

Selection of Reference Genes for Transcription Studies Considering Co-Regulation and Average Transcriptional Stability: Case Study on Adventitious Root Induction in Olive (Olea europaea L.) Microshoots


Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis of SHAM Effects on Adventitious Rooting
2.2. Biological Material for Transcript Quantification Analyses
2.3. Nucleic Acid Extraction
2.4. Reverse Transcription of mRNA
2.5. Selection of Candidate Genes as Internal Reference Genes
2.6. Design and Testing of Primers
2.7. Cloning of Amplified Fragments and Verification of Obtained Amplicons
2.8. Quantitative Polymerase Chain Reaction (qPCR)
2.9. Additional Test for gDNA Contamination
2.10. Calculation of PCR Efficiency
2.11. Data Analysis
3. Results and Discussion
3.1. Selection of Genes as Candidates for Reference Genes and Amplification Tests
3.2. Calculation of Cqs
3.3. Estimation of Transcript Level Variations
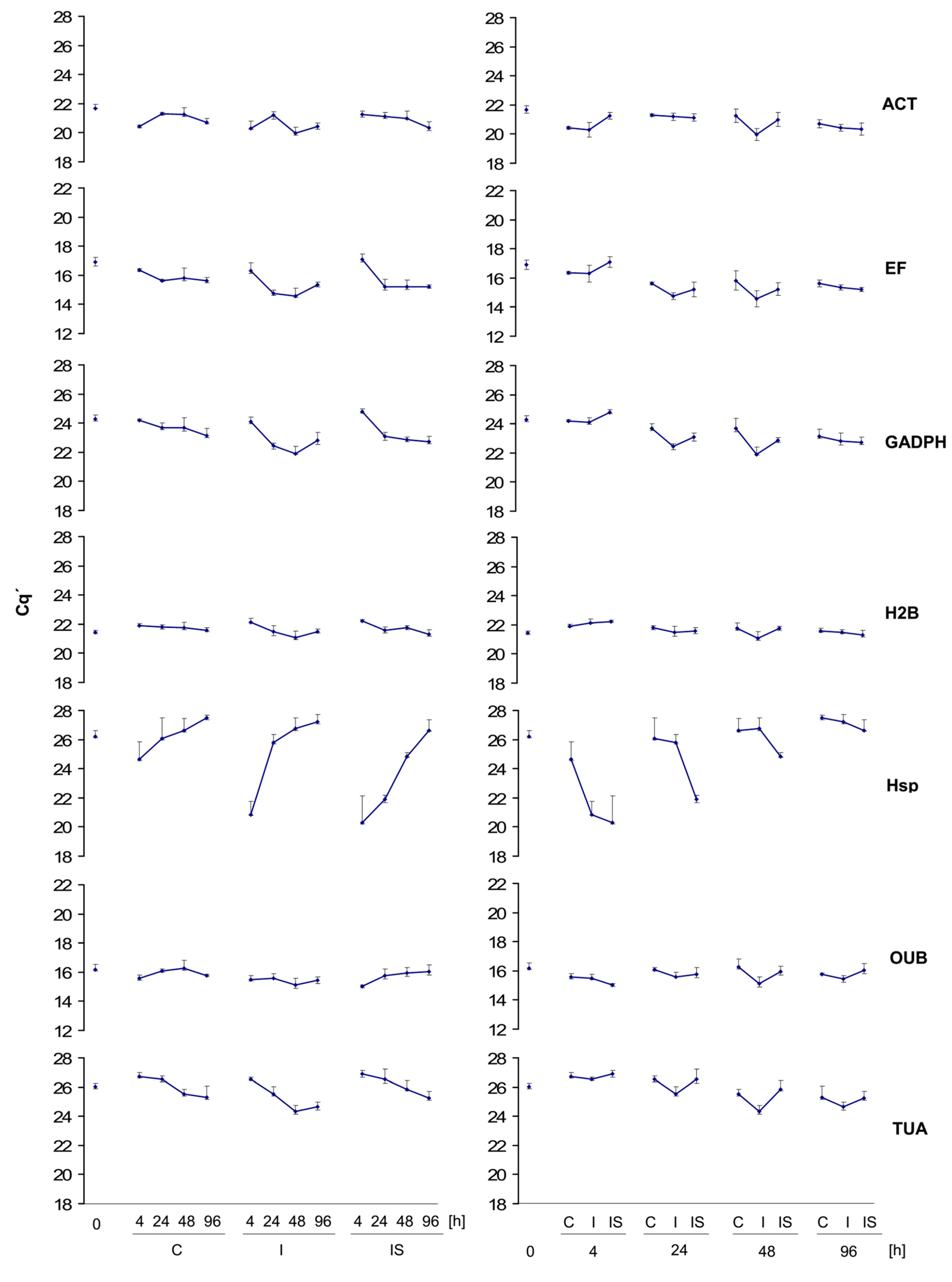
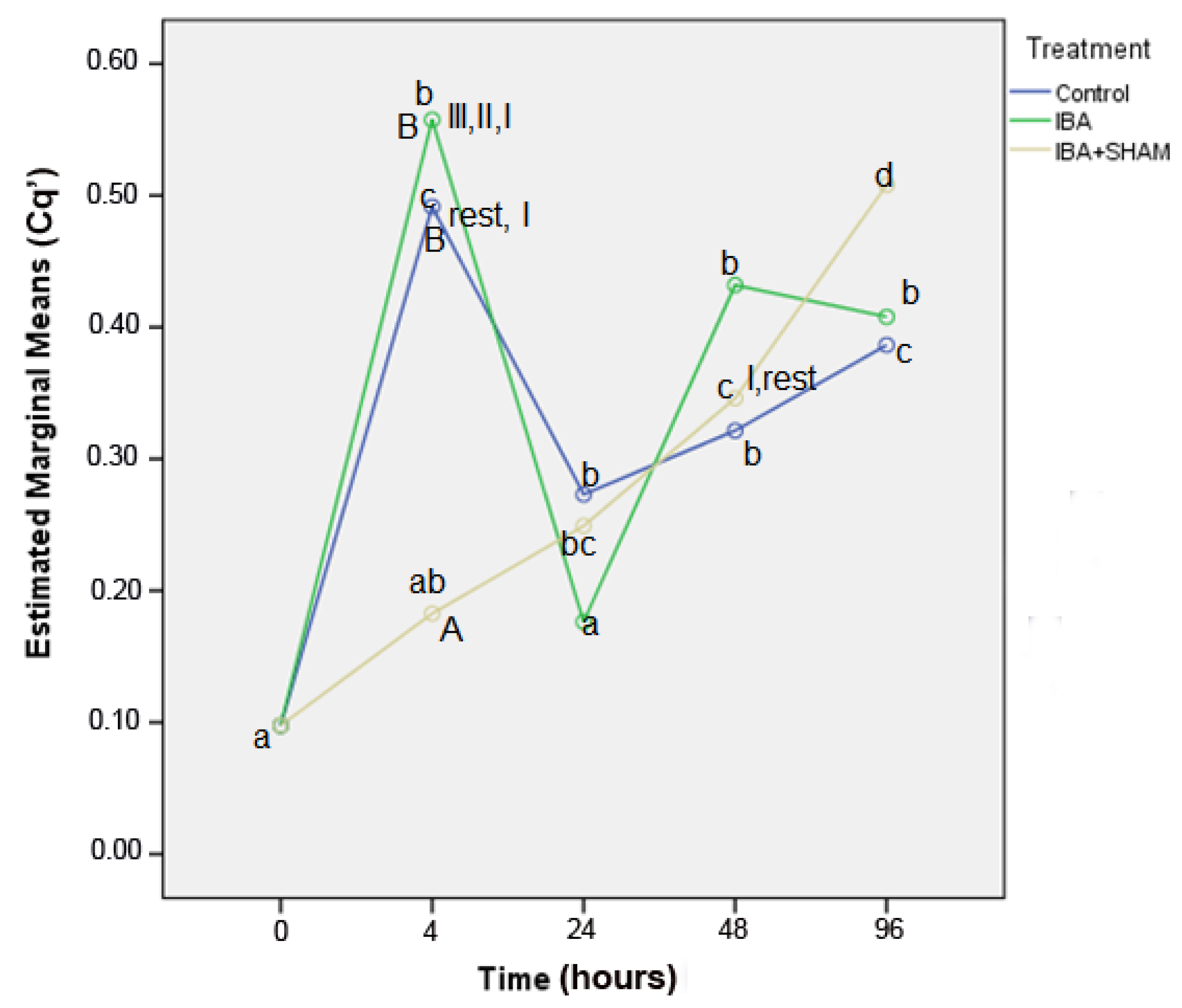
3.3.1. Displaying of Cq’ Means and Standard Deviation for Experimental Groups
3.3.2. Calculation of Transcriptional Gene Stability Values
3.4. Ranking of Candidate Housekeeping Genes According to Their Stability
3.5. Discarding of Transcriptionally Unstable Genes
3.6. Re-Rankings of Remaining Genes
3.7. Ranking with Software Methods as Support and Comparison with CV/F Ranking
3.8. Selection of RG Sets
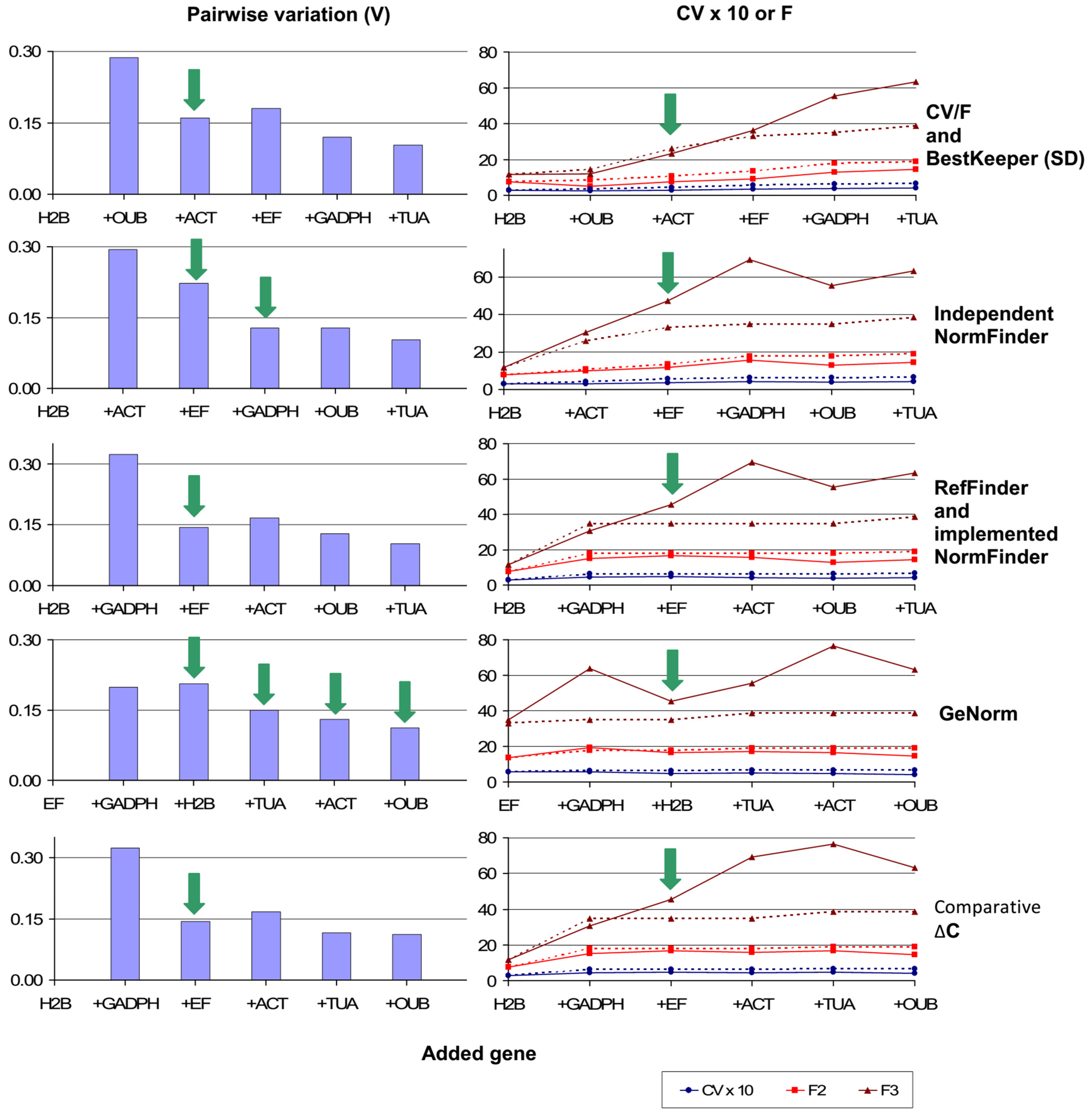
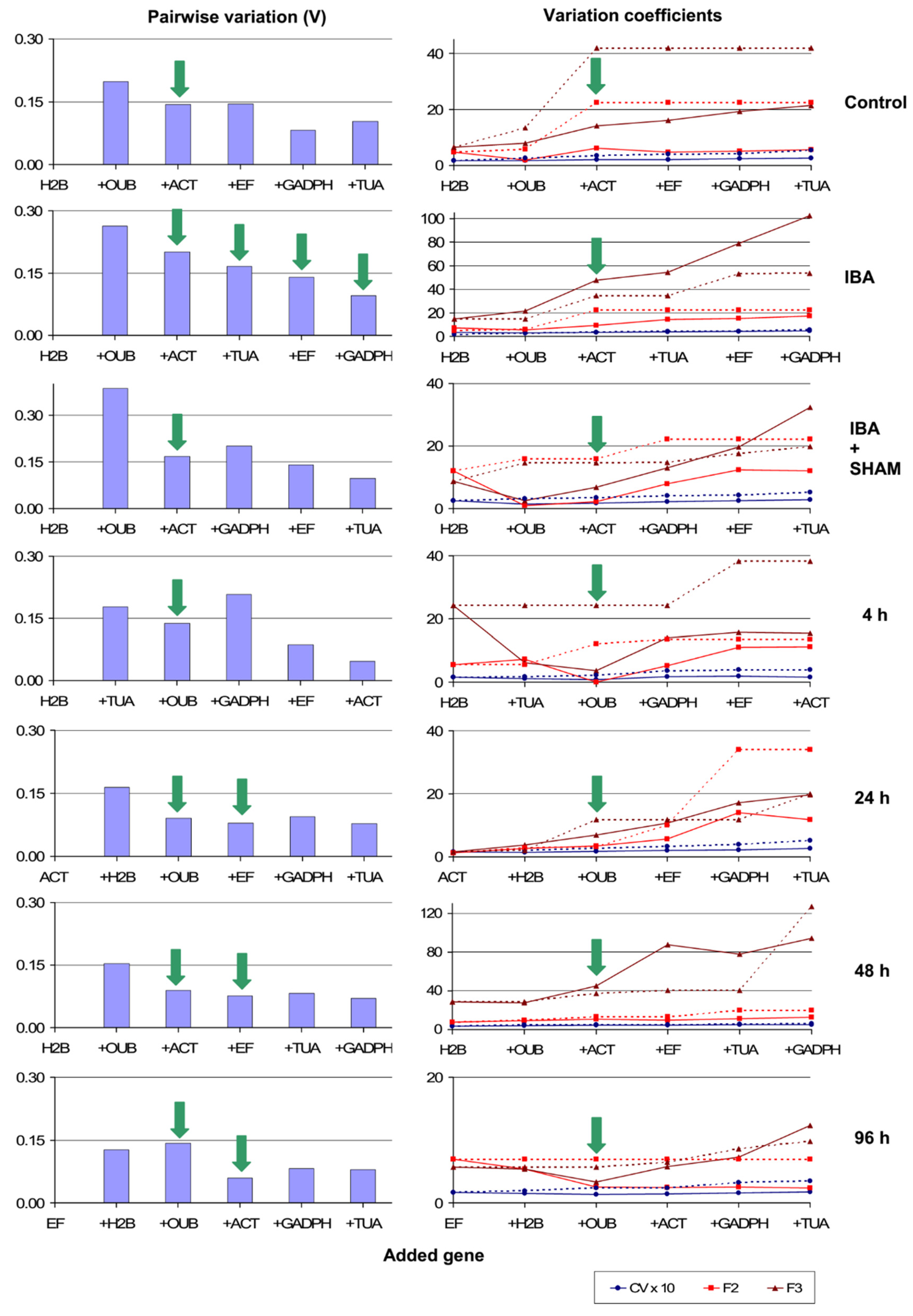
3.8.1. Calculation of Pairwise Variations between Possible NFs
3.8.2. Determination of Representative Candidate NFs
3.8.3. Assessment of Congruent Stability and Selection of NFs for Each Gene Ranking Method
- Ideally, important variability parameters (CV and F) for NFs should be lower than those of the CHG composing the NF, having the maximum values for such parameters (see Figure 2 right). Otherwise, this worse-ranked CHG may be unnecessarily contributing to additional NF instability.
- In this regard, as in general, when selecting NFs, the ranking provided by overall variability (CV) should ideally have priority over that given by inter-group variability (F).
- F would be more representative of the whole experimental panel if it spanned a higher number of factors: in the present case, F3 should have more priority than F2.
- Economic criteria may be additionally taken into account when two or more NFs may be similarly valid according to all above criteria, consequently selecting the RG set with the minimum number of CHGs.
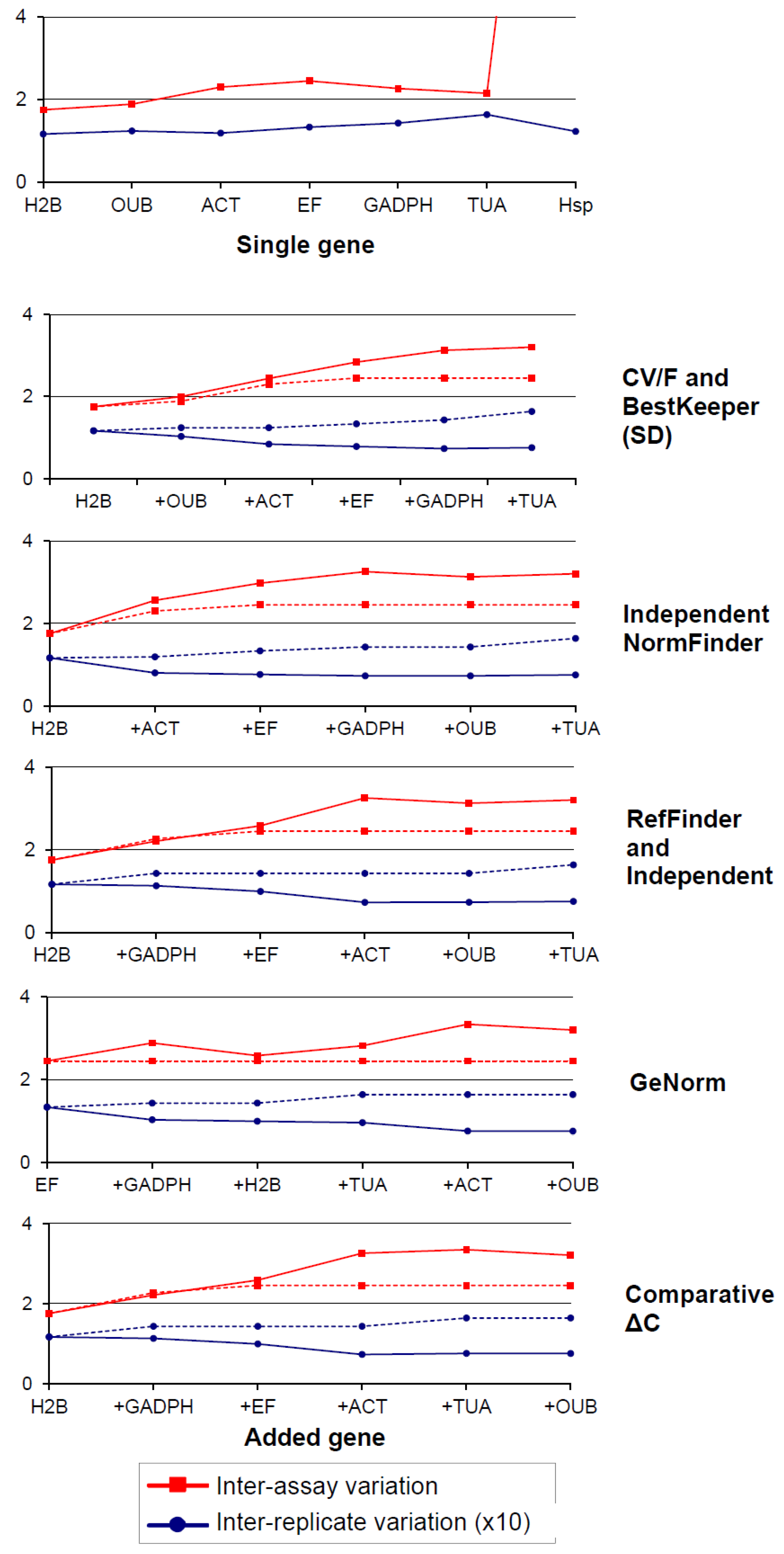
3.9. Error Compensation Versus Stability: Inspection of the Quality of the Selected NFs
3.10. Determination of the Optimal Normalization Factor for the Complete Bi- and Trifactorial Panels
3.11. Normalization Factors for More Specific Experimental Conditions
3.12. Evaluation of NF3 by Comparison with NF2
3.12.1. NF2/NF3 Comparison by Normalizing a Stable Target Gene
3.12.2. NF2/NF3 Comparison and Validation by Normalizing an Unstable Gene
3.12.3. Combined Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AOX | Alternative oxidase |
| BKr | BestKeeper Pearson coefficient of correlation |
| BKS | BestKeeper regarding standard deviation |
| CHG | Candidate housekeeping gene |
| CoD | Comparative ΔCq |
| Cq | Quantification cycle |
| Cq’ | Corrected quantification cycle |
| CV | Coefficient of total variation (for data on relative transcript accumulation) |
| DMSO | Dimethyl sulfoxide |
| F | Inter-group variation (for data on relative transcript accumulation) |
| gNo | geNorm |
| IBA | Indole-3-butyric acid |
| NF | Normalization factor |
| NFi | NormFinder |
| qPCR | Quantitative polymerase chain reaction |
| RFi | RefFinder |
| RG | Reference gene |
| RT | Reverse transcription |
