
Transcriptome Data Analysis Applied to Grapevine Growth Stage Identification
 ,
,  , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phenological Classification
2.2. RNA-Seq Data Processing and Quantification
2.3. Differential Gene Expression Analysis
2.4. Gene Ontology
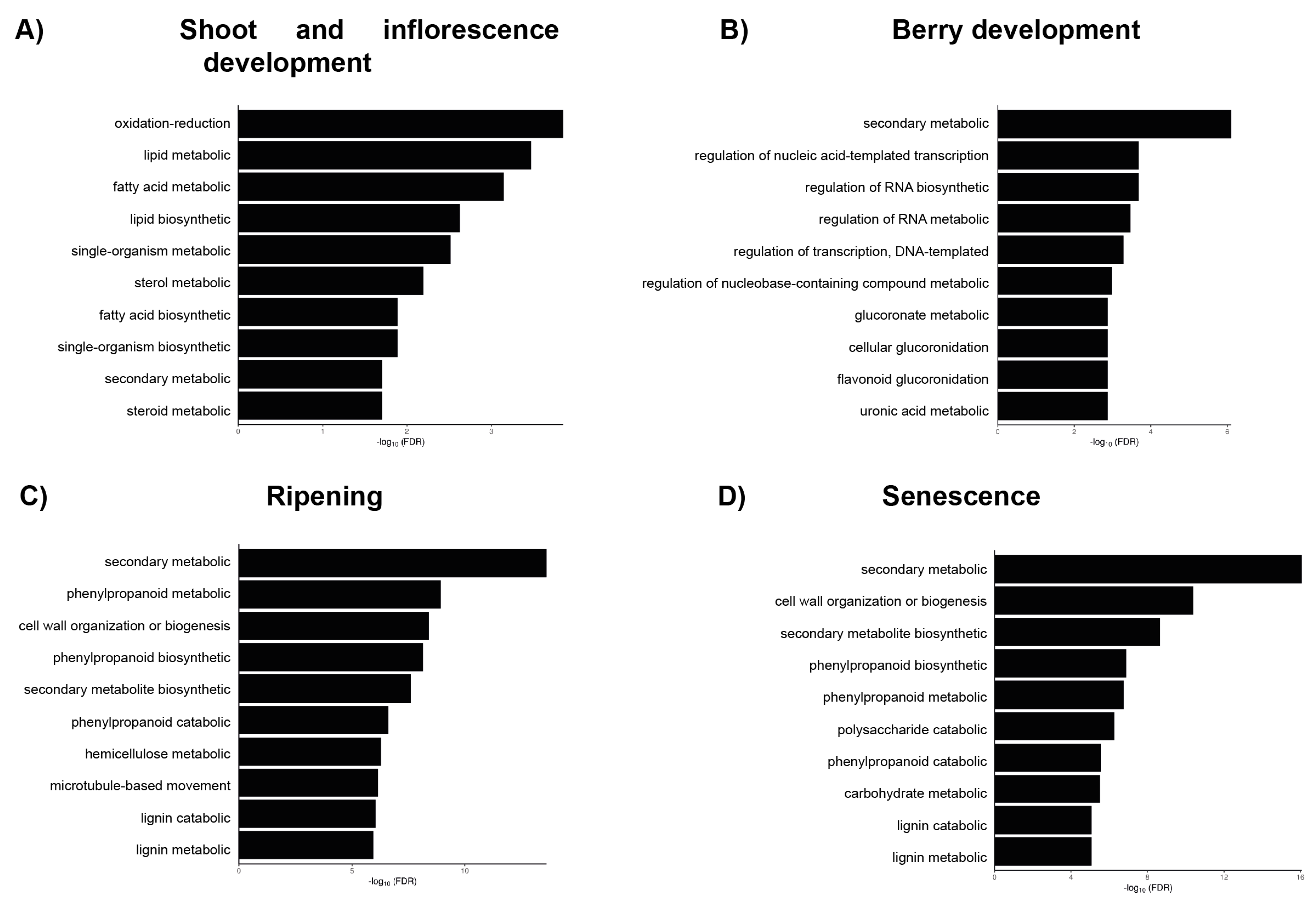
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Alturki, S.M. Phenology and production of Hassaoui grapevines as affected by climate anomalies in Al Ahsa region. Saudi J. Biol. Sci. 2022, 29, 1175–1184. [Google Scholar] [CrossRef]
- Schwartz, M.D. Phenology: An Integrative Environmental Science; Springer Science & Business Media: Berlin, Germany, 2003. [Google Scholar]
- Fenner, M. The phenology of growth and reproduction in plants. Perspect. Plant Ecol. Evol. Syst. 1998, 1, 78–91. [Google Scholar] [CrossRef]
- Hudson, I.L.; Keatley, M.R. Phenological Research: Methods for Environmental and Climate Change Analysis; Springer Science & Business Media: Berlin, Germany, 2009. [Google Scholar]
- Lorenz, D.; Eichhorn, K.; Bleiholder, H.; Klose, R.; Meier, U.; Weber, E. Growth Stages of the Grapevine: Phenological growth stages of the grapevine (Vitis vinifera L. ssp. vinifera)—Codes and descriptions according to the extended BBCH scale. Aust. J. Grape Wine Res. 1995, 1, 100–103. [Google Scholar] [CrossRef]
- Coombe, B. Growth stages of the grapevine: Adoption of a system for identifying grapevine growth stages. Aust. J. Grape Wine Res. 1995, 1, 104–110. [Google Scholar] [CrossRef]
- Sakamoto, T.; Yokozawa, M.; Toritani, H.; Shibayama, M.; Ishitsuka, N.; Ohno, H. A crop phenology detection method using time-series MODIS data. Remote Sens. Environ. 2005, 96, 366–374. [Google Scholar] [CrossRef]
- Xin, J.; Yu, Z.; van Leeuwen, L.; Driessen, P.M. Mapping crop key phenological stages in the North China Plain using NOAA time series images. Int. J. Appl. Earth Obs. Geoinf. 2002, 4, 109–117. [Google Scholar] [CrossRef]
- Dorjsuren, M.; Liou, Y.A.; Cheng, C.H. Time series MODIS and in situ data analysis for Mongolia drought. Remote Sens. 2016, 8, 509. [Google Scholar] [CrossRef]
- Moghimi, A.; Pourreza, A.; Zuniga-Ramirez, G.; Williams, L.E.; Fidelibus, M.W. A novel machine learning approach to estimate grapevine leaf nitrogen concentration using aerial multispectral imagery. Remote Sens. 2020, 12, 3515. [Google Scholar] [CrossRef]
- Albanai, J.A. Building a bridge between MODIS and VIIRS in measuring chlorophyll-a through linear regression: A case study in the Arabian Gulf. Arab. J. Geosci. 2023, 16, 1–9. [Google Scholar] [CrossRef]
