
Genome-Wide Association Studies of Plant Architecture-Related Traits in the Chinese Soybean Mini Core Collection

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Field Trials and Trait Measurement
2.3. Phenotypic Data Analysis
2.4. Genotyping and Linkage Disequilibrium (LD) Analysis
2.5. Population Structure and Kinship
2.6. Genome-Wide Association Analysis
2.7. Prediction and Annotation of Candidate Genes
3. Results
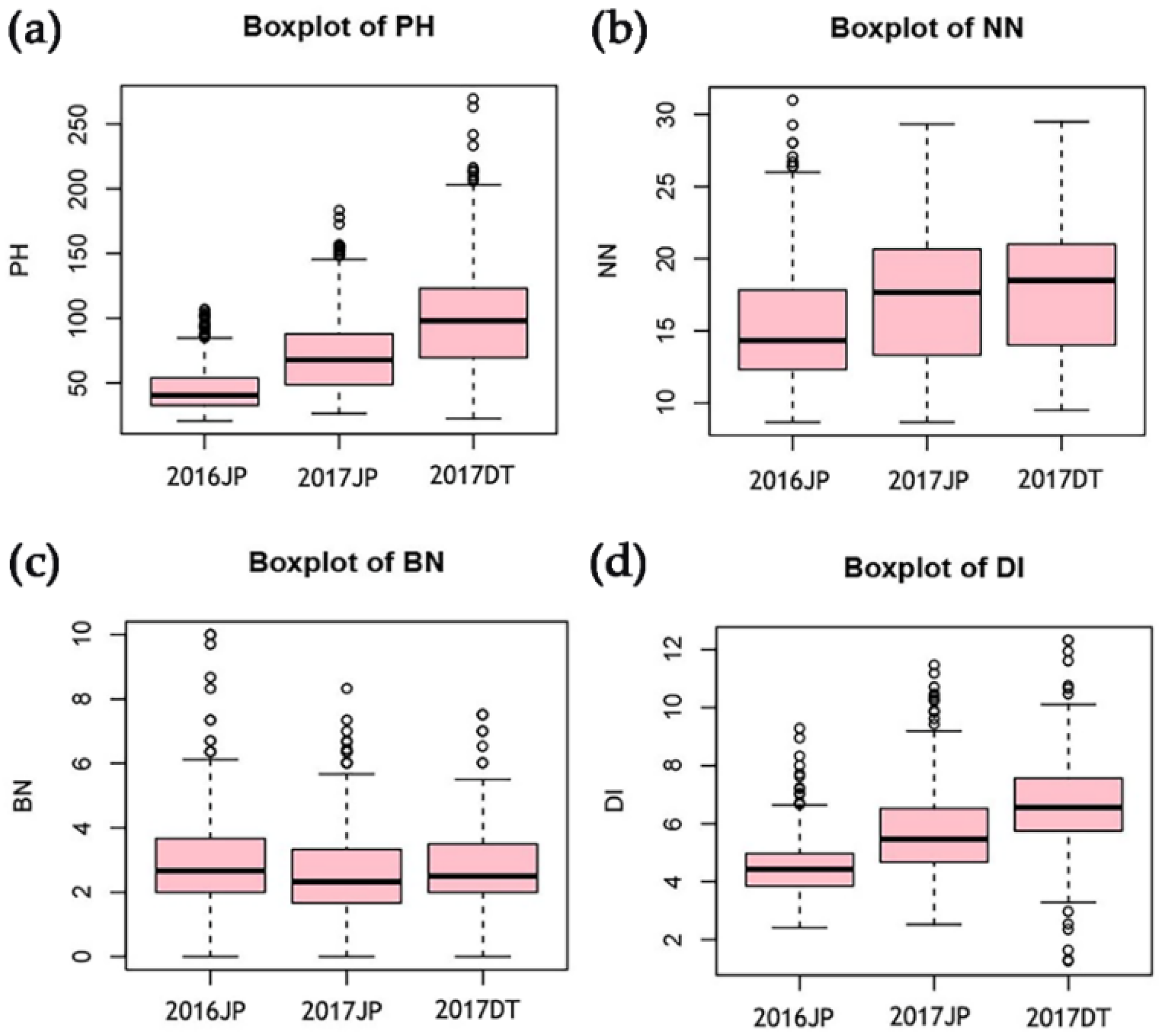
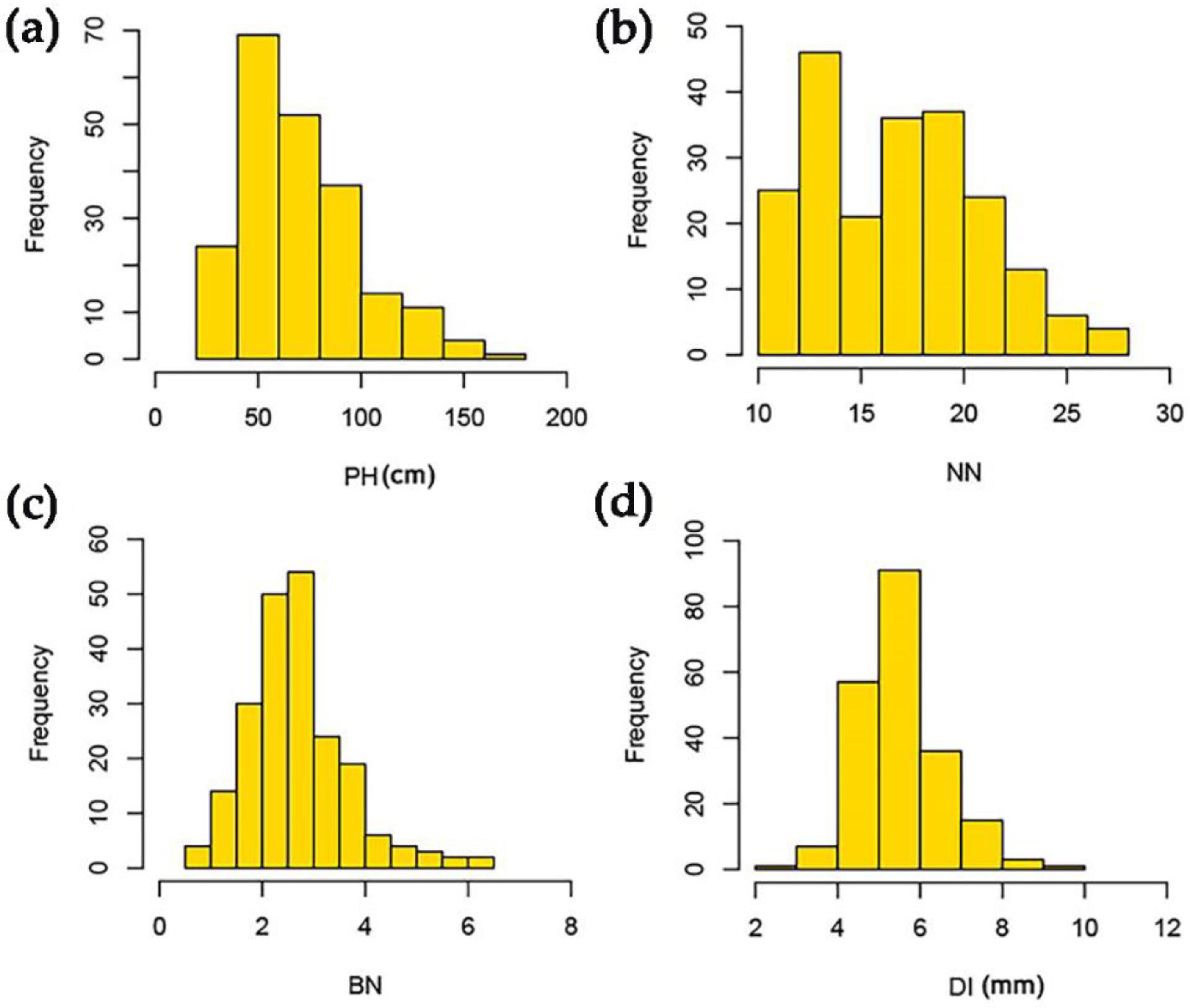
3.1. Phenotypic Variation in the Four Plant Architecture-Related Traits
3.2. Genetic Diversity, Linkage Disequilibrium, and Population Structure
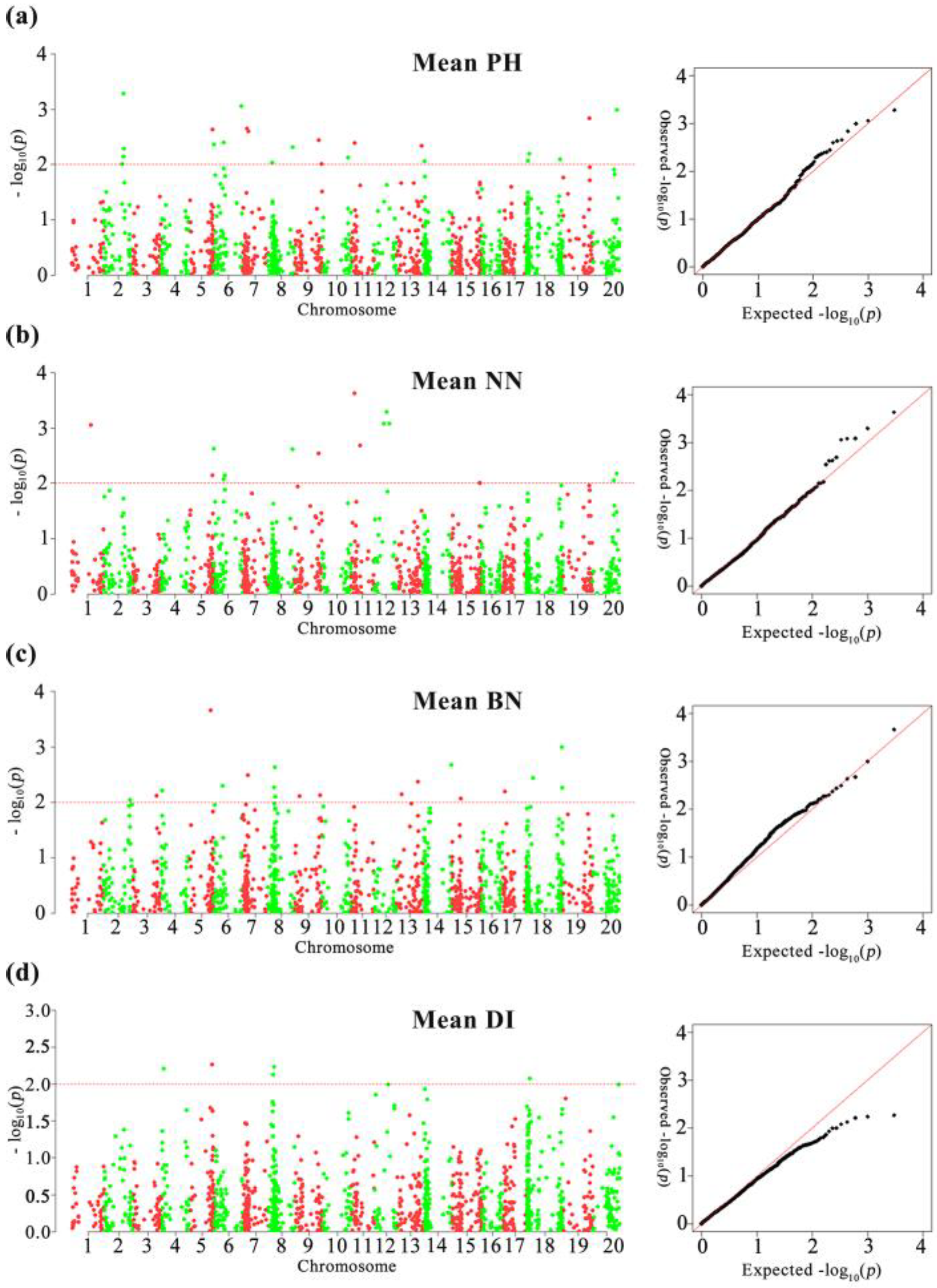
3.3. GWAS of the Four Plant Architecture-Related Traits
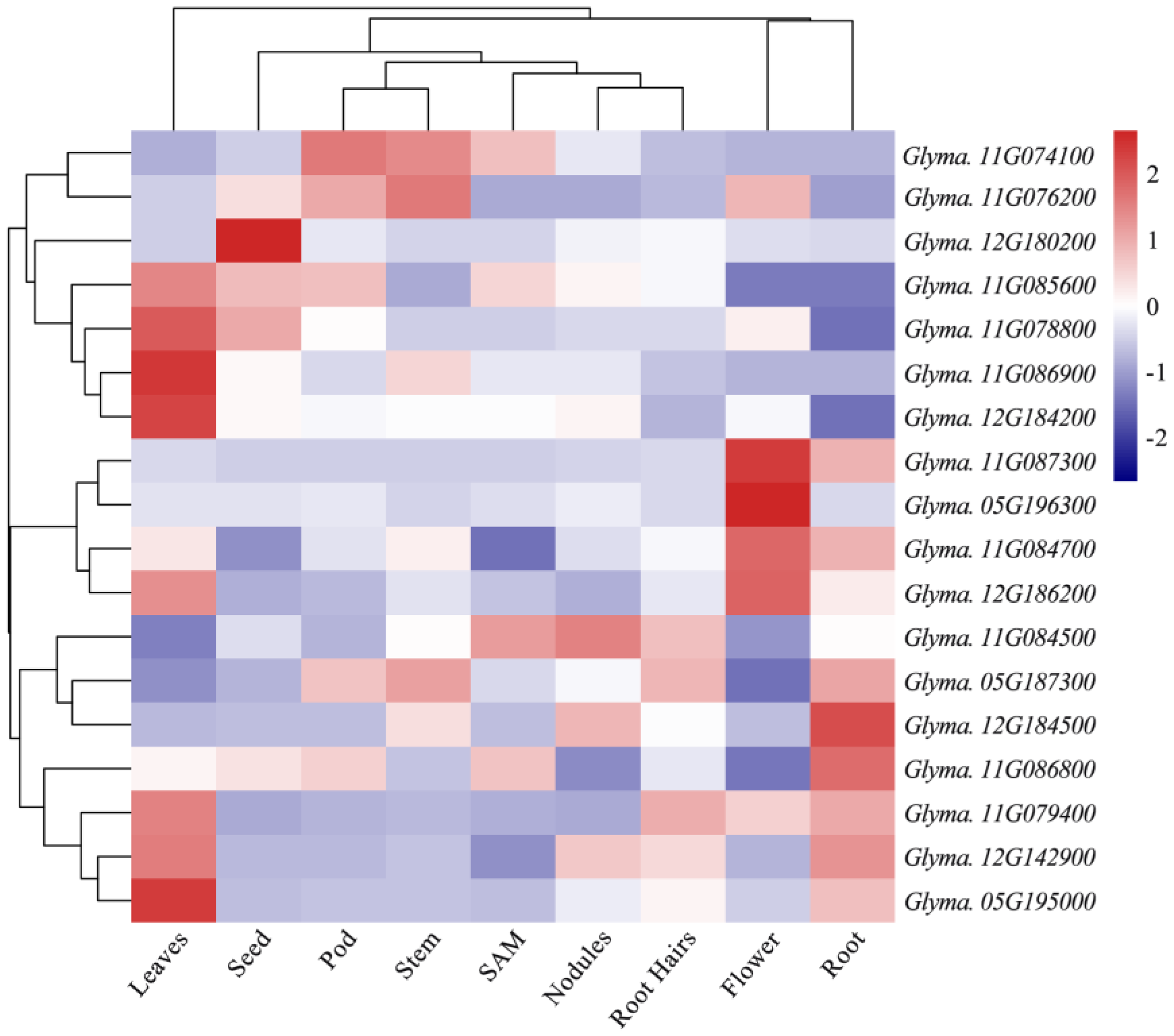
3.4. Functional Annotation of Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Qin, J.; Zhang, J.A.; Liu, D.; Yin, C.C.; Wang, F.M.; Chen, P.Y.; Chen, H.; Ma, J.B.; Zhang, B.; Xu, J.; et al. iTRAQ-based analysis of developmental dynamics in the soybean leaf proteome reveals pathways associated with leaf photosynthetic rate. Mol. Genet. Genomics 2016, 291, 1595–1605. [Google Scholar] [CrossRef]
- Kim, S.W.; Less, J.F.; Wang, L.; Yan, T.H.; Kiron, V.; Kaushik, S.J.; Lei, X.G. Meeting Global Feed Protein Demand: Challenge, Opportunity, and Strategy. Annu. Rev. Anim. Biosci. 2019, 7, 221–243. [Google Scholar] [CrossRef]
- Donald, C.M. The breeding of crop ideotypes. Euphytica 1968, 17, 385–403. [Google Scholar] [CrossRef]
- Huyghe, C. Genetics and genetic modifications of plant architecture in grain legumes: A review. Agronomie 1998, 18, 383–411. [Google Scholar] [CrossRef]
- Sarlikioti, V.; de Visser, P.H.B.; Buck-Sorlin, G.H.; Marcelis, L.F.M. How plant architecture affects light absorption and photosynthesis in tomato: Towards an ideotype for plant architecture using a functional–structural plant model. Ann. Bot. 2011, 108, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.C.; Xu, Y.C.; Li, K.; Peng, Y.; Zhan, W.; Li, W.Q.; Li, L.; Yan, J.B. The Genetic Basis of Plant Architecture in 10 Maize Recombinant Inbred Line Populations. Plant Physiol. 2017, 175, 858–873. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.L.; Ding, W.T.; Xue, D.; Li, X.N.; Zhou, Y.; Shen, J.C.; Feng, J.Y.; Guo, N.; Qiu, L.J.; Xing, H.; et al. Genome-wide association studies of plant architecture-related traits and 100-seed weight in soybean landraces. BMC Genom. Data 2021, 22, 1–14. [Google Scholar] [CrossRef]
- Hill, W.G. Understanding and using quantitative genetic variation. Philos. Trans. R. Soc. B 2010, 365, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Liu, Z.Z.; Zhang, J.; Liu, S.Y.; Qu, J.; Guan, S.Y.; Pan, L.D.; Wang, D.; Liu, J.W.; Wang, P.W. Analysis of quantitative trait loci for main plant traits in soybean. GMR Genet. Mol. Res. 2015, 14, 6101–6109. [Google Scholar] [CrossRef]
- Zhang, H.R.; Hao, D.R.; Sitoe, H.M.; Yin, Z.T.; Hu, Z.B.; Zhang, G.Z.; Yu, D.Y. Genetic dissection of the relationship between plant architecture and yield component traits in soybean (Glycine max) by association analysis across multiple environments. Plant Breed. 2015, 134, 564–572. [Google Scholar] [CrossRef]