| SHAM | Salicylhydroxamic acid |
| TG | Target gene |
References
- Bustin, S.A. Quantitative Real-Time RT-PCR—A Perspective. J. Mol. Endocrinol. 2005, 34, 597–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, M.L.; Medrano, J.F. Real-Time PCR for MRNA Quantification. Biotechniques 2005, 39, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-Time RT-PCR Normalisation; Strategies and Considerations. Genes Immun. 2005, 6, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dundas, J.; Ling, M. Reference Genes for Measuring MRNA Expression. Theory Biosci. 2012, 131, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.T.; Poolakkalody, N.J.; Shah, J.M. Plant Reference Genes for Development and Stress Response Studies. J. Biosci. 2018, 43, 173–187. [Google Scholar] [CrossRef]
- Kortner, T.M.; Valen, E.C.; Kortner, H.; Marjara, I.S.; Krogdahl, Å.; Bakke, A.M. Candidate Reference Genes for Quantitative Real-Time PCR (QPCR) Assays during Development of a Diet-Related Enteropathy in Atlantic Salmon (Salmo salar L.) and the Potential Pitfalls of Uncritical Use of Normalization Software Tools. Aquaculture 2011, 318, 355–363. [Google Scholar] [CrossRef]
- Brunner, A.M.; Yakovlev, I.A.; Strauss, S.H. Validating Internal Controls for Quantitative Plant Gene Expression Studies. BMC Plant Biol. 2004, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, M.R.; Ruedell, C.M.; Ricachenevsky, F.K.; Sperotto, R.A.; Pasquali, G.; Fett-Neto, A.G. Reference Gene Selection for Quantitative Reverse Transcription-Polymerase Chain Reaction Normalization during in Vitro Adventitious Rooting in Eucalyptus Globulus Labill. BMC Mol. Biol. 2010, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Kozera, B.; Rapacz, M. Reference Genes in Real-Time PCR. J. Appl. Genet. 2013, 54, 391–406. [Google Scholar] [CrossRef] [Green Version]
- Torres, S.; Lama, C.; Mantecón, L.; Flemetakis, E.; Infante, C. Selection and Validation of Reference Genes for Quantitative Real-Time PCR in the Green Microalgae Tetraselmis Chui. PLoS ONE 2021, 16, e0245495. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, C.; You, Y.; Liang, W.; Wang, N.; MA, F.; Li, C. Validation of Reference Genes for QRT-PCR Analysis in Peel and Flesh of Six Apple Cultivars (Malus domestica) at Diverse Stages of Fruit Development. Sci. Hortic. 2019, 244, 165–171. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. GeNorm Software Manual. 2007. Available online: https://www.gene-quantification.com/geNorm_manual.pdf (accessed on 1 June 2022).
- Salama, M.; Zahran, M.; Hassan, M. Comparing the Rooting Ability of Some Olive Cultivars Propagated by Leafy Cuttings under Mist. Ann. Agric Sci. Fac. Agric. Ain. Shams. Univ. 1987, 32, 577–590. [Google Scholar]
- Qrunfleh, M.M.; Rushdi, Y.; Musmar, T.; Rushdi, L. Root Formation in Cuttings of “Nabali” Olives Treated with Uniconazole and Indole Butyric Acid. Dirasat 1994, 21, 71–79. [Google Scholar]
- Fabbri, A. The Effect of Various Anatomical Characteristics on the Rooting of Cuttings in Olives, Cv. Frangivento. Riv. Della Ortoflorofruttic. Ital. 1980, 64, 325–335. [Google Scholar]
- Bakr, E.; Selim, H.; Nour, G.; Gabr, M. Developmental Anatomy of Adventitious Roots on Stem Cuttings of “Wetaken” Olive Cultivar. Egypt. J. Hort. 1977, 4, 91–97. [Google Scholar]
- Santos-Macedo, E.; Vieira, C.; Carrizo, D.; Porfirio, S.; Hegewald, H.; Arnholdt-Schmitt, B.; Calado, M.L.; Peixe, A. Adventitious Root Formation in Olive (Olea europaea L.) Microshoots: Anatomical Evaluation and Associated Biochemical Changes in Peroxidase and Polyphenol Oxidase Activities. J. Hortic. Sci. Biotechnol. 2013, 88, 53–59. [Google Scholar] [CrossRef]
