
Modern Approaches for the Genetic Improvement of Rice, Wheat and Maize for Abiotic Constraints-Related Traits: A Comparative Overview


Abstract
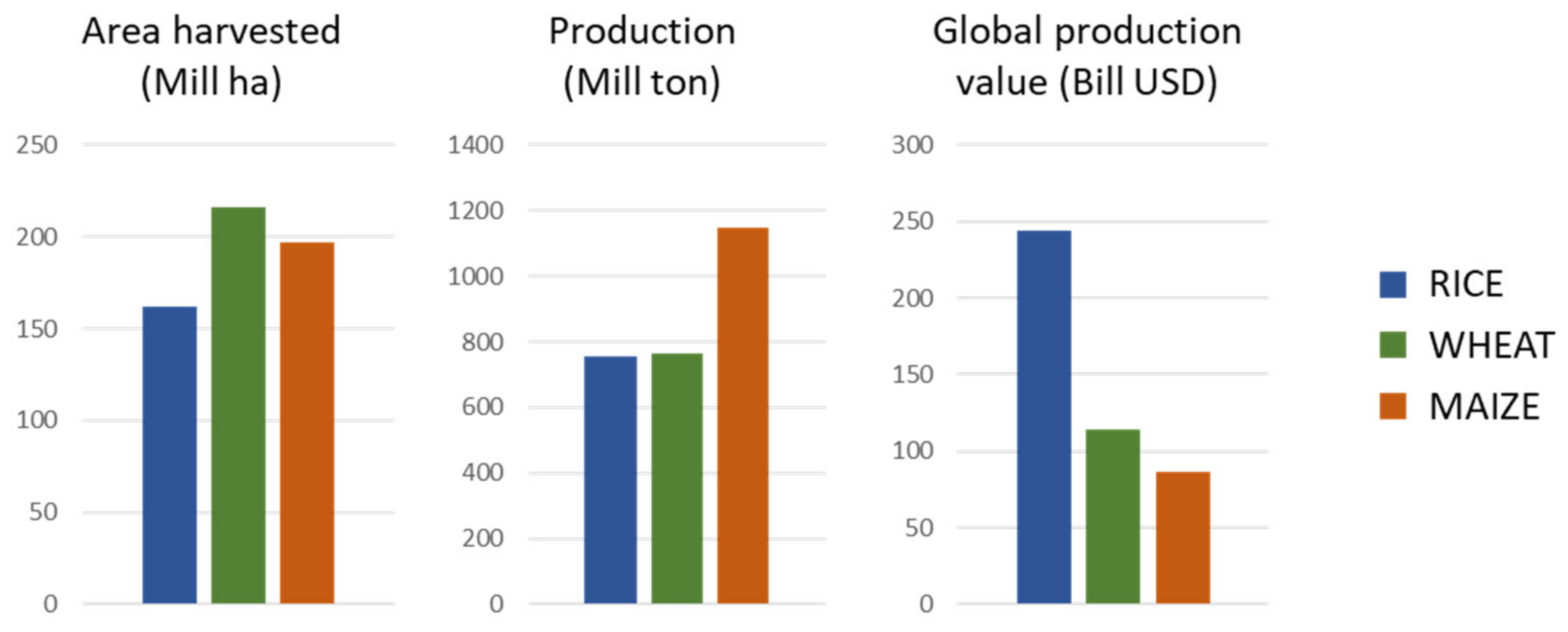
:1. Introduction
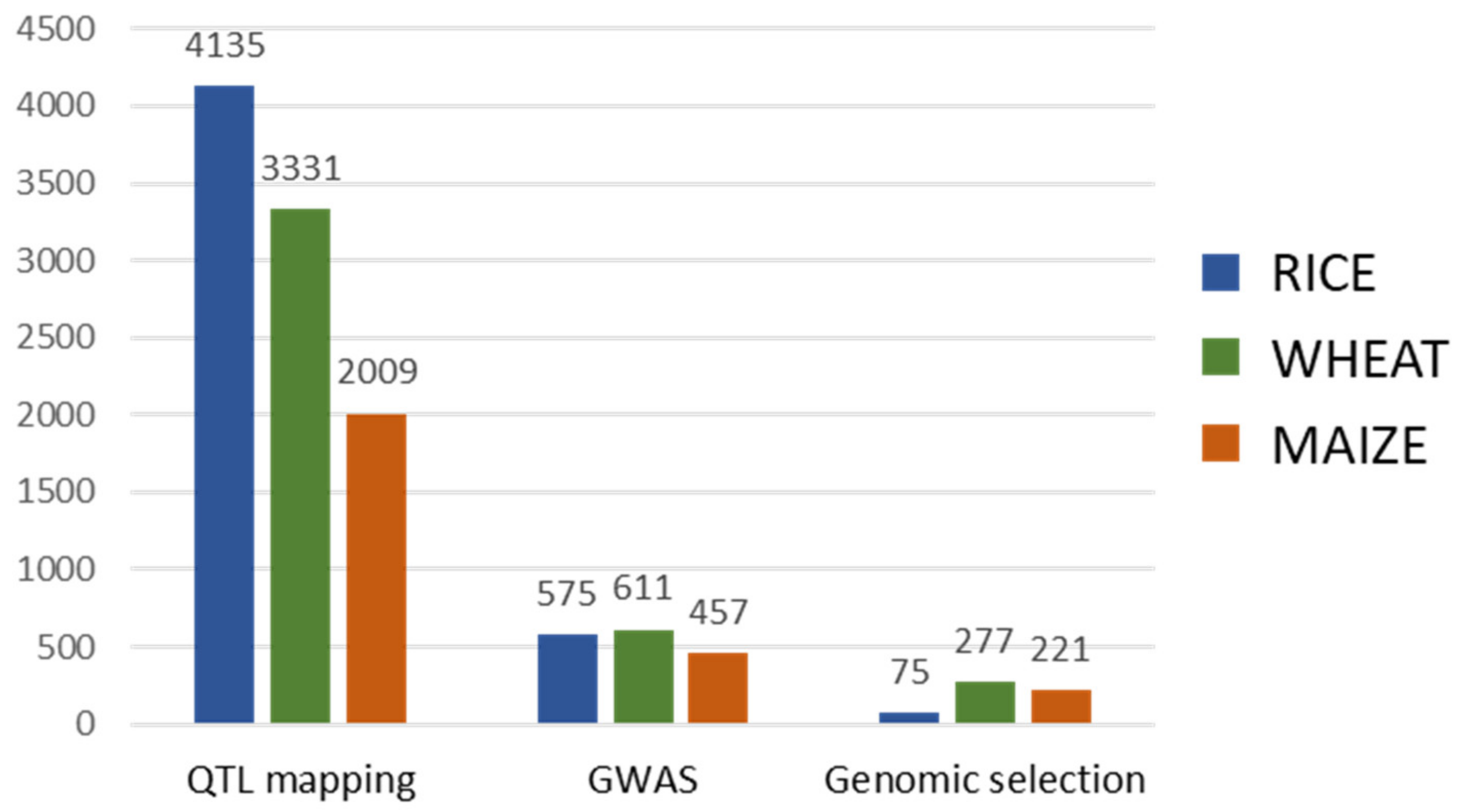
2. Marker-Assisted Breeding
2.1. Marker-Assisted Selection
- (a)
- A suitable marker system. The relatively recent development of high-throughput Next Generation Sequencing (NGS) platforms has revolutionized the field, enabling and generalizing the use of single nucleotide polymorphisms (SNPs) as molecular markers. Earlier marker systems not based on NGS methods had several disadvantages related to expensiveness, time-consumption, or reproducibility.
- (b)
- The development of genetic or physical maps, where the marker–trait associations can be contextualized at a genome level and the most suited markers can be chosen for MAS. High-density genetic linkage maps, based on the segregation of markers and genes in experimental populations, have been built in most economically important plant species for MAS applications. However, because crop genome sequences are available, fine physical maps have become a popular alternative, mainly because of their faster development and almost unlimited resolution (i.e., at the base-pair level).
- (c)
- The identification of marker–trait associations. As mentioned, the genetic linkage between the trait of interest and the marker is a key aspect for marker-assisted breeding. The success in breeding programs can only be guaranteed if markers are tightly linked to the genes or QTLs, or closely associated with the target traits.