- Gupta, P.K.; Rustgi, S.; Kulwal, P.L. Linkage disequilibrium and association studies in higher plants: Present status and future prospects. Plant Mol. Biol. 2005, 57, 461–485. [Google Scholar] [CrossRef]
- Sehgal, D.; Singh, R.; Rajpal, V.R. Quantitative trait loci mapping in plants: Concepts and approaches. In Molecular Breeding for Sustainable Crop Improvement; Rajpal, V.R., Rao, S.R., Raina, S.N., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 2, pp. 31–59. [Google Scholar]
- Wang, Z.; Tian, Z.X. Genomics progress will facilitate molecular breeding in soybean. Sci. China Life Sci. 2015, 58, 813–815. [Google Scholar] [CrossRef] [Green Version]
- Flint-Garcia, S.A.; Thuillet, A.C.; Yu, J.M.; Pressoir, G.; Romero, S.M.; Mitchell, S.E.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Maize association population: A high-resolution platform for quantitative trait locus dissection. Plant J. 2005, 44, 1054–1064. [Google Scholar] [CrossRef]
- Atwell, S.; Huang, Y.S.; Vilhjalmsson, B.J.; Willems, G.; Horton, M.; Li, Y.; Meng, D.Z.; Platt, A.; Tarone, A.M.; Hu, T.T.; et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Y.Q.; Xie, W.B.; Gong, L.; Lu, K.; Wang, W.S.; Li, Y.; Liu, X.Q.; Zhang, H.Y.; Dong, H.X.; et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef]
- Guo, Z.F.; Chen, D.J.; Alqudah, A.M.; Roder, M.S.; Ganal, M.W.; Schnurbusch, T. Genome-wide association analyses of 54 traits identified multiple loci for the determination of floret fertility in wheat. New Phytol. 2017, 214, 257–270. [Google Scholar] [CrossRef]
- Fang, C.; Ma, Y.M.; Wu, S.W.; Liu, Z.; Wang, Z.; Yang, R.; Hu, G.H.; Zhou, Z.K.; Yu, H.; Zhang, M.; et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 2017, 18, 161. [Google Scholar] [CrossRef]
- Zhang, J.P.; Song, Q.J.; Cregan, P.B.; Nelson, R.L.; Wang, X.Z.; Wu, J.X.; Jiang, G.L. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genom. 2015, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.G.; Guo, C.Y.; Sun, F.L.; Zhang, J.S.; Wang, Z.L.; Kong, J.J.; He, Q.Y.; Sharmin, R.A.; Zhao, T.J. Genome-Wide Association Studies for Dynamic Plant Height and Number of Nodes on the Main Stem in Summer Sowing Soybeans. Front. Recent Dev. Plant Sci. 2018, 9, 1184. [Google Scholar] [CrossRef] [Green Version]
- Shim, S.; Ha, J.; Kim, M.Y.; Choi, M.S.; Kang, S.T.; Jeong, S.C.; Moon, J.K.; Lee, S.H. GmBRC1 is a Candidate Gene for Branching in Soybean (Glycine max (L.) Merrill). Int. J. Mol. Sci. 2019, 20, 135. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.M.; Fang, Y.H.; Li, X.Y.; Zeng, K.; Chen, H.F.; Zhang, H.B.; Yang, H.L.; Cao, D.; Hao, Q.N.; Yuan, S.L.; et al. Identification of soybean drought-tolerant genotypes and loci correlated with agronomic traits contributes new candidate genes for breeding. Plant Mol. Biol. 2020, 102, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.M.; Wang, Y.P.; Ren, H.X.; Du, W.G.; Yang, X.Y.; Wang, D.L.; Cheng, Y.X.; Zhao, J.M.; Gai, J.Y. Exploring the QTL–allele constitution of main stem node number and its differentiation among maturity groups in a Northeast China soybean population. Crop Sci. 2020, 60, 1223–1238. [Google Scholar] [CrossRef]
