
Primary Mapping and Analysis of the CmARM14 Candidate Gene for Mature Fruit Abscission in Melon
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Inheritance Analysis
2.2. Identification of MFA
2.3. Construction of Bulks for Preliminary Mapping
2.4. Whole-Genome Sequencing of Parental Lines and Bulk SLAF-Seq
2.5. Fine Mapping
2.6. qRT–PCR
2.7. Subcellular Localization
2.8. Candidate Gene Analysis
3. Results
3.1. The F3-57 Family Constitutes a Population Whose Members Segregate for Single Gene at the mfa10.1 Locus
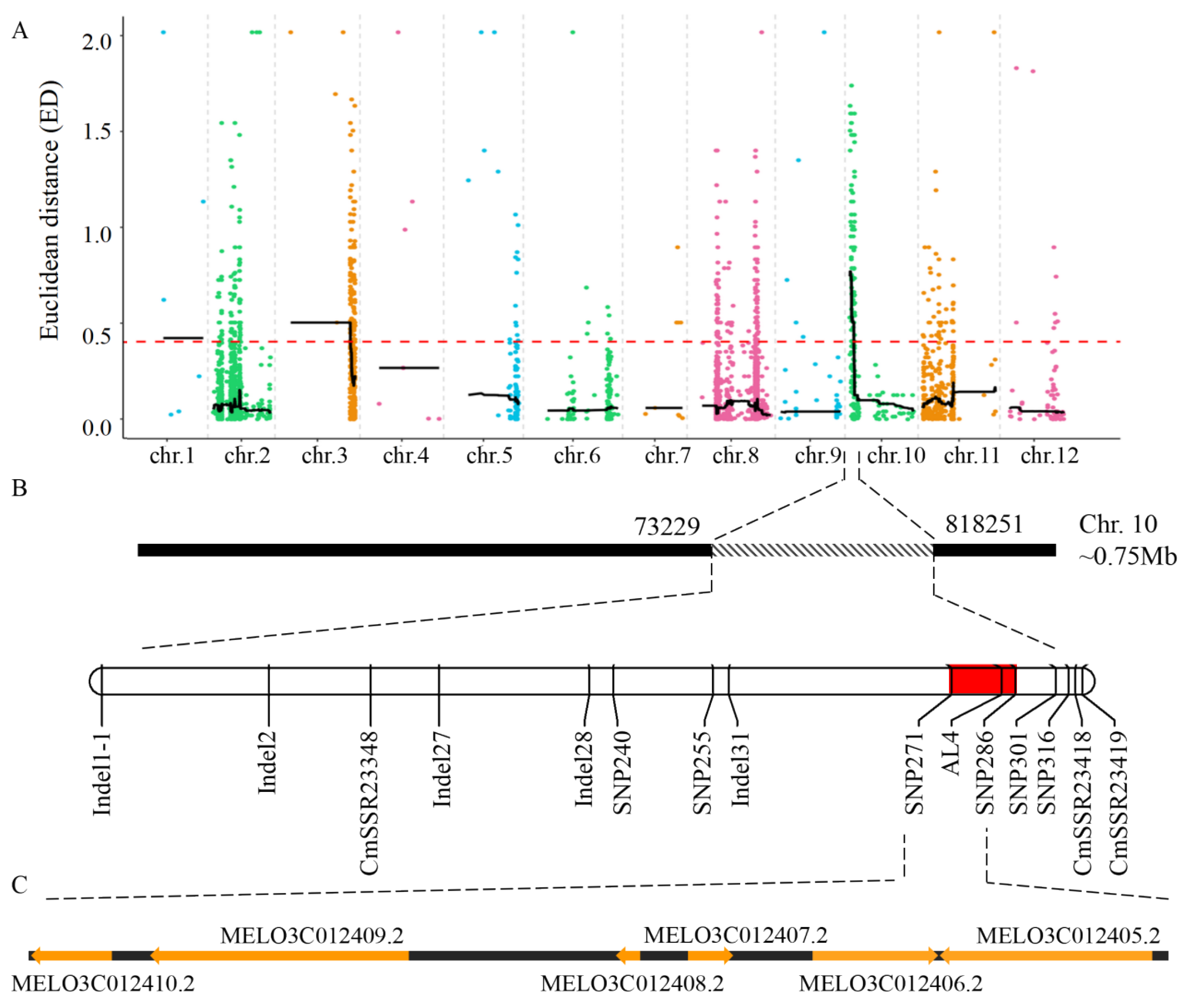
3.2. Primary Mapping of the mfa10.1 Locus by the SLAF-BSA Method
3.3. Fine Mapping of mfa10.1 and Analysis of the Candidate Gene CmARM14
3.4. Subcellular Localization of CmARM14
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ji, Y.; Qu, Y.; Jiang, Z.; Yan, J.; Chu, J.; Xu, M.; Su, X.; Yuan, H.; Wang, A. The mechanism for brassinosteroids suppressing climacteric fruit ripening. Plant Physiol. 2021, 185, 1875–1893. [Google Scholar] [PubMed]
- Gil-Amado, J.A.; Gomez-Jimenez, M.C. Transcriptome analysis of mature fruit abscission control in olive. Plant Cell Physiol. 2013, 54, 244–269. [Google Scholar] [PubMed] [Green Version]
- Schuster, M.; van der Hoorn, R.A.L. Plant Biology: Distinct New Players in Processing Peptide Hormones during Abscission. Curr Biol. 2020, 30, R715–R717. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Chun, J.P.; Tucker, M.L. Transcriptional Regulation of Abscission Zones. Plants 2019, 8, 154. [Google Scholar]
- Liu, D.; Li, J.; Li, Z.; Pei, Y. Hydrogen sulfide inhibits ethylene-induced petiole abscission in tomato (Solanum lycopersicum L.). Hortic. Res. 2020, 7, 14. [Google Scholar]
- Moreno, E.; Obando, J.M.; Dos-Santos, N.; Fernández-Trujillo, J.P.; Monforte, A.J.; Garcia-Mas, J. Candidate genes and QTLs for fruit ripening and softening in melon. Theor. Appl. Genet. 2008, 116, 589–602. [Google Scholar]
- Vegas, J.; Garcia-Mas, J.; Monforte, A.J. Interaction between QTLs induces an advance in ethylene biosynthesis during melon fruit ripening. Theor. Appl. Genet. 2013, 126, 1531–1544. [Google Scholar] [CrossRef]
- Périn, C.; Gomez-Jimenez, M.; Hagen, L.; Dogimont, C.; Pech, J.C.; Latché, A.; Pitrat, M.; Lelièvre, J.M. Molecular and genetic characterization of a non-climacteric phenotype in melon reveals two loci conferring altered ethylene response in fruit. Plant Physiol. 2002, 129, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Takada, K.; Kanazawa, K.; Takatuka, K. Studies on the breeding of melon for resistance to powdery mildew. II. Inheritance of resistance to powdery mildew and correlation of resistance to other characters. Bull Veg. Ornamental. Crops Res. Stn. 1975, A2, 11–31. [Google Scholar]