- Porfirio, S.; Calado, M.L.; Noceda, C.; Cabrita, M.J.; da Silva, M.G.; Azadi, P.; Peixe, A. Tracking Biochemical Changes during Adventitious Root Formation in Olive (Olea europaea L.). Sci. Hortic. 2016, 204, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Santos Macedo, E.; Sircar, D.; Cardoso, H.G.; Peixe, A.; Arnholdt-Schmitt, B. Involvement of Alternative Oxidase (AOX) in Adventitious Rooting of Olea europaea L. Microshoots Is Linked to Adaptive Phenylpropanoid and Lignin Metabolism. Plant Cell Rep. 2012, 31, 1581–1590. [Google Scholar] [CrossRef]
- Arnholdt-Schmitt, B.; Costa, J.; de Melo, D. Alternative Respiration: Less Energy-Efficient but Highly Effective. Trends Plant Sci. 2006, 6, 281–287. [Google Scholar] [CrossRef]
- Arnholdt-Schmitt, B.; Santos Macedo, E.; Peixe, A.; Cardoso, H.; Cordeiro, A. AOX—A Potential Functional Marker for Efficient Rooting in Olive Shoot Cuttings. In Proceedings of the Second International Seminar Olivebioteq, Marsala Mazara del Vallo, Italy, 5–10 November 2006; Volume 1, pp. 249–254. [Google Scholar]
- Santos-Macedo, E.; Cardoso, H.; Hernández, A.; Peixe, A.; Polidoros, A.; Ferreira, A.; Cordeiro, A.; Arnholdt-Schmitt, B. Physiologic Responses and Gene Diversity Indicate Olive Alternative Oxidase as a Potential Source for Markers Involved in Efficient Adventitious Root Induction. Physiol. Plant. 2009, 137, 532–552. [Google Scholar] [CrossRef]
- Peixe, A.; Serras, M.; Campos, C.; Zavattieri, M.; Dias, M. Adventitious Rooting in Olive (Olea europaea L.) Cuttings: A Histological Evaluation. Rev. Ciênc. Agrár. Soc. Ciênc. Agrár. Port. 2007, 30, 476–482. [Google Scholar]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ul Haq, S.; Khan, A.; Ali, M.; Khattak, A.M.; Gai, W.X.; Zhang, H.X.; Wei, A.M.; Gong, Z.H. Heat Shock Proteins: Dynamic Biomolecules to Counter Plant Biotic and Abiotic Stresses. Int. J. Mol. Sci. 2019, 20, 5321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, A.; Yu, H.; Li, X.; Cui, H.; Chen, J.; Huang, P. Selection and Validation of Reference Genes for RT-QPCR Analysis in Aegilops Tauschii (Coss.) under Different Abiotic Stresses. Int. J. Mol. Sci. 2021, 22, 11017. [Google Scholar] [CrossRef]
- Chen, J.; Huang, Z.; Huang, H.; Wei, S.; Liu, Y.; Jiang, C.; Zhang, J.; Zhang, C. Selection of Relatively Exact Reference Genes for Gene Expression Studies in Goosegrass (Eleusine indica) under Herbicide Stress. Sci. Rep. 2017, 7, 46494. [Google Scholar] [CrossRef]
- Wang, B.; Duan, H.; Chong, P.; Su, S.; Shan, L.; Yi, D.; Wang, L.; Li, Y. Systematic Selection and Validation of Suitable Reference Genes for Quantitative Real-Time PCR Normalization Studies of Gene Expression in Nitraria Tangutorum. Sci. Rep. 2020, 10, 15891. [Google Scholar] [CrossRef]
- Nicot, N.; Hausman, J.-F.; Hoffmann, L.; Evers, D. Housekeeping Gene Selection for Real-Time RT-PCR Normalization in Potato during Biotic and Abiotic Stress. J. Exp. Bot. 2005, 56, 2907–2914. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Q.-L.; Wang, X.-T.; Yang, X.-Z.; Li, X.-P.; Yuan, M.-L. Selection of Reference Genes for QRT-PCR and Expression Analysis of High-Altitude-Related Genes in Grassland Caterpillars (Lepidoptera: Erebidae: Gynaephora) along an Altitude Gradient. Ecol. Evol. 2017, 7, 9054–9065. [Google Scholar] [CrossRef]
- Ashish, S.; Bhure, S.K.; Harikrishna, P.; Ramteke, S.S.; Muhammed Kutty, V.H.; Shruthi, N.; Ravi Kumar, G.V.P.P.S.; Manish, M.; Ghosh, S.K.; Mihir, S. Identification and Evaluation of Reference Genes for Accurate Gene Expression Normalization of Fresh and Frozen-Thawed Spermatozoa of Water Buffalo (Bubalus bubalis). Theriogenology 2017, 92, 6–13. [Google Scholar] [CrossRef]
- Nardeli, S.M.; Matta, B.P.; Saad, C.F.; Reinert, F.; Peixoto, R.S.; Alves-Ferreira, M. Early Heat Shock Protein Response and Selection of Reference Genes in Arabidopsis Thaliana Seedlings Subjected to Marine Fuel Contamination. Water Air Soil Pollut. 2017, 228, 101. [Google Scholar] [CrossRef]