- Fahim, A.M.; Pan, L.Y.; Li, C.Y.; He, J.B.; Xing, G.N.; Wang, W.B.; Zhang, F.K.; Li, N.; Gai, J.Y. QTL-allele system of main stem node number in recombinant inbred lines of soybean (Glycine max) using association versus linkage mapping. Plant Breed. 2021, 140, 870–883. [Google Scholar] [CrossRef]
- Huang, J.; Guo, N.; Li, Y.H.; Sun, J.T.; Hu, G.J.; Zhang, H.P.; Li, Y.F.; Zhang, X.; Zhao, J.M.; Xing, H.; et al. Phenotypic evaluation and genetic dissection of resistance to Phytophthora sojae in the Chinese soybean mini core collection. BMC Genet. 2016, 17, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhao, J.M.; Bu, Y.P.; Xue, D.; Liu, Z.X.; Li, X.N.; Huang, J.; Guo, N.; Wang, H.T.; Xing, H.; et al. Genome-Wide Association Studies of Soybean Seed Hardness in the Chinese Mini Core Collection. Plant Mol. Biol. Rep. 2018, 36, 605–617. [Google Scholar] [CrossRef]
- Li, X.N.; Zhou, Y.; Bu, Y.P.; Wang, X.F.; Zhang, Y.M.; Guo, N.; Zhao, J.M.; Xing, H. Genome-wide association analysis for yield-related traits at the R6 stage in a Chinese soybean mini core collection. Genes Genom. 2021, 43, 897–912. [Google Scholar] [CrossRef]
- Chen, Q.S.; Zhang, Z.C.; Liu, C.Y.; Xin, D.W.; Qiu, H.M.; Shan, D.P.; Shan, C.Y.; Hu, G.H. QTL Analysis of Major Agronomic Traits in Soybean. Agric. Sci. China 2007, 6, 399–405. [Google Scholar] [CrossRef]
- SAS Institute. Base SAS 9.4 Procedures Guide: Statistical Procedures; SAS Institute: Cary, NC, USA, 2013. [Google Scholar]
- Nyquist, W.E.; Baker, R.J. Estimation of heritability and prediction of selection response in plant populations. Crit. Rev. Plant Sci. 1991, 10, 235–322. [Google Scholar] [CrossRef]
- Li, Y.H.; Li, W.; Zhang, C.; Yang, L.A.; Chang, R.Z.; Gaut, B.S.; Qiu, L.J. Genetic diversity in domesticated soybean (Glycine max) and its wild progenitor (Glycine soja) for simple sequence repeat and single-nucleotide polymorphism loci. New Phytol. 2010, 188, 242–253. [Google Scholar] [CrossRef]
- Li, Y.H.; Reif, J.C.; Ma, Y.S.; Hong, H.L.; Liu, Z.X.; Chang, R.Z.; Qiu, L.J. Targeted association mapping demonstrating the complex molecular genetics of fatty acid formation in soybean. BMC Genom. 2015, 16, 841. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Kan, G.Z.; Hu, Z.B.; Cheng, H.; Zhang, Y.; Wang, Q.; Wang, H.; Yang, Y.M.; Li, H.Y.; Hao, D.R.; et al. Use of single nucleotide polymorphisms and haplotypes to identify genomic regions associated with protein content and water-soluble protein content in soybean. Theor. Appl. Genet. 2014, 127, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. bioRxiv 2014. in preprint. [Google Scholar]
- Cichy, K.A.; Wiesinger, J.A.; Mendoza, F.A. Genetic diversity and genome-wide association analysis of cooking time in dry bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 2015, 128, 1555–1567. [Google Scholar] [CrossRef]
- Liu, K.J.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.E.; Orf, J.H.; Liu, L.J.; Dong, Z.M.; Rajcan, I. Genetic basis of soybean adaptation to North American vs. Asian mega-environments in two independent populations from Canadian × Chinese crosses. Theor. Appl. Genet. 2013, 126, 1809–1823. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.S.; Li, W.B.; Zhang, Z.C.; Chen, Q.S.; Ning, H.L.; Qiu, L.J.; Sun, G.L. Quantitative trait loci analysis for the developmental behavior of Soybean (Glycine max L. Merr.). Theor. Appl. Genet. 2006, 112, 665–673. [Google Scholar] [CrossRef]