- Pereira, L.; Santo Domingo, M.; Ruggieri, V.; Argyris, J.; Phillips, M.A.; Zhao, G.; Lian, Q.; Xu, Y.; He, Y.; Huang, S.; et al. Genetic dissection of climacteric fruit ripening in a melon population segregating for ripening behavior. Hortic. Res. 2020, 7, 187. [Google Scholar]
- Yao, N.; Wang, L.; Yan, H.; Liu, Y.; Lu, B.R. Mapping quantitative trait loci (QTL) determining seed-shattering in weedy rice: Evolution of seed shattering in weedy rice through de-domestication. Euphytica 2015, 204, 513–522. [Google Scholar]
- Nakano, T.; Kimbara, J.; Fujisawa, M.; Kitagawa, M.; Ihashi, N.; Maeda, H.; Kasumi, T.; Ito, Y. Macrocalyx and Jointless interact in the transcriptional regulation of tomato fruit abscission zone development. Plant Physiol. 2012, 158, 439–450. [Google Scholar] [PubMed] [Green Version]
- Galpaz, N.; Gonda, I.; Shem-Tov, D.; Barad, O.; Tzuri, G.; Lev, S.; Fei, Z.; Xu, Y.; Mao, L.; Jiao, C.; et al. Deciphering genetic factors that determine melon fruit-quality traits using RNA-Seq-based high-resolution QTL and eQTL mapping. Plant J. 2018, 94, 169–191. [Google Scholar]
- Xu, Y.Y.; Liu, S.R.; Gan, Z.M.; Zeng, R.F.; Zhang, J.Z.; Hu, C.G. High-Density Genetic Map Construction and Identification of QTLs Controlling Leaf Abscission Trait in Poncirus trifoliata. Int. J. Mol. Sci. 2021, 22, 5723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liu, C.; Cheng, H.; Kan, G.; Cui, S.; Meng, Q.; Gai, J.; Yu, D. Quantitative trait loci associated with soybean tolerance to low phosphorus stress based on flower and pod abscission. Plant Breed. 2010, 129, 243–249. [Google Scholar]
- Celton, J.M.; Kelner, J.J.; Martinez, S.; Bechti, A.; Khelifi Touhami, A.; James, M.J.; Durel, C.E.; Laurens, F.; Costes, E. Fruit self-thinning: A trait to consider for genetic improvement of apple tree. PLoS ONE 2014, 9, e91016. [Google Scholar]
- Sheng, Y.Y.; Yang, L.M.; Dai, D.Y.; Zhang, J.X.; Wang, L.; Wang, D.; Cai, Y.; Tian, L.M. Cytological Observation of Fruit Peduncle Abscission Zone and Preliminary Mapping of Mature Fruit Abscission AL3 gene in Melon. Acta Hortic. Sin. 2022, 49, 341–351. [Google Scholar]
- Luan, F.S.; Sheng, Y.Y.; Wang, Y.H.; Jack, E.S. Performance of melon hybrids derived from parents of diverse geographic Origins. Euphytica 2010, 173, 1–16. [Google Scholar]
- Wei, W.; Chen, J.Y.; Zeng, Z.X.; Kuang, J.F.; Lu, W.J.; Shan, W. The Ubiquitin E3 Ligase MaLUL2 Is Involved in High Temperature-Induced Green Ripening in Banana Fruit. Int. J. Mol. Sci. 2020, 21, 9386. [Google Scholar] [CrossRef]