- Assab, E.; Rampino, P.; Mita, G.; Perrotta, C. Heat Shock Response in Olive (Olea europaea L.) Twigs: Identification and Analysis of a CDNA Coding a Class I Small Heat Shock Protein. Plant Biosyst. Int. J. Deal. All Asp. Plant Biol. 2011, 145, 419–425. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.; Maniatis, T. Molecular Cloning. In A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2012. [Google Scholar]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. QBase Relative Quantification Framework and Software for Management and Automated Analysis of Real-Time Quantitative PCR Data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, C.; Jensen, J.; Ørntoft, T. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, N. A Technique Whose Time Has Come. Science 2002, 296, 557–559. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.H.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.B.; Moorman, A.F.M. Amplification Efficiency: Linking Baseline and Bias in the Analysis of Quantitative PCR Data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of Stable Housekeeping Genes, Differentially Regulated Target Genes and Sample Integrity: BestKeeper--Excel-Based Tool Using Pair-Wise Correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of Housekeeping Genes for Gene Expression Studies in Human Reticulocytes Using Real-Time PCR. BMC Mol. Biol. 2006, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Fan, C.; Li, H.; Zhang, Q.; Fu, Y.-F. Evaluation of Putative Reference Genes for Gene Expression Normalization in Soybean by Quantitative Real-Time RT-PCR. BMC Mol. Biol. 2009, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- De Jonge, H.J.M.; Fehrmann, R.S.N.; de Bont, E.S.J.M.; Hofstra, R.M.W.; Gerbens, F.; Kamps, W.A.; de Vries, E.G.E.; van der Zee, A.G.J.; te Meerman, G.J.; ter Elst, A. Evidence Based Selection of Housekeeping Genes. PLoS ONE 2007, 2, e898. [Google Scholar] [CrossRef] [Green Version]
- Boda, E.; Pini, A.; Hoxha, E.; Parolisi, R.; Tempia, F. Selection of Reference Genes for Quantitative Real-Time RT-PCR Studies in Mouse Brain. J. Mol. Neurosci. 2009, 37, 238–253. [Google Scholar] [CrossRef]
- Zampieri, M.; Ciccarone, F.; Guastafierro, T.; Bacalini, M.; Calabrese, R.; Moreno-Villanueva, M.; Realea, A.; Chevannec, M.; Bürkleb, A.; Caiafa, P. Validation of Suitable Internal Control Genes for Expression Studies in Aging. Mech. Ageing Dev. 2010, 131, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, V.K.; Sampathkumar, N.K.; Massaad, C.; Grenier, J. Optimal Use of Statistical Methods to Validate Reference Gene Stability in Longitudinal Studies. PLoS ONE 2019, 14, e0219440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanlou, K.M.; van Bockstaele, E. A Critique of Widely Used Normalization Software Tools and an Alternative Method to Identify Reliable Reference Genes in Red Clover (Trifolium pratense L.). Planta 2012, 236, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- de Santis, C.; Smith-Keune, C.; Jerry, D.R. Normalizing RT-QPCR Data: Are We Getting the Right Answers? An Appraisal of Normalization Approaches and Internal Reference Genes from a Case Study in the Finfish Lates Calcarifer. Mar. Biotechnol. 2011, 13, 170–180. [Google Scholar] [CrossRef]
- Zsóri, K.; Muszbek, L.; Csiki, Z.; Shemirani, A. Validation of Reference Genes for the Determination of Platelet Transcript Level in Healthy Individuals and in Patients with the History of Myocardial Infarction. Int. J. Mol. Sci. 2013, 14, 3456–3466. [Google Scholar] [CrossRef]
- Axtner, J.; Sommer, S. Validation of Internal Reference Genes for Quantitative Real-Time PCR in a Non-Model Organism, the Yellow-Necked Mouse, Apodemus Flavicollis. BMC Res. Notes 2009, 2, 264. [Google Scholar] [CrossRef] [Green Version]