- Kim, K.S.; Diers, B.W.; Hyten, D.L.; Mian, M.A.R.; Shannon, J.G.; Nelson, R.L. Identification of positive yield QTL alleles from exotic soybean germplasm in two backcross populations. Theor. Appl. Genet. 2012, 125, 1353–1369. [Google Scholar] [CrossRef] [Green Version]
- Guzman, P.S.; Diers, B.W.; Neece, D.J.; Martin, S.K.S.; Leroy, A.R.; Grau, C.R.; Hughes, T.J.; Nelson, R.L. QTL Associated with Yield in Three Backcross-Derived Populations of Soybean. Crop Sci. 2007, 47, 111–122. [Google Scholar] [CrossRef]
- Orf, J.H.; Chase, K.; Jarvik, T.; Mansur, L.M.; Cregan, P.B.; Adler, F.R.; Lark, K.G. Genetics of Soybean Agronomic Traits: I. Comparison of Three Related Recombinant Inbred Populations. Crop Sci. 1999, 39, 1642–1651. [Google Scholar] [CrossRef] [Green Version]
- Kabelka, E.A.; Diers, B.W.; Fehr, W.R.; LeRoy, A.R.; Baianu, I.C.; You, T.; Neece, D.J.; Nelson, R.L. Putative Alleles for Increased Yield from Soybean Plant Introductions. Crop Sci. 2004, 44, 784–791. [Google Scholar] [CrossRef]
- Palomeque, L.; Liu, L.J.; Li, W.B.; Hedges, B.; Cober, E.R.; Rajcan, I. QTL in mega-environments: II. Agronomic trait QTL co-localized with seed yield QTL detected in a population derived from a cross of high-yielding adapted × high-yielding exotic soybean lines. Theor. Appl. Genet. 2009, 119, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Gai, J.Y.; Wang, Y.J.; Wu, X.L.; Chen, S.Y. A comparative study on segregation analysis and QTL mapping of quantitative traits in plants—with a case in soybean. Front. Agric. China 2007, 1, 1–7. [Google Scholar] [CrossRef]
- Liu, W.X.; Kim, M.Y.; Van, K.; Lee, Y.-H.; Li, H.; Liu, X.H.; Lee, S.H. QTL Identification of Yield-Related Traits and their Association with Flowering and Maturity in Soybean. J. Crop Sci. Biotechnol. 2011, 14, 65–70. [Google Scholar] [CrossRef]
- Wang, D.; Graef, G.L.; Procopiuk, A.M.; Diers, B.W. Identification of putative QTL that underlie yield in interspecific soybean backcross populations. Theor. Appl. Genet. 2004, 108, 458–467. [Google Scholar] [CrossRef]
- Reinprecht, Y.; Poysa, V.W.; Yu, K.F.; Rajcan, I.; Ablett, G.R.; Pauls, K.P. Seed and agronomic QTL in low linolenic acid, lipoxygenase-free soybean (Glycine max (L.) Merrill) germplasm. Genome 2006, 49, 1510–1527. [Google Scholar] [CrossRef] [PubMed]
- Mansur, L.M.; Lark, K.G.; Kross, H.; Oliveira, A. Interval mapping of quantitative trait loci for reproductive, morphological, and seed traits of soybean (Glycine max L.). Theor. Appl. Genet. 1993, 86, 907–913. [Google Scholar] [CrossRef]
- Lee, S.H.; Bailey, M.A.; Mian, M.A.R.; Shipe, E.R.; Ashley, D.A.; Parrott, W.A.; Hussey, R.S.; Boerma, H.R. Identification of quantitative trait loci for plant height, lodging, and maturity in a soybean population segregating for growth habit. Theor. Appl. Genet. 1996, 92, 516–523. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.L.; Wang, H.W.; Liu, S.X.; Ferjani, A.; Li, J.S.; Yan, J.B.; Yang, X.H.; Qin, F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat. Genet. 2016, 48, 1233–1241. [Google Scholar] [CrossRef]