- Corbacho, J.; Romojaro, F.; Pech, J.C.; Latché, A.; Gomez-Jimenez, M.C. Transcriptomic events involved in melon mature-fruit abscission comprise the sequential induction of cell-wall degrading genes coupled to a stimulation of endo and exocytosis. PLoS ONE 2013, 8, e58363. [Google Scholar]
- Li, Q.; Serio, R.J.; Schofield, A.; Liu, H.; Rasmussen, S.R.; Hofius, D.; Stone, S.L. Arabidopsis RING-type E3 ubiquitin ligase XBAT35.2 promotes proteasome-dependent degradation of ACD11 to attenuate abiotic stress tolerance. Plant J. 2020, 104, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.; Lim, C.W.; Lee, S.C. The pepper RING-type E3 ligase, CaATIR1, positively regulates abscisic acid signalling and drought response by modulating the stability of CaATBZ1. Plant Cell Environ. 2020, 43, 1911–1924. [Google Scholar] [CrossRef] [PubMed]
- Ling, Q.; Sadali, N.M.; Soufi, Z.; Zhou, Y.; Huang, B.; Zeng, Y.; Rodriguez-Concepcion, M.; Jarvis, R.P. The chloroplast-associated protein degradation pathway controls chromoplast development and fruit ripening in tomato. Nat. Plants 2021, 7, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Shim, K.C.; Kim, S.H.; Jeon, Y.A.; Lee, H.S.; Adeva, C.; Kang, J.W.; Kim, H.J.; Tai, T.H.; Ahn, S.N. A RING-Type E3 Ubiquitin Ligase, OsGW2, Controls Chlorophyll Content and Dark-Induced Senescence in Rice. Int. J. Mol. Sci. 2020, 21, 1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Seo, P.J. The E3 ubiquitin ligase HOS1 is involved in ethylene regulation of leaf expansion in Arabidopsis. Plant Signal Behav. 2015, 10, e1003755. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.; Lian, X.; Cheng, J.; Zeng, W.; Zheng, X.; Wang, W.; Ye, X.; Li, J.; Li, Z.; Zhang, L.; et al. Genome-wide identification and transcriptome profiling reveal that E3 ubiquitin ligase genes relevant to ethylene, auxin and abscisic acid are differentially expressed in the fruits of melting flesh and stony hard peach varieties. BMC Genom. 2019, 20, 892. [Google Scholar]
- An, J.P.; Liu, X.; Song, L.Q.; You, C.X.; Wang, X.F.; Hao, Y.J. Functional Characterization of the Apple RING E3 Ligase MdMIEL1 in Transgenic Arabidopsis. Hortic. Plant J. 2017, 3, 53–59. [Google Scholar] [CrossRef]
- Sun, J.; Yuan, C.; Wang, M.; Ding, A.; Chai, G.; Sun, Y.; Zhou, G.; Yang, D.; Kong, Y. MUD1, a RING-v E3 ubiquitin ligase, has an important role in the regulation of pectin methylesterification in Arabidopsis seed coat mucilage. Plant Physiol. Biochem. 2021, 168, 230–238. [Google Scholar]
- Sun, H.; Li, J.; Li, X.; Lv, Q.; Chen, L.; Wang, B.; Li, L. RING E3 ubiquitin ligase TaSADR1 negatively regulates drought resistance in transgenic Arabidopsis. Plant Physiol. Biochem. 2022, 170, 255–265. [Google Scholar]