- Goulao, L.; Fortunato, A.; Ramalho, J. Selection of Reference Genes for Normalizing Quantitative Real-Time PCR Gene Expression Data with Multiple Variables in Coffea Spp. Plant Mol. Biol. Rep. 2012, 30, 741–759. [Google Scholar] [CrossRef]
- Le, D.T.; Aldrich, D.L.; Valliyodan, B.; Watanabe, Y.; van Ha, C.; Nishiyama, R.; Guttikonda, S.K.; Quach, T.N.; Gutierrez-Gonzalez, J.J.; Tran, L.S.P.; et al. Evaluation of Candidate Reference Genes for Normalization of Quantitative RT-PCR in Soybean Tissues under Various Abiotic Stress Conditions. PLoS ONE 2012, 7, e46487. [Google Scholar] [CrossRef]
- Reid, K.E.; Olsson, N.; Schlosser, J.; Peng, F.; Lund, S.T. An Optimized Grapevine RNA Isolation Procedure and Statistical Determination of Reference Genes for Real-Time RT-PCR during Berry Development. BMC Plant Biol. 2006, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.Y.; Seo, P.J.; Yang, M.S.; Xiang, F.; Park, C.M. Exploring Valid Reference Genes for Gene Expression Studies in Brachypodium Distachyon by Real-Time PCR. BMC Plant Biol. 2008, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Maroufi, A.; Van Bockstaele, E.; De Loose, M. Validation of Reference Genes for Gene Expression Analysis in Chicory (Cichorium intybus) Using Quantitative Real-Time PCR. BMC Mol. Biol. 2010, 11, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marum, L.; Miguel, A.; Ricardo, C.P.; Miguel, C. Reference Gene Selection for Quantitative Real-Time PCR Normalization in Quercus Suber. PLoS ONE 2012, 7, e35113. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines:Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustin, S.A.; Beaulieu, J.-F.; Huggett, J.; Jaggi, R.; Kibenge, F.S.B.; Olsvik, P.A.; Penning, L.C.; Toegel, S. MIQE Précis: Practical Implementation of Minimum Standard Guidelines for Fluorescence-Based Quantitative Real-Time PCR Experiments. BMC Mol. Biol. 2010, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Derveaux, S.; Vandesompele, J.; Hellemans, J. How to Do Successful Gene Expression Analysis Using Real-Time PCR. Methods 2010, 50, 227–230. [Google Scholar] [CrossRef]
- Chervoneva, I.; Li, Y.; Schulz, S.; Croker, S.; Wilson, C.; Waldman, S.A.; Hyslop, T. Selection of Optimal Reference Genes for Normalization in Quantitative RT-PCR. BMC Bioinform. 2010, 11, 253. [Google Scholar] [CrossRef] [Green Version]
- Lyng, M.B.; Laenkholm, A.-V.; Pallisgaard, N.; Ditzel, H.J. Identification of Genes for Normalization of Real-Time RT-PCR Data in Breast Carcinomas. BMC Cancer 2008, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Wan, Q.; Chen, S.; Shan, Z.; Yang, Z.; Chen, L.; Zhang, C.; Yuan, S.; Hao, Q.; Zhang, X.; Qiu, D.; et al. Stability Evaluation of Reference Genes for Gene Expression Analysis by RT-QPCR in Soybean under Different Conditions. PLoS ONE 2017, 12, e0189405. [Google Scholar] [CrossRef] [Green Version]
- Nonis, A.; Vezzaro, A.; Ruperti, B. Evaluation of RNA Extraction Methods and Identification of Putative Reference Genes for Real-Time Quantitative Polymerase Chain Reaction Expression Studies on Olive (Olea europaea L.) Fruits. J. Agric. Food Chem. 2012, 60, 6855–6865. [Google Scholar] [CrossRef]
- Resetic, T.; Stajner, N.; Bandelj, D.; Javornik, B.; Jakse, J. Validation of Candidate Reference Genes in RT-QPCR Studies of Developing Olive Fruit and Expression Analysis of Four Genes Involved in Fatty Acids Metabolism. Mol. Breed. 2013, 32, 211–222. [Google Scholar] [CrossRef]