- Wang, M.; Li, W.Z.; Fang, C.; Xu, F.; Liu, Y.C.; Wang, Z.; Yang, R.; Zhang, M.; Liu, S.L.; Lu, S.J.; et al. Parallel selection on a dormancy gene during domestication of crops from multiple families. Nat. Genet. 2018, 50, 1435–1441. [Google Scholar] [CrossRef]
- Li, N.; Lin, B.; Wang, H.; Li, X.M.; Yang, F.F.; Ding, X.H.; Yan, J.B.; Chu, Z.H. Natural variation in ZmFBL41 confers banded leaf and sheath blight resistance in maize. Nat. Genet. 2019, 51, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.J.; Dong, L.D.; Fang, C.; Liu, S.L.; Kong, L.P.; Cheng, Q.; Chen, L.Y.; Su, T.; Nan, H.Y.; Zhang, D.; et al. Stepwise selection on homeologous PRR genes controlling flowering and maturity during soybean domestication. Nat. Genet. 2020, 52, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Frankel, O.H.; Brown, A.H.D. Current plant genetic resources—A critical appraisal. In Genetics: New Frontiers; Oxford & IBH Publishing: New Delhi, India, 1984; Volume IV, pp. 1–11. [Google Scholar]
- Brown, A.H.D. Core collections: A practical approach to genetic resources management. Genome 1989, 31, 818–824. [Google Scholar] [CrossRef]
- Zhang, P.; Zhong, K.Z.; Shahid, M.Q.; Tong, H.H. Association Analysis in Rice: From Application to Utilization. Front. Recent Dev. Plant Sci. 2016, 7, 1202. [Google Scholar] [CrossRef] [Green Version]
- Schlappi, M.R.; Jackson, A.K.; Eizenga, G.C.; Wang, A.J.; Chu, C.C.; Shi, Y.; Shimoyama, N.; Boykin, D.L. Assessment of Five Chilling Tolerance Traits and GWAS Mapping in Rice Using the USDA Mini-Core Collection. Front. Plant Sci. 2017, 8, 957. [Google Scholar] [CrossRef] [Green Version]
- Oz, M.; Karasu, A.; Goksoy, A.T.; Turan, Z.M. Interrelationships of Agronomical Characteristics in Soybean (Glycine max) Grown in Different Environments. Int. J. Agric. Biol. 2009, 11, 85–88. [Google Scholar]
- Asanome, N.; Ikeda, T. Effect of Branch Direction’s Arrangement on Soybean Yield and Yield Components. J. Agron. Crop Sci. 1998, 181, 95–102. [Google Scholar] [CrossRef]
- Guillaumot, D.; Lopez-Obando, M.; Baudry, K.; Avon, A.; Rigaill, G.; de Longevialle, A.F.; Broche, B.; Takenaka, M.; Berthome, R.; De Jaeger, G.; et al. Two interacting PPR proteins are major Arabidopsis editing factors in plastid and mitochondria. Proc. Natl. Acad. Sci. USA 2017, 114, 8877–8882. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.Y.; Liu, M.; Xiao, Z.Y.; Ren, X.M.; Zhao, H.L.; Gong, D.M.; Liang, K.; Tan, Z.D.; Shao, Y.Q.; Qiu, F.Z. ZmSMK9, a pentatricopeptide repeat protein, is involved in the cis-splicing of nad5, kernel development and plant architecture in maize. Plant Sci. 2019, 288, 110205. [Google Scholar] [CrossRef]
- Schuler, M.A.; Werck-Reichhart, D. Functional Genomics of P450S. Annu. Rev. Plant Biol. 2003, 54, 629–667. [Google Scholar] [CrossRef]