- Xu, W.; Wooster, M.J. Sentinel-3 SLSTR active fire (AF) detection and FRP daytime product-Algorithm description and global intercomparison to MODIS, VIIRS and landsat AF data. Sci. Remote Sens. 2023, 7, 100087. [Google Scholar] [CrossRef]
- Parker, A.K.; Hofmann, R.W.; Van Leeuwen, C.; McLachlan, A.R.; Trought, M.C. Manipulating the leaf area to fruit mass ratio alters the synchrony of total soluble solids accumulation and titratable acidity of grape berries. Aust. J. Grape Wine Res. 2015, 21, 266–276. [Google Scholar] [CrossRef]
- Chuine, I.; Régnière, J. Process-based models of phenology for plants and animals. Annu. Rev. Ecol. Evol. Syst. 2017, 48, 159–182. [Google Scholar] [CrossRef]
- Gildor, T.; Smadar, B.T.D.L. Comparative studies of gene expression kinetics: Methodologies and insights on development and evolution. Front. Genet. 2018, 9, 339. [Google Scholar] [CrossRef]
- Tornielli, G.B.; Sandri, M.; Fasoli, M.; Amato, A.; Pezzotti, M.; Zuccolotto, P.; Zenoni, S. A molecular phenology scale of grape berry development. Hortic. Res. 2023, 10, uhad048. [Google Scholar] [CrossRef]
- Leiboff, S.; Hake, S. Reconstructing the transcriptional ontogeny of maize and sorghum supports an inverse hourglass model of inflorescence development. Curr. Biol. 2019, 29, 3410–3419. [Google Scholar] [CrossRef]
- Meyer, D.H.; Schumacher, B. BiT age: A transcriptome-based aging clock near the theoretical limit of accuracy. Aging Cell 2021, 20, e13320. [Google Scholar] [CrossRef]
- Kudoh, H. Molecular phenology in plants: In natura systems biology for the comprehensive understanding of seasonal responses under natural environments. New Phytol. 2016, 210, 399–412. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar]
- Zenoni, S.; Ferrarini, A.; Giacomelli, E.; Xumerle, L.; Fasoli, M.; Malerba, G.; Bellin, D.; Pezzotti, M.; Delledonne, M. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol. 2010, 152, 1787–1795. [Google Scholar] [CrossRef]
- Lijavetzky, D.; Cabezas, J.A.; Ibáñez, A.; Rodríguez, V.; Martínez-Zapater, J.M. High throughput SNP discovery and genotyping in grapevine (Vitis vinifera L.) by combining a re-sequencing approach and SNPlex technology. BMC Genom. 2007, 8, 424. [Google Scholar] [CrossRef]
- Salmaso, M.; Faes, G.; Segala, C.; Stefanini, M.; Salakhutdinov, I.; Zyprian, E.; Toepfer, R.; Grando, M.S.; Velasco, R. Genome diversity and gene haplotypes in the grapevine (Vitis vinifera L.), as revealed by single nucleotide polymorphisms. Mol. Breed. 2005, 14, 385–395. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Troggio, M.; Cartwright, D.A.; Cestaro, A.; Pruss, D.; Pindo, M.; FitzGerald, L.M.; Vezzulli, S.; Reid, J.; et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE 2007, 2, e1326. [Google Scholar] [CrossRef]