- Ray, D.L.; Johnson, J.C. Validation of Reference Genes for Gene Expression Analysis in Olive (Olea europaea) Mesocarp Tissue by Quantitative Real-Time RT-PCR. BMC Res. Notes 2014, 7, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hürkan, K.; Sezer, F.; Özbilen, A.; Taşkın, K.M. Identification of Reference Genes for Real-Time Quantitative Polymerase Chain Reaction Based Gene Expression Studies on Various Olive (Olea europaea L.) Tissues. J. Hortic. Sci. Biotechnol. 2018, 93, 644–651. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 22 Days after Induction | 28 Days after Induction | Assay (Rooting Capacity) | ||||
|---|---|---|---|---|---|---|
| Control | IBA | IBA + SHAM | Control | IBA | IBA + SHAM | |
| 0 (50) | 60 (50) | 0 (50) | - | - | - | I |
| 0 (25) | 38 (40) | 0 (40) | - | - | - | II |
| 0 (25) | 0 (50) | 0 (25) | 0 (25) | 32 (25) | 0 (25) | III |
| Name | Abbreviation | Role | Blasted Sequence (NCBI) | Blast Identities and Gaps | Found O. europaea Accession Used for Primer Design | Source of accession | Sequenced Amplicon with Forward Primer and Reverse Complementary of Reverse Primer (Alignment Mismatches in Cursive) | Tm of Amplicon | Efficiency | Alignment Mismatches (No Gaps Found) |
|---|---|---|---|---|---|---|---|---|---|---|
| Beta-actin | ACT | Microfilament component | AF545569.1 (act1) | act1 mRNA, partial CDS | TTGCTCTCGACTATGAACAGGATCTTGAGACTGCCAAGAGTAGCTCATCTGTTGAGAAAAACTATGAATTGCCAGATGGACAGGTTATTACTATTGGGGCCGAGAG | 76.81 | 1.904 | 0/106 | ||
| Elongation factor 1-alpha | EF | Translational elongation | AM946404.1 | Elongation factor partial gene, exons 1–2 | TTTTGAGGGTGACAACATGATTGAGAGGTCCACCAACCTCGACTGGTACAAGGGCCCAACCCTG | 77.02 | 1.868 | 0/64 | ||
| Glyceraldehyde-3-phosphate dehydrogenase | GAPDH | Glycolisis enzyme | NM_106601.3 (A. thaliana GAPCP1 mRNA, complete CDS) | 447/553 (81%), 0/553 (0%) | FL684222.1 | cv. Leccino fruitlet cDNA | CGACCTTGAGTCACCAACAAAATCATTGGAGACAACGTCTTCATCAGTGTAGCCGAGGATGC | 76.03 | 1.920 | 0/62 |
| Histone H2B | H2B | Chromatin structure | NC_003076.8 At5g59910 (A. thaliana HTB4) | 328/408 (80%), 3/408 (1%) | GO244518.1 | cDNA library from leaves and fruits | AAGCGTCTAGGCTTGCAAGGTACAACAAGAAGCCTACGATTACTTCTCGGGAGATTCAGACTGC | 76.62 | 1.935 | 0/64 |
| Small heat shock protein 18.3 | Hsp | Stress response | FN554869.1 | mRNA for putative class I Hsp18.3, cv. Cellina di Nardo | ACTTGGCACCGCATGGAGAGGAGCGCCGGAAAATTCCTTCGCCGGTTCAGG | 78.02 | 1.917 | 1/51 | ||
| Polyubiquitin | OUB | Protein degradation | AF429430.1 | OUB2 mRNA, complete CDS | AGGCATCCCACCAGACCAACAGAGGCTCATTTTCGCTGGTAAACAGTTTGAGGATGGTCTTAGTTTGGCTGACTATAACATTCAGAAGGAGTCCACACTCCACTTCGTGTTGAGGCTTCGCGGT | 81.29 | 1.855 | 11/124 | ||
| Alpha- tubulin | TUA | Microtubule structure | EF506517.1 (O. europaea putative alpha -tubulin mRNA) | 237/282 (84%), 4/282 (1%) | GO245051.1 | cDNA library from leaves and fruits partial cds | GTGCATTCCTTCACTGGTATGTGGGTGAGGGCATGGAGGAAGGAAAATTCTCAAAGGCTAAAGAGG | 75.68 | 1.916 | 1/66 |
| All Replicates | Time × Treatment | Time × Treatment × Assay | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RFi Algorithms | CV | F2 | Independent NFi | F3 | Independent NFi | ||||||
| BKr2 | BKS | CoD | GNo | NFi | RFi | ||||||
| All tested housekeeping genes | EF (0.67) | H2B (0.34) | H2B (1.07) | EF/GAPDH (0.59) | OUB (0.24) | H2B (1.57) | H2B (0.29) | Hsp (4.84) | OUB (0.24) | H2B (11.66) | OUB (0.27) |
| ACT (0.57) | OUB (0.38) | ACT (1.12) | H2B (0.32) | OUB (2.45) | OUB (0.34) | H2B (7.70) | H2B (0.26) | OUB (14.59) | H2B (0.28) | ||
| OUB (0.55) | ACT (0.58) | OUB (1.14) | H2B (0.69) | ACT (0.38) | EF (2.83) | ACT (0.43) | OUB (8.49) | ACT (0.31) | ACT (26.05) | ACT (0.33) | |
| GAPDH (0.54) | EF (0.68) | EF (1.16) | TUA (0.74) | EF (0.66) | ACT (3.08) | EF (0.56) | ACT (10.79) | EF (0.45) | TUA (33.03) | EF (0.46) | |
| TUA (0.43) | GAPDH (0.76) | GAPDH (1.17) | ACT (0.79) | GAPDH (0.76) | GAPDH (3.34) | GAPDH (0.63) | EF (13.53) | GAPDH (0.49) | EF (34.86) | GAPDH (0.5) | |
