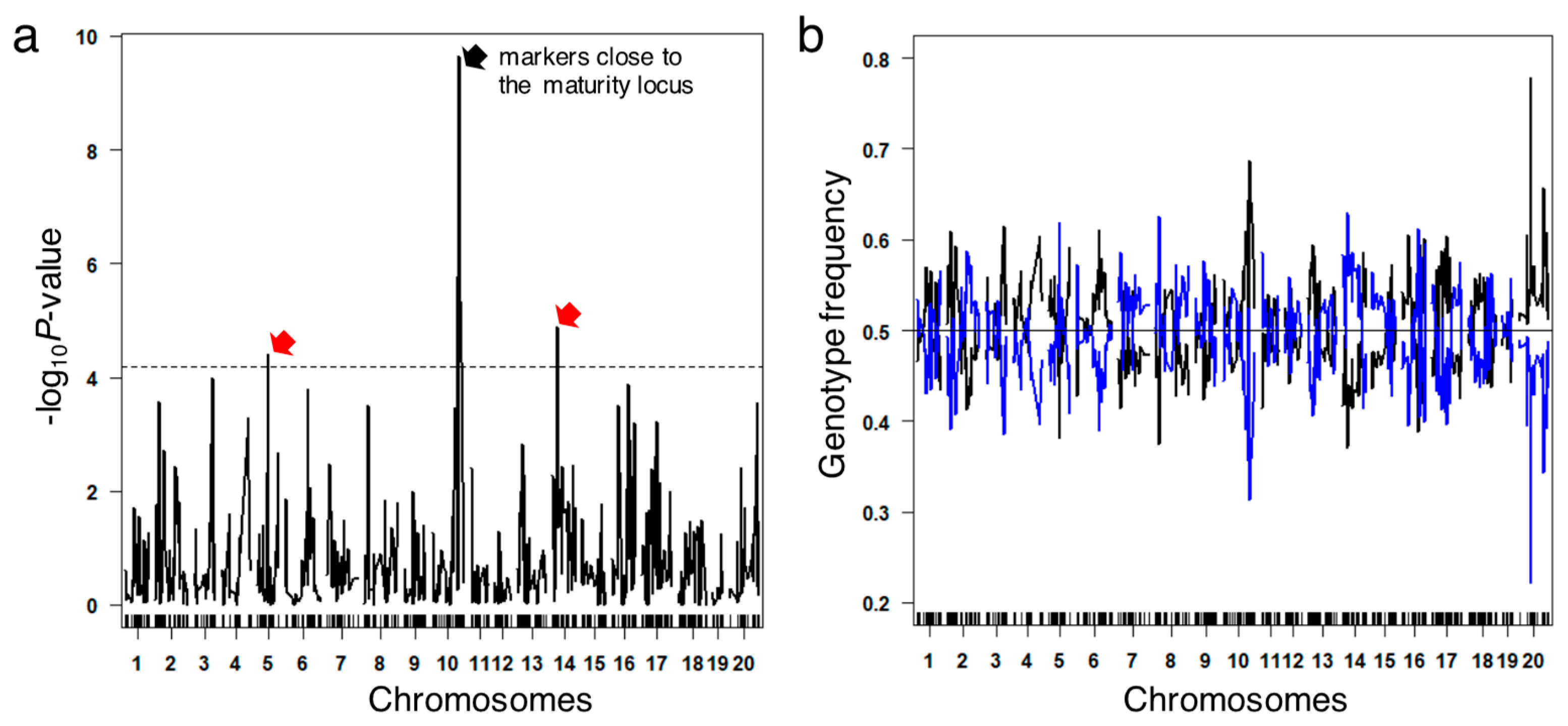
3.3. Validating Effects by Genome Structure
3.4. Correcting the Order of Markers
3.5. Segregation Analysis
3.6. Construction of the Genetic Map
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Yu, J.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Holland, J.B.; McMullen, M.D.; Buckler, E.S. Genetic design and statistical power of nested association mapping in maize. Genetics 2008, 178, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Arends, D.; Prins, P.; Jansen, R.C.; Broman, K.W. R/qtl: High-throughput multiple QTL mapping. Bioinformatics 2010, 26, 2990–2992. [Google Scholar] [CrossRef] [PubMed]
- Broman, K.W.; Wu, H.; Sen, S.; Churchill, G.A. R/qtl: QTL mapping in experimental crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broman, K.W.; Sen, S. A Guide to QTL Mapping with R/qtl; Springer: New York, NY, USA, 2009. [Google Scholar]
- Ihaka, R.; Gentleman, R.R. A language for data analysis and graphics. J. Comp. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Weiss, M.G.; Stevenson, T.M. Registration of soybean varieties, V. Agron. J. 1955, 47, 541–543. [Google Scholar] [CrossRef]
- Johnson, H.W. Registration of soybean varieties, VI. Agron. J. 1958, 50, 690–691. [Google Scholar] [CrossRef]
- Fan, J.G.; Oliphant, A.; Shen, R.; Kermani, B.G.; Garcia, F.; Gunderson, K.L.; Hansen, M.; Steemers, F.; Butler, S.L.; Deloukas, P. Highly parallel SNP genotyping. Cold Spring Harb. Symp. Quant. Biol. 2003, 68, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Akkaya, M.S.; Shoemaker, R.C.; Specht, J.E.; Bhagwat, A.A.; Cregan, P.B. Integration of simple sequence repeat DNA markers into a soybean linkage map. Crop Sci. 1995, 35, 1439–1445. [Google Scholar] [CrossRef]
- Hyten, D.L.; Choi, I.Y.; Song, Q.; Specht, J.E.; Carter, T.E.; Shoemaker, R.C.; Hwang, E.Y.; Matukumalli, L.K.; Cregan, P.B. A high density integrated genetic linkage map of soybean and the development of a 1536 universal soy linkage panel for quantitative trait locus mapping. Crop Sci. 2010, 50, 960–968. [Google Scholar] [CrossRef]
- Palmer, R.G.; Kilen, T.C. Quantitative Genetics. In Soybeans: Improvement, Production, and Uses, 2nd ed.; Wilcox, J.R., Ed.; American Society of Agronomy: Madison, WI, USA, 1987; Volume 5, pp. 135–209. [Google Scholar]