- Serrano-Bueno, G.; de Los Reyes, P.; Chini, A.; Ferreras-Garrucho, G.; de Medina-Hernández, V.S.; Boter, M.; Solano, R.; Valverde, F. Regulation of floral senescence in Arabidopsis by coordinated action of constans and jasmonate signaling. Mol. Plant. 2022, 15, 1710–1724. [Google Scholar]
- Chen, Y.Y. Preliminary analysis of the function of melon ubiquitin ligase gene CmRMA1H1 in fruit ripening. Inner. Mongolia. Univ. 2020. [Google Scholar] [CrossRef]
- Min, J.H.; Ju, H.W.; Yang, K.Y.; Chung, J.S.; Cho, B.H.; Kim, C.S. Heterologous expression of the gourd E3 ubiquitin ligase gene LsRZF1 compromises the drought stress tolerance in Arabidopsis thaliana. Plant Physiol. Biochem. 2014, 77, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Meng, X.; Guo, D.; Yang, S.; Zhang, G.; Liang, Z. Grapevine U-Box E3 Ubiquitin Ligase VlPUB38 Negatively Regulates Fruit Ripening by Facilitating Abscisic-Aldehyde Oxidase Degradation. Plant Cell Physiol. 2021, 61, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Y.; Shan, W.; Yang, T.W.; Wu, C.J.; Liu, X.C.; Chen, J.Y.; Lu, W.J.; Li, Z.G.; Deng, W.; Kuang, J.F. MaMYB4 is a negative regulator and a substrate of RING-type E3 ligases MaBRG2/3 in controlling banana fruit ripening. Plant J. 2022, 110, 1651–1669. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, Z.Q.; Qin, G.Z.; Tian, S.P. Effects of brassinosteroids on postharvest disease and senescence of jujube fruit in storage. Postharvest Biol. Technol. 2010, 56, 50–55. [Google Scholar] [CrossRef]
- Zhu, T.; Tan, W.R.; Deng, X.G.; Zheng, T.; Zhang, D.W.; Lin, H.H. Effects of brassinosteroids on quality attributes and ethylene synthesis in postharvest tomato fruit. Postharvest Biol. Technol. 2015, 100, 196–204. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Marker Name | Chromosome | Genome Position | Forward Sequence | Reverse Sequence |
|---|---|---|---|---|---|
| 1 | Indel1-1 | 10 | 194,740 … 195,031 | ACGTTGCAAGTGTGCCTTATC | GTGAGTGAGGTAGGTGTGGA |
| 2 | Indel2 | 10 | 234,249 … 234,583 | TTTAGGTTTGTGAGGAAGAAGG | TCCCCCCAATAAAGTAAAACA |
| 3 | CmSSR23348 | 10 | 234,705 … 235,730 | CATTGATCCAAATGTTACCCAA | GGGAGGGGCTATGGGTTAT |
| 4 | Indel27 | 10 | 563,304 … 563,535 | TGGCCTTTTTGACTGCTCTT | TAAATCTCCTACCGTTCCCG |
| 5 | Indel28 | 10 | 575,734 … 575,971 | AAGAATGTATGGATTAAAAGGGTTT | TCTTTCGCTCATGGAAGTCA |
| 6 | SNP240 | 10 | 579,866 … 580,121 | AACTCCTTCCTTTTCCCGTC | GCATCAAAACCATTTTTCTTTG |
| 7 | SNP255 | 10 | 608,958 … 609,170 | AGTTTTGTCAACCACACCCA | TTTCTCCGATTTCTTTCCATTC |
| 8 | Indel31 | 10 | 616,081 … 616,308 | TCGAAGATGGTCCATTAGGG | TTGGAAGCAAAGCAACTCCT |
| 9 | SNP271 | 10 | 650,203 … 650,387 | CCCTCCCTCCCTCTAAATGA | GGATGTTGCCTTGAAAAAGC |