| H2B (0.38) | TUA (0.80) | TUA (1.23) | OUB (0.82) | TUA (0.88) | TUA (5.42) | TUA (0.67) | TUA (17.89) | TUA (0.58) | GAPDH (38.6) | TUA (0.56) | |
| Hsp (0.30) | Hsp (2.01) | Hsp (2.79) | Hsp (1.38) | Hsp (2.74) | Hsp (7.00) | Hsp (3.04) | GAPDH (19.02) | Hsp (1.43) | Hsp (1237.12) | Hsp (1.37) | |
| Best 6 tested housekeeping genes | EF (0.86) | H2B (0.34) | H2B (0.74) | EF/GAPDH(0.58) | H2B (0.42) | H2B (1.32) | H2B(0.29) | H2B (7.70) | H2B (0.21) | H2B (11.66) | H2B (0.21) |
| GAPDH (0.85) | OUB (0.38) | GAPDH (0.80) | GAPDH (0.57) | GAPDH (2.11) | OUB (0.34) | OUB (8.49) | ACT (0.28) | OUB (14.59) | ACT (0.30) | ||
| TUA (0.80) | ACT(0.58) | EF (0.82) | H2B (0.70) | EF (0.58) | EF (2.45) | ACT (0.43) | ACT (10.79) | EF (0.31) | ACT (26.05) | EF (0.32) | |
| H2B (0.64) | EF (0.68) | ACT (0.82) | TUA (0.74) | ACT (0.59) | ACT (3.94) | EF (0.56) | EF (13.53) | GAPDH (0.32) | TUA (33.03) | GAPDH (0.32) | |
| ACT (0.60) | GAPDH (0.76) | TUA (0.86) | ACT (0.79) | TUA (0.64) | OUB (4.56) | GAPDH (0.63) | TUA (17.89) | OUB (0.34) | EF (34.86) | OUB (0.36) | |
| OUB (0.37) | TUA (0.80) | OUB (0.88) | OUB (0.82) | OUB (0.70) | TUA (4.95) | TUA (0.67) | GAPDH (19.02) | TUA (0.37) | GAPDH (38.6) | TUA (0.37) | |
| All Replicates (Treatment) | Time | Time × Assay | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Level | BKS | CoD | GNo | NFi | RFi | CV | F | NFi | F | NFi |
| Control Treatment | H2B | ACT | ACT/ OUB | H2B | ACT (1.57) | H2B (0.18) | Hsp (3.85) | OUB (0.22) | H2B (6.6) | H2B (0.29) |
| OUB | H2B | ACT | H2B (1.57) | OUB (0.26) | H2B (4.78) | H2B (0.26) | OUB (13.55) | OUB (0.29) | ||
| ACT | OUB | H2B | OUB | OUB (2.06) | ACT (0.36) | OUB (5.86) | ACT (0.32) | EF (24.01) | ACT (0.3) | |
| EF | EF | EF | EF | EF (4.00) | EF (0.41) | GAPDH (7.27) | GAPDH (0.47) | GAPDH (27.98) | GAPDH (0.5) | |
| GAPDH | GAPDH | GAPDH | GAPDH | GAPDH (5.00) | GAPDH (0.42) | EF (8.93) | EF (0.5) | TUA (40.48) | EF (0.52) | |
| TUA | TUA | TUA | TUA | TUA (6.00) | TUA (0.54) | TUA (9.63) | TUA (0.64) | ACT (42.01) | TUA (0.62) | |
| Hsp | Hsp | Hsp | Hsp | Hsp (7.00) | Hsp (1.32) | ACT (22.53) | Hsp (1.01) | Hsp (442.64) | Hsp (1.08) | |
| IBA | OUB | OUB | EF/GAPDH | OUB | OUB (1.32) | H2B (0.32) | OUB (5.51) | OUB (0.39) | OUB (13.16) | OUB (0.43) |
| H2B | GAPDH | GAPDH | GAPDH (2.21) | OUB (0.34) | H2B (7.13) | H2B (0.44) | H2B (14.69) | H2B (0.44) | ||
| ACT | H2B | OUB | H2B | H2B (2.91) | ACT (0.49) | ACT (12.54) | TUA (0.47) | TUA (29.5) | TUA (0.46) | |
| EF | TUA | H2B | TUA | EF (3.16) | TUA (0.62) | Hsp (12.54) | EF (0.48) | ACT (34.59) | EF (0.51) | |
| TUA | EF | TUA | EF | TUA (4.47) | EF (0.64) | EF (16.73) | ACT (0.49) | EF (53.02) | GAPDH (0.52) | |
| GAPDH | ACT | ACT | ACT | ACT (5.05) | GAPDH (0.67) | GAPDH (20.47) | GAPDH (0.49) | GAPDH (53.55) | ACT (0.53) | |
| Hsp (2.27) | TUA (24.77) | Hsp (2664.02) | ||||||||
| IBA + SHAM | H2B | H2B | H2B/TUA | H2B | H2B (1.00) | H2B (0.25) | Hsp (4.04) | ACT (0.31) | H2B (8.68) | H2B (0.33) |
| OUB | ACT | ACT | TUA (2.45) | OUB (0.32) | ACT (7.51) | H2B (0.34) | ACT (13.65) | ACT (0.34) | ||
| ACT | TUA | ACT | TUA | ACT (2.45) | ACT (0.35) | TUA (8.69) | TUA (0.5) | OUB (14.68) | TUA (0.51) | |
| TUA | GAPDH | GAPDH | GAPDH | GAPDH (4.23) | GAPDH (0.42) | H2B (12.06) | GAPDH (0.53) | GAPDH (14.69) | GAPDH (0.51) | |
| GAPDH | EF | EF | EF | OUB (4.56) | EF (0.42) | EF (13.78) | EF (0.63) | EF (17.63) | EF (0.62) | |
| EF | OUB | OUB | OUB | EF (5.23) | TUA (0.53) | OUB (15.85) | OUB (0.66) | TUA (19.87) | OUB (0.67) | |
| Hsp (1.97) | GAPDH (22.17) | Hsp (1107.17) | ||||||||
| 4 h | GAPDH | GAPDH | EF|GAPDH | GAPDH | GAPDH (1.00) | H2B (0.16) | Hsp (3.19) | H2B (0.2) | TUA (4.11) | H2B (0.18) |
| OUB | EF | TUA | EF (2.06) | TUA (0.18) | TUA (3.92) | GAPDH (0.2) | OUB (15.79) | GAPDH (0.22) | ||