- Licausi, F.; Giorgi, F.M.; Zenoni, S.; Osti, F.; Pezzotti, M.; Perata, P. Genomic and transcriptomic analysis of the AP2/ERF superfamily in Vitis vinifera. BMC Genom. 2010, 11, 719. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, M.; Li, X.; Jiu, S.; Wang, C.; Fang, J. Genome-Wide Analysis of the Sucrose Synthase Gene Family in Grape (Vitis vinifera): Structure, Evolution, and Expression Profiles. Genes 2017, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Hirai, M.Y.; Yano, M.; Goodenowe, D.B.; Kanaya, S.; Kimura, T.; Awazuhara, M.; Arita, M.; Fujiwara, T.; Saito, K. Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2004, 101, 10205–10210. [Google Scholar] [CrossRef]
- Knowles, D.G.; Röder, M.; Merkel, A.; Guigó, R. Grape RNA-Seq analysis pipeline environment. Bioinformatics 2013, 29, 614–621. [Google Scholar] [CrossRef]
- Wang, L.; Li, H.; Li, J.; Li, G.; Zahid, M.S.; Li, D.; Ma, C.; Xu, W.; Song, S.; Li, X.; et al. Transcriptome analysis revealed the expression levels of genes related to abscisic acid and auxin biosynthesis in grapevine (Vitis vinifera L.) under root restriction. Front. Plant Sci. 2022, 13, 959693. [Google Scholar] [CrossRef]
- Sweetman, C.; Wong, D.C.; Ford, C.M.; Drew, D.P. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genom. 2012, 13, 691. [Google Scholar] [CrossRef] [PubMed]
- Walls, R.L.; Cooper, L.; Elser, J.; Gandolfo, M.A.; Mungall, C.J.; Smith, B.; Stevenson, D.W.; Jaiswal, P. The plant ontology facilitates comparisons of plant development stages across species. Front. Plant Sci. 2019, 10, 631. [Google Scholar] [CrossRef]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update—Simplifying data submissions. Nucleic Acids Res. 2014, 43, D1113–D1116. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Bioinform 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 18 February 2024).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417. [Google Scholar] [CrossRef] [PubMed]
- Altimiras, F.; Uszczynska-Ratajczak, B.; Camara, F.; Vlasova, A.; Palumbo, E.; Newhouse, S.; Deacon, R.M.; Farias, L.A.; Hurley, M.J.; Loyola, D.E.; et al. Brain transcriptome sequencing of a natural model of Alzheimer’s disease. Front. Aging Neurosci. 2017, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Soneson, C.; Robinson, M.D. Importing transcript abundance datasets with tximport. F1000Research 2005, 1, 5. [Google Scholar]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Bolser, D.; Staines, D.M.; Pritchard, E.; Kersey, P. Ensembl plants: Integrating tools for visualizing, mining, and analyzing plant genomics data. In Plant Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2016; pp. 115–140. [Google Scholar]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016, 45, D1040–D1045. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- Cramer, G.R.; Ghan, R.; Schlauch, K.A.; Tillett, R.L.; Heymann, H.; Ferrarini, A.; Delledonne, M.; Zenoni, S.; Fasoli, M.; Pezzotti, M. Transcriptomic analysis of the late stages of grapevine (Vitis vinifera cv. Cabernet sauvignon) berry ripening reveals significant induction of ethylene signaling and flavor pathways in the skin. BMC Plant Biol. 2014, 14, 370. [Google Scholar] [CrossRef]
- Ruberti, C.; Barizza, E.; Bodner, M.; La Rocca, N.; De Michele, R.; Carimi, F.; Schiavo, F.L.; Zottini, M. Mitochondria change dynamics and morphology during grapevine leaf senescence. PLoS ONE 2014, 9, e102012. [Google Scholar] [CrossRef] [PubMed]
- Castellarin, S.; Bavaresco, L.; Falginella, L.; Gonçalves, M.V.Z.; Di Gaspero, G. Phenolics in grape berry and key antioxidants. In The Biochemistry of the Grape Berry; Bentham Science Publishers: Sharjah, United Arab Emirates, 2012. [Google Scholar]
- Savoi, S.; Wong, D.C.; Arapitsas, P.; Miculan, M.; Bucchetti, B.; Peterlunger, E.; Fait, A.; Mattivi, F.; Castellarin, S.D. Transcriptome and metabolite profiling reveals that prolonged drought modulates the phenylpropanoid and terpenoid pathway in white grapes (Vitis vinifera L.). BMC Plant Biol. 2016, 16, 67. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; Gutierrez, R.L.; Dimopoulos, N.; Gambetta, G.; Castellarin, S. Combined physiological, transcriptome, and cis-regulatory element analyses indicate that key aspects of ripening, metabolism, and transcriptional program in grapes (Vitis vinifera L.) are differentially modulated accordingly to fruit size. BMC Genom. 2016, 17, 416. [Google Scholar] [CrossRef] [PubMed]
- Poret, M.; Chandrasekar, B.; van der Hoorn, R.A.; Avice, J.C. Characterization of senescence-associated protease activities involved in the efficient protein remobilization during leaf senescence of winter oilseed rape. Plant Sci. 2016, 246, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Jackman, R.L.; Stanley, D.W. Perspectives in the textural evaluation of plant foods. Trends Food Sci. Technol. 1995, 6, 187–194. [Google Scholar] [CrossRef]
- Lijavetzky, D.; Carbonell-Bejerano, P.; Grimplet, J.; Bravo, G.; Flores, P.; Fenoll, J.; Hellín, P.; Oliveros, J.C.; Martínez-Zapater, J.M. Berry flesh and skin ripening features in Vitis vinifera as assessed by transcriptional profiling. PLoS ONE 2012, 7, e39547. [Google Scholar] [CrossRef]
- Shangguan, L.; Mu, Q.; Fang, X.; Zhang, K.; Jia, H.; Li, X.; Bao, Y.; Fang, J. RNA-Sequencing Reveals Biological Networks during Table Grapevine (‘Fujiminori’) Fruit Development. PLoS ONE 2017, 12, e0170571. [Google Scholar] [CrossRef]