| 10 | SNP286 | 10 | 685,250 … 685,509 | CCGTCGCGTTCGTTTTATTA | AAAAATGAAATTGGCAGCACTT |
| 11 | SNP301 | 10 | 719,646 … 719,884 | AACCCCTCAATAACCCAACC | GAGTGAAGCAAACACCACGA |
| 12 | SNP316 | 10 | 738,058 … 738,298 | GGCCCCATAATTGAGAAAAA | ATTGCATGCATGTGGATGAT |
| 13 | CmSSR23418 | 10 | 742,790 … 743,815 | ATTTGATTTTTGCAAAGCGG | CAAAATTGGGGAGTATAGTGACAA |
| 14 | CmSSR23419 | 10 | 761,037 … 762,060 | TTGGTAGGCTAAAAGATCGTCC | CAAAAGACACATCATGAGGGC |
| Clean Reads | Clean Base | Q30 (%) | GC (%) | Total Reads | Mapped (%) | Properly Mapped (%) | |
|---|---|---|---|---|---|---|---|
| ZT00091 | 45,492,988 | 13,622,302,478 | 92.88 | 38.03 | 90,985,976 | 95.04 | 87.53 |
| M2-10 | 42,680,203 | 12,780,598,726 | 93.03 | 36.94 | 85,360,406 | 97.18 | 89.48 |
| MFA BULK | 36,547,360 | 10,943,443,284 | 91.68 | 36.13 | 73,094,720 | 98.77 | 93.12 |
| None MFA BULK | 31,846,911 | 9,529,678,748 | 93.74 | 37.01 | 63,693,822 | 96.78 | 90.76 |
| Chromosome | ED Analysis Method | Index Analysis Method | Combination | Gene Number | ||||
|---|---|---|---|---|---|---|---|---|
| Genome Start | Genome End | Genome Start | Genome End | Genome Start | Genome End | |||
| SNP methods | 10 | 23,044 | 2,057,566 | 23,044 | 1,558,140 | 23,044 | 1,558,140 | 206 |
| INDEL methods | 10 | 73,229 | 1,779,001 | 73,229 | 818,251 | 73,229 | 818,251 | 111 |
| Combination region | 10 | 73,229 | 818,251 | 111 | ||||
| Melon Gene ID | Gene Annotation | Physical Location |
|---|---|---|
| MELO3C012410.2 | thioredoxin-like 3-2, chloroplastic isoform X8 | 650,442 … 652,744 |
| MELO3C012409.2 | Amino acid transporter family protein | 654,175 … 661,881 |
| MELO3C012408.2 | Unknown protein | 668,489 … 669,000 |
| MELO3C012407.2 | SKP1-like protein 12 | 670,514 … 671,673 |
| MELO3C012406.2 | RING-type E3 ubiquitin transferase | 674,353 … 677,946 |
| MELO3C012405.2 | Vacuolar protein-sorting-associated protein 33-like protein | 676,286 … 685,240 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, D.; Wang, L.; Zhang, J.; Qin, H.; Liu, H.; Sheng, Y. Primary Mapping and Analysis of the CmARM14 Candidate Gene for Mature Fruit Abscission in Melon. Agronomy 2022, 12, 3117. https://doi.org/10.3390/agronomy12123117
Dai D, Wang L, Zhang J, Qin H, Liu H, Sheng Y. Primary Mapping and Analysis of the CmARM14 Candidate Gene for Mature Fruit Abscission in Melon. Agronomy. 2022; 12(12):3117. https://doi.org/10.3390/agronomy12123117
Chicago/Turabian StyleDai, Dongyang, Ling Wang, Junming Zhang, Haojie Qin, Huiying Liu, and Yunyan Sheng. 2022. "Primary Mapping and Analysis of the CmARM14 Candidate Gene for Mature Fruit Abscission in Melon" Agronomy 12, no. 12: 3117. https://doi.org/10.3390/agronomy12123117