| EF | TUA | TUA | EF | TUA (2.91) | OUB (0.22) | EF (5.28) | TUA (0.2) | GAPDH (20.15) | EF (0.3) | |
| TUA | H2B | H2B | H2B | H2B (4.43) | GAPDH (0.25) | H2B (5.51) | EF (0.28) | EF (21.49) | TUA (0.3) | |
| ACT | ACT | ACT | ACT | OUB (4.56) | EF (0.37) | ACT (8.01) | ACT (0.33) | H2B (24.26) | ACT (0.35) | |
| H2B | OUB | OUB | OUB | ACT (5.00) | ACT (0.39) | OUB (12.15) | OUB (0.45) | ACT (38.25) | OUB (0.45) | |
| Hsp (1.53) | GAPDH (13.54) | Hsp (1408.8) | ||||||||
| 1 d | H2B | EF | EF|OUB | EF | EF (1.41) | ACT (0.15) | ACT (1.22) | EF (0.15) | ACT (1.58) | EF (0.22) |
| ACT | OUB | OUB | OUB (1.86) | H2B (0.2) | H2B (2.85) | OUB (0.17) | H2B (1.98) | OUB (0.23) | ||
| OUB | ACT | ACT | ACT | ACT (2.71) | OUB (0.27) | OUB (3.15) | H2B (0.25) | GAPDH (8.59) | H2B (0.27) | |
| EF | TUA | TUA | TUA | H2B (3.34) | EF (0.33) | TUA (7.6) | ACT (0.3) | EF (10.25) | ACT (0.32) | |
| GAPDH | H2B | H2B | H2B | TUA (4.43) | GAPDH (0.39) | EF (10.14) | TUA (0.31) | OUB (11.74) | TUA (0.37) | |
| TUA | GAPDH | GAPDH | GAPDH | GAPDH (5.73) | TUA (0.52) | GAPDH (34.04) | GAPDH (0.34) | TUA (20.03) | GAPDH (0.39) | |
| Hsp (1.19) | Hsp (145.76) | Hsp (41.6) | ||||||||
| 2d | H2B | H2B | ACT/OUB | H2B | H2B (1.32) | H2B (0.37) | EF (6.19) | OUB (0.19) | OUB (18.46) | H2B (0.23) |
| OUB | OUB | OUB | OUB (1.68) | OUB (0.49) | H2B (7.79) | H2B (0.22) | H2B (28.88) | OUB (0.26) | ||
| ACT | ACT | H2B | ACT | ACT (2.28) | ACT (0.52) | OUB (9.76) | EF (0.23) | TUA (33.74) | ACT (0.29) | |
| GAPDH | GAPDH | GAPDH | GAPDH | GAPDH (4) | EF (0.53) | ACT (13.31) | ACT (0.26) | ACT (37.23) | EF (0.35) | |
| TUA | EF | EF | EF | EF (5.23) | TUA (0.59) | GAPDH (17.13) | GAPDH (0.43) | EF (40.52) | GAPDH (0.37) | |
| EF | TUA | TUA | TUA | TUA (5.73) | GAPDH (0.61) | TUA (19.76) | TUA (0.49) | GAPDH (126.8) | TUA (0.46) | |
| Hsp | Hsp | Hsp | Hsp | Hsp (7.00) | Hsp (0.7) | Hsp (29.99) | Hsp (1.09) | Hsp (291.95) | Hsp (1.11) | |
| 4d | H2B | H2B | ACT/H2B | H2B | H2B (1.00) | EF (0.16) | GAPDH (1.21) | ACT (0.12) | OUB (4.12) | H2B (0.14) |
| EF | ACT | ACT | ACT (2.00) | H2B (0.19) | H2B (2.16) | H2B (0.14) | H2B (5.36) | EF (0.21) | ||
| OUB | EF | EF | EF | EF (2.71) | OUB (0.24) | ACT (2.29) | EF (0.15) | EF (5.66) | ACT (0.25) | |
| ACT | GAPDH | GAPDH | GAPDH | GAPDH (4.23) | ACT (0.24) | TUA (2.68) | GAPDH (0.21) | ACT (6.45) | GAPDH (0.35) | |
| GAPDH | OUB | OUB | OUB | OUB (4.4) | GAPDH (0.32) | Hsp (5.65) | OUB (0.32) | GAPDH (8.63) | OUB (0.4) | |
| TUA | TUA | TUA | TUA | TUA (6.00) | TUA (0.35) | EF (6.92) | TUA (0.34) | TUA (9.82) | TUA (0.44) | |
| Hsp | Hsp | Hsp | Hsp | Hsp (7.00) | Hsp (0.48) | OUB (6.99) | Hsp (0.37) | Hsp (33.83) | Hsp (0.5) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noceda, C.; Peixe, A.; Arnholdt-Schmitt, B. Selection of Reference Genes for Transcription Studies Considering Co-Regulation and Average Transcriptional Stability: Case Study on Adventitious Root Induction in Olive (Olea europaea L.) Microshoots. Agronomy 2022, 12, 3201. https://doi.org/10.3390/agronomy12123201
Noceda C, Peixe A, Arnholdt-Schmitt B. Selection of Reference Genes for Transcription Studies Considering Co-Regulation and Average Transcriptional Stability: Case Study on Adventitious Root Induction in Olive (Olea europaea L.) Microshoots. Agronomy. 2022; 12(12):3201. https://doi.org/10.3390/agronomy12123201
Chicago/Turabian StyleNoceda, Carlos, Augusto Peixe, and Birgit Arnholdt-Schmitt. 2022. "Selection of Reference Genes for Transcription Studies Considering Co-Regulation and Average Transcriptional Stability: Case Study on Adventitious Root Induction in Olive (Olea europaea L.) Microshoots" Agronomy 12, no. 12: 3201. https://doi.org/10.3390/agronomy12123201