- Miedes, E.; Zarra, I.; Hoson, T.; Herbers, K.; Sonnewald, U.; Lorences, E. Xyloglucan endotransglucosylase and cell wall extensibility. J. Plant Physiol. 2011, 168, 196–203. [Google Scholar] [CrossRef]
- García, J.; Pope, C.; Altimiras, F. A Distributed-Means Segmentation Algorithm Applied to Lobesia botrana Recognition. Complexity 2017, 2017, 5137317. [Google Scholar] [CrossRef]
- Thiéry, D.; Monceau, K.; Moreau, J. Different emergence phenology of European grapevine moth (Lobesia botrana, Lepidoptera: Tortricidae) on six varieties of grapes. Bull. Entomol. Res. 2014, 104, 277–287. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Major Stage | Detailed Stage | Baillod and Baggiolini | Extended BBCH | Eichhorn and Lorenz | Modified E-L |
|---|---|---|---|---|---|
| Shoot and inflorescence development | A | 0 | 1 | 1 | |
| 1 | 2 | 2 | |||
| B | 3 | ||||
| 5 | 3 | 3 | |||
| Budburst | C | 7 | 5 | 4 | |
| D | 9 | 5 | |||
| E | 11 | 7 | 7 | ||
| 12 | 9 | 9 | |||
| 13 | |||||
| 14 | 11 | ||||
| Shoots 10 cm | F | 15, 53 | 12 | 12 | |
| 16 | 13 | ||||
| 14 | |||||
| G | 55 | 15 | 15 | ||
| 19 | 16 | ||||
| H | 57 | 17 | 17 | ||
| 18 | |||||
| Flowering | Flowering begins | 60 | 19 | 19 | |
| 61 | 20 | ||||
| 63 | 21 | 21 | |||
| Full bloom | I | 65 | 23 | 23 | |
| 68 | 25 | 25 | |||
| 69 | 26 | 26 | |||
| Berry development | Setting | J | 71 | 27 | 27 |
| 73 | 29 | 29 | |||
| Berries pea size | K | 75 | 31 | 31 | |
| 77 | 33 | 32 | |||
| L | 79 | 33 | |||
| Ripening | 34 | ||||
| Veraison | M | 81 | 35 | 35 | |
| 36 | |||||
| 37 | |||||
| Harvest | N | 89 | 38 | 38 | |
| 39 | |||||
| Senescence | O | 91 | 41 | 41 | |
| 92 | |||||
| P | 93 | 43 | 43 | ||
| 95 | |||||
| 97 | 47 | 47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altimiras, F.; Pavéz, L.; Pourreza, A.; Yañez, O.; González-Rodríguez, L.; García, J.; Galaz, C.; Leiva-Araos, A.; Allende-Cid, H. Transcriptome Data Analysis Applied to Grapevine Growth Stage Identification. Agronomy 2024, 14, 613. https://doi.org/10.3390/agronomy14030613
Altimiras F, Pavéz L, Pourreza A, Yañez O, González-Rodríguez L, García J, Galaz C, Leiva-Araos A, Allende-Cid H. Transcriptome Data Analysis Applied to Grapevine Growth Stage Identification. Agronomy. 2024; 14(3):613. https://doi.org/10.3390/agronomy14030613
Chicago/Turabian StyleAltimiras, Francisco, Leonardo Pavéz, Alireza Pourreza, Osvaldo Yañez, Lisdelys González-Rodríguez, José García, Claudio Galaz, Andrés Leiva-Araos, and Héctor Allende-Cid. 2024. "Transcriptome Data Analysis Applied to Grapevine Growth Stage Identification" Agronomy 14, no. 3: 613. https://doi.org/10.3390/agronomy14030613