- Yang, C.M.; Yan, J.; Jiang, S.Q.; Li, X.; Min, H.W.; Wang, X.F.; Hao, D.Y. Resequencing 250 soybean accessions: New insights into genes associated with agronomic traits and genetic networks. bioRxiv 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Hagen, G.; Guilfoyle, T. Auxin-responsive gene expression: Genes, promoters and regulatory factors. Plant Mol. Biol. 2002, 49, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Bi, Y.M.; Zhu, T.; Rothstein, S.J. SAUR39, a Small Auxin-Up RNA Gene, Acts as a Negative Regulator of Auxin Synthesis and Transport in Rice. Plant Physiol. 2009, 151, 691–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.Z.; Jin, S.X.; Liu, X.Y.; Tan, J.F.; Yang, X.Y.; Zhang, X.L. Overexpression of Arabidopsis cyclin D2;1 in cotton results in leaf curling and other plant architectural modifications. Plant Cell, Tissue Organ Cult. 2012, 110, 261–273. [Google Scholar] [CrossRef]
- Raina, M.; Kumar, A.; Yadav, N.; Kumari, S.; Yusuf, M.A.; Mustafiz, A.; Kumar, D. StCaM2, a calcium binding protein, alleviates negative effects of salinity and drought stress in tobacco. Plant Mol. Biol. 2021, 106, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, R.; Werr, W. Pattern Formation in the Monocot Embryo as Revealed by NAM and CUC3 Orthologues from Zea mays L. Plant Mol. Biol. 2005, 58, 669–685. [Google Scholar] [CrossRef]
- Zhong, R.Q.; Richardson, E.A.; Ye, Z.H. Two NAC domain transcription factors, SND1 and NST1, function redundantly in regulation of secondary wall synthesis in fibers of Arabidopsis. Planta 2007, 225, 1603–1611. [Google Scholar] [CrossRef]
- Jiang, D.G.; Chen, W.T.; Dong, J.F.; Li, J.; Yang, F.; Wu, Z.C.; Zhou, H.; Wang, W.S.; Zhuang, C.X. Overexpression of miR164b-resistant OsNAC2 improves plant architecture and grain yield in rice. J. Exp. Bot. 2018, 69, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Ding, C.H.; Lin, X.H.; Zuo, Y.; Yu, Z.L.; Baerson, S.R.; Pan, Z.Q.; Zeng, R.S.; Song, Y.Y. Transcription factor OsbZIP49 controls tiller angle and plant architecture through the induction of indole-3-acetic acid-amido synthetases in rice. Plant J. 2021, 108, 1346–1364. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Environments | Mean | SD | Min | Max | Significance | h2 (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| FG | FE | FG×E | |||||||
| PH | 2016JP | 45.43 | 16.58 | 21.64 | 101 | 113.37 ** | 7305.66 ** | 13.19 ** | 89.25 |
| 2017JP | 73.17 | 30.66 | 27.5 | 177.67 | |||||
| 2017DT | 100.72 | 40.34 | 24 | 249.33 | |||||
| Mean | 69.7 | 28.06 | 28.06 | 165.52 | |||||
| NN | 2016JP | 15.36 | 3.98 | 9.11 | 29.4 | 82.87 ** | 627.76 ** | 5.39 ** | 93.92 |
| 2017JP | 17.43 | 4.41 | 9.33 | 27.67 | |||||
| 2017DT | 18 | 4.23 | 10 | 28.83 | |||||
| Mean | 16.75 | 4 | 10.15 | 27.6 | |||||
| BN | 2016JP | 2.82 | 1.27 | 0.33 | 9.89 | 13.62 ** | 20.63 ** | 4.22 ** | 69.97 |
| 2017JP | 2.54 | 1.23 | 0 | 7.33 | |||||
| 2017DT | 2.68 | 1.18 | 0.17 | 6.67 | |||||
| Mean | 2.68 | 0.99 | 0.67 | 6.28 | |||||
| DI | 2016JP | 4.51 | 0.83 | 2.9 | 8.85 | 22.41 ** | 1797.21 ** | 4.87 ** | 73.05 |
| 2017JP | 5.72 | 1.24 | 3.36 | 10.66 | |||||
| 2017DT | 6.69 | 1.42 | 2.37 | 11.8 | |||||
| Mean | 5.56 | 1 | 2.94 | 9.29 | |||||
| Traits | PH | NN | BN | DI |
|---|---|---|---|---|
| PH | 1 | |||
| NN | 0.878 ** | 1 | ||
| BN | 0.487 ** | 0.507 ** | 1 | |
| DI | 0.542 ** | 0.522 ** | 0.422 ** | 1 |
| Trait | Markers | Chromosome | Position (bp) | Allelic | Environments | −log10(p) | R2 (%) | References |
|---|---|---|---|---|---|---|---|---|
| PH | Q-02-0147081 | 2 | 29,025,631 | A/C | 2016JP/2017JP/Mean | 2.01–3.15 | 3.22–6.04 | [37,38,39] |