- Kosambi, D.D. The estimation of map distances from recombination values. Ann. Eugen. 1944, 12, 172–175. [Google Scholar] [CrossRef]
- Lander, E.S.; Green, P.; Abrahamson, J.; Barlow, A.; Daly, M.J.; Lincoln, S.E.; Newburg, L. Mapmaker: An interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1987, 1, 174–181. [Google Scholar] [CrossRef]
- Xu, S. Theoretical basis of the Beavis effect. Genetics 2003, 165, 2259–2268. [Google Scholar] [PubMed]
- Eddy, S.R. What is a hidden Markov model? Nat. Biotechnol. 2004, 22, 1315–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, R.L. Two major genes for time of flowering and maturity in soybeans. Crop Sci. 1971, 11, 242–244. [Google Scholar] [CrossRef]
- Darvasi, A.; Weinreb, A.; Minke, V.; Weller, J.I.; Soller, M. Detecting marker-QTL linkage and estimating QTL gene effect and map location using a saturated genetic map. Genetics 1993, 134, 943–951. [Google Scholar] [PubMed]
- Darvasi, A.; Soller, M. A simple method to calculate resolving power and confidence interval of QTL map location. Behav. Genet. 1997, 27, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Basten, C.J.; Zeng, Z.B. Windows QTL Cartographer 2.51; Department of Statistics, North Carolina State University: Raleigh, NC, USA, 2010. [Google Scholar]
- Utz, H.F.; Melchinger, A.E. PLABQTL: A program for composite interval mapping of QTL. J. Quant. Trait Loci. 1996, 2, 1–5. [Google Scholar]
- Yang, J.; Hu, C.; Hu, H.; Yu, R.; Xia, Z.; Ye, X.; Zhu, J. QTLNetwork: Mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 2008, 24, 721–723. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, S.; Lee, T.G. Correcting Pervasive Errors in Genotypic Datasets to Develop Genetic Maps. Agronomy 2019, 9, 196. https://doi.org/10.3390/agronomy9040196
Hwang S, Lee TG. Correcting Pervasive Errors in Genotypic Datasets to Develop Genetic Maps. Agronomy. 2019; 9(4):196. https://doi.org/10.3390/agronomy9040196
Chicago/Turabian StyleHwang, Sadal, and Tong Geon Lee. 2019. "Correcting Pervasive Errors in Genotypic Datasets to Develop Genetic Maps" Agronomy 9, no. 4: 196. https://doi.org/10.3390/agronomy9040196