| Q-02-0158174 | 2 | 31,120,457 | A/C | 2016JP/2017JP/Mean | 2.14–3.07 | 3.39–5.54 | ||
| Q-02-0159217 | 2 | 31,236,629 | A/G | 2016JP/2017JP/ 2017DT/Mean | 2.07–4.14 | 4.15–8.19 | ||
| Q-02-0164155 | 2 | 31,724,101 | A/G | 2016JP/2017JP/Mean | 2.29–3.27 | 3.68–5.72 | ||
| Q-05-0207994 | 5 | 40,057,732 | A/G | 2016JP/2017JP/Mean | 2.27–2.64 | 3.91–4.38 | [28] | |
| BARC-014527-01571 | 6 | 656,098 | C/T | 2016JP/2017DT/Mean | 2.13–2.69 | 5.18–5.97 | [40] | |
| Q-06-0128380 | 6 | 17,613,829 | A/C | 2016JP/2017JP/Mean | 2.08–2.4 | 4.54–5.3 | [37,41,42,43] | |
| BARC-021425-04104 | 6 | 48,403,137 | A/C | 2016JP/2017JP/Mean | 2.27–3.06 | 5.22–6.76 | [44,45] | |
| Map-1255 | 7 | 7,190,281 | A/T | 2017JP/Mean | 2.66–3.46 | 4.42–6.6 | [46] | |
| BARC-041421-07980 | 9 | 44,020,453 | A/T | 2016JP/2017JP/ 2017DT/Mean | 2.02–3.44 | 4.89–7.96 | ||
| BARC-028861-06032 | 9 | 48,977,064 | A/G | 2017JP/2017DT/Mean | 2.01–2.2 | 4.24–5.23 | ||
| Map-1899 | 10 | 44,738,812 | A/C | 2016JP/2017DT | 2.25–2.31 | 3.62–4.75 | ||
| BARC-017097-02199 | 11 | 6,030,644 | A/G | 2016JP/2017JP/ 2017DT/Mean | 2.14–4.62 | 5.08–10.48 | [28] | |
| Map-2516 | 13 | 43,492,402 | A/C | 2016JP/Mean | 2.34–2.52 | 3.79–4.18 | [10] | |
| Q-14-0026827 | 14 | 3,164,755 | A/C | 2017JP/2017DT/Mean | 2.07–2.54 | 3.25–5.26 | ||
| Q-18-0016625 | 18 | 1,921,789 | A/T | 2017DT/Mean | 2.05–2.07 | 4.64–5.79 | [47] | |
| Q-18-0040883 | 18 | 4,329,135 | A/G | 2016JP/Mean | 2.2–2.3 | 3.5–3.71 | [38] | |
| Map-3990 | 19 | 45,340,653 | A/G | 2016JP/Mean | 2.25–2.84 | 5.12–6.34 | [48,49] | |
| Q-20-0260630 | 20 | 43,442,146 | A/C | 2017DT/Mean | 2.04–2.99 | 4.09–5.12 | ||
| NN | Map-0076 | 1 | 33,989,570 | C/G | 2016JP/2017DT/Mean | 2.06–5.05 | 5.62–11.27 | |
| BARC-014287-01306 | 5 | 4,279,362 | C/T | 2017JP/2017DT | 2.77–3.37 | 6.5–9.44 | ||
| BARC-014527-01571 | 6 | 656,098 | C/T | 2016JP/Mean | 2.63–2.88 | 5.88–6.35 | ||
| Q-06-0128380 | 6 | 17,613,829 | A/C | 2017JP/Mean | 2.08–2.61 | 4.55–5.96 | ||
| Q-08-0277446 | 8 | 43,136,899 | A/T | 2017JP/2017DT/Mean | 2.11–2.62 | 3.89–4.38 | ||
| BARC-041421-07980 | 9 | 44,020,453 | A/T | 2017DT/Mean | 2.22–2.54 | 5.75–6.19 | ||
| BARC-028861-06032 | 9 | 48,977,064 | A/G | 2017JP/2017DT | 2.08–2.26 | 4.59–4.84 | ||
| BARC-017097-02199 | 11 | 6,030,644 | A/G | 2016JP/2017JP/Mean | 2.23–3.63 | 5.21–8.23 | [28] | |
| Map-2026 | 11 | 25,164,685 | A/T | 2016JP/2017JP/ 2017DT/Mean | 2.12–3.04 | 3.57–6.56 | ||
| Map-2211 | 12 | 17,971,162 | A/G | 2016JP/2017JP/ 2017DT/Mean | 2.47–3.11 | 6.61–6.83 | ||
| Map-2213 | 12 | 18,455,502 | A/C | 2016JP/2017JP/ 2017DT/Mean | 2.2–3.4 | 6.2–7.76 | ||
| Map-2218 | 12 | 23,616,522 | C/G | 2017JP/Mean | 2.92–3.08 | 5.23–5.36 | ||
| Q-15-0364055 | 15 | 49,261,407 | A/T | 2017DT/Mean | 2.02–2.33 | 3.17–4.72 | ||
| Q-15-0364629 | 15 | 49,325,995 | A/C | 2017DT/Mean | 2–2.33 | 3.14–4.73 | ||
| BARC-030259-06840 | 20 | 38,262,189 | A/G | 2016JP/2017JP/Mean | 2.06–2.08 | 3.31–3.51 | ||
| BN | BARC-014639-01604 | 5 | 37,602,081 | A/T | 2017JP/Mean | 3.67–4.2 | 8.42–10.1 | [9] |
| Q-07-0088101 | 7 | 8,771,610 | A/G | 2016JP/Mean | 2.25–2.49 | 3.85–4.34 | ||
| BARC-013587-01169 | 8 | 10,563,212 | C/G | 2017JP/Mean | 2.02–2.28 | 4.74–5.15 | ||
| Q-08-0094591 | 8 | 12,454,445 | A/G | 2017DT/Mean | 2.01–2.64 | 3.65–4.48 | ||
| Map-2491 | 13 | 37,273,176 | A/T | 2016JP/Mean | 2.37–2.41 | 4.09–4.11 | ||
| BARC-039561-07508 | 14 | 48,880,009 | C/T | 2017JP/Mean | 2.68–3.99 | 4.64–7.82 | ||
| BARC-016029-02040 | 15 | 15,125,233 | A/G | 2017JP/Mean | 2.07–2.19 | 4.7–5.23 | ||
| BARC-018645-03217 | 17 | 4,097,240 | A/G | 2017JP/Mean | 2.2–2.36 | 5.04–5.67 | ||
| DI | Q-05-0193181 | 5 | 42,009,549 | A/C | 2017DT/Mean | 2.08–2.27 | 3.61–3.81 | |
| Q-08-0059708 | 8 | 8,402,455 | A/G | 2017JP/Mean | 2.13–2.33 | 4.66–5.29 | ||
| Map-1899 | 10 | 44,738,812 | A/C | 2016JP/2017JP | 2.16–2.3 | 3.52–3.8 | ||
| Map-2223 | 12 | 34,510,897 | A/G | 2017JP/2017DT | 2.24–2.78 | 4.98–7.09 | ||
| Q-15-0012218 | 15 | 1,858,944 | A/G | 2016JP/2017DT | 2.26–2.34 | 5.28–5.73 | ||
| Q-18-0040883 | 18 | 4,329,135 | A/G | 2017JP/Mean | 2.08–2.27 | 3.24–3.74 |
| Traits | Candidate Genes | Homologous Gene | Function Annotation |
|---|---|---|---|
| PH | Glyma.11g074100 | AT4G36220 | Cytochrome P450 (CYP84A1, FAH1) |
| Glyma.11g076200 | AT4G38860 | SAUR-like auxin-responsive protein family | |
| Glyma.11g078800 | AT1G80550 | Pentatricopeptide repeat (PPR) superfamily protein | |
| Glyma.11g079400 | AT1G75900 | GDSL-like Lipase/Acylhydrolase superfamily protein | |
| Glyma.11g084500 | AT1G20610 | Cyclin B2;3 | |
| Glyma.11g084700 | AT2G17200 | Ubiquitin family protein | |
| Glyma.11g085600 | AT1G18485 | Pentatricopeptide repeat (PPR) superfamily protein | |
| Glyma.11g086800 | AT2G17525 | Pentatricopeptide repeat (PPR) superfamily protein | |
| Glyma.11g086900 | AT2G17525 | Pentatricopeptide repeat (PPR) superfamily protein | |
| Glyma.11g087300 | AT5G65980 | Auxin efflux carrier family protein | |
| NN | Glyma.12g142900 | AT4G27280 | Calcium-binding EF-hand family protein |
| BN | Glyma.05g187300 | AT5G64740 | Cellulose synthase 6 |
| Glyma.05g195000 | AT1G01720 | NAC domain transcriptional regulator superfamily protein | |
| Glyma.05g196300 | AT2G46690 | SAUR-like auxin-responsive protein family | |
| DI | Glyma.12g180200 | AT5G22810 | GDSL-like Lipase/Acylhydrolase superfamily protein |
| Glyma.12g184200 | AT3G62890 | Pentatricopeptide repeat (PPR) superfamily protein | |
| Glyma.12g184500 | AT1G08320 | bZIP transcription factor family protein | |
| Glyma.12g186200 | AT5G22380 | NAC domain containing protein 90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, W.; Zhang, X.; Liu, D.; Li, C.; Wang, C.; Sun, R.; Jin, X.; Guo, N.; Zhao, J.; Xing, H. Genome-Wide Association Studies of Plant Architecture-Related Traits in the Chinese Soybean Mini Core Collection. Agronomy 2022, 12, 817. https://doi.org/10.3390/agronomy12040817
Ding W, Zhang X, Liu D, Li C, Wang C, Sun R, Jin X, Guo N, Zhao J, Xing H. Genome-Wide Association Studies of Plant Architecture-Related Traits in the Chinese Soybean Mini Core Collection. Agronomy. 2022; 12(4):817. https://doi.org/10.3390/agronomy12040817
Chicago/Turabian StyleDing, Wentao, Xiaoli Zhang, Dandan Liu, Chen Li, Congcong Wang, Ruidong Sun, Xiangpei Jin, Na Guo, Jinming Zhao, and Han Xing. 2022. "Genome-Wide Association Studies of Plant Architecture-Related Traits in the Chinese Soybean Mini Core Collection" Agronomy 12, no. 4: 817. https://doi.org/10.3390/agronomy12040817