2.2. Genomic Selection
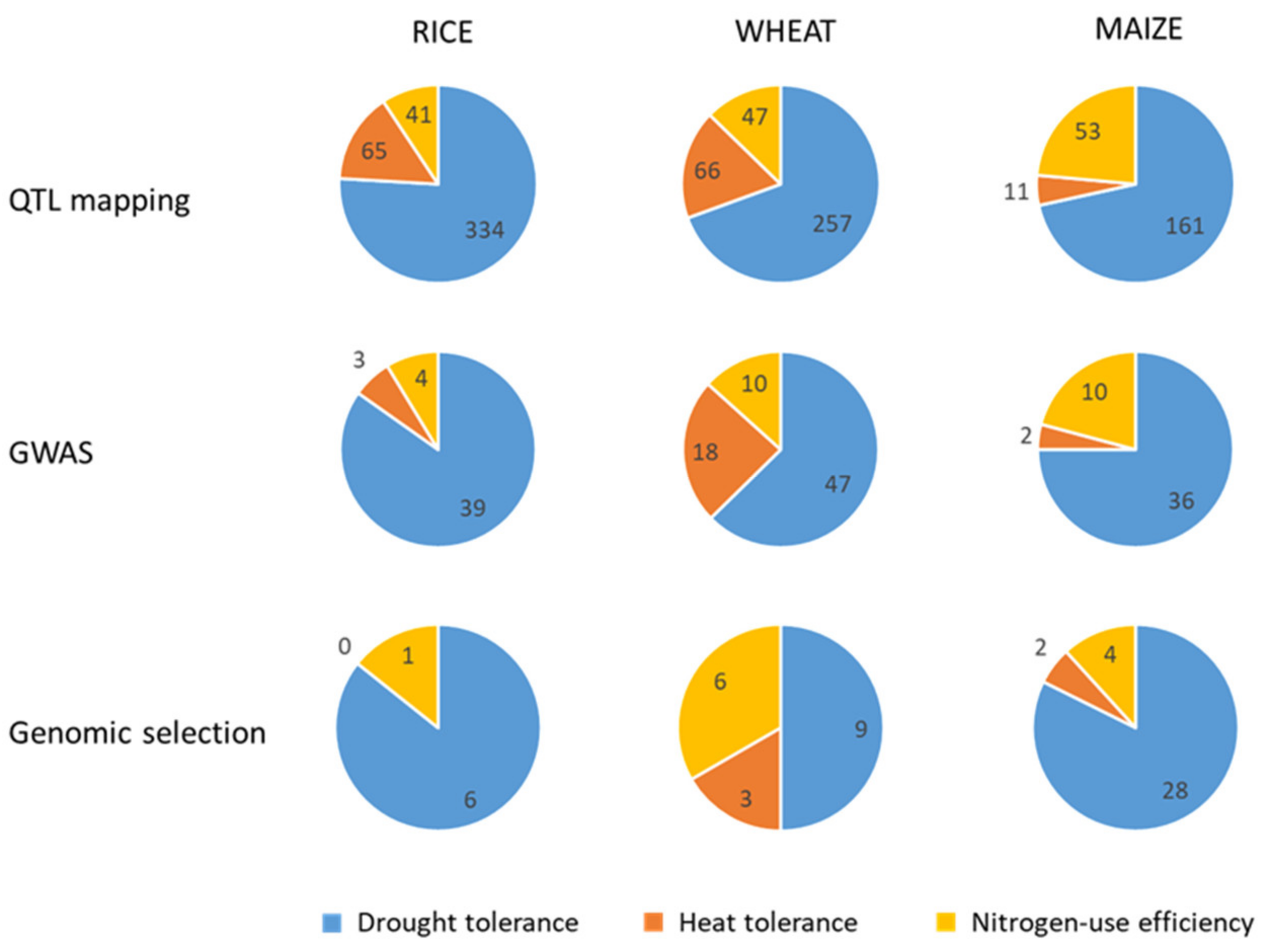
2.3. Marker-Assisted Breeding Oriented to Crop Improvement for Abiotic Limitations’ Response
2.3.1. Rice
2.3.2. Wheat
2.3.3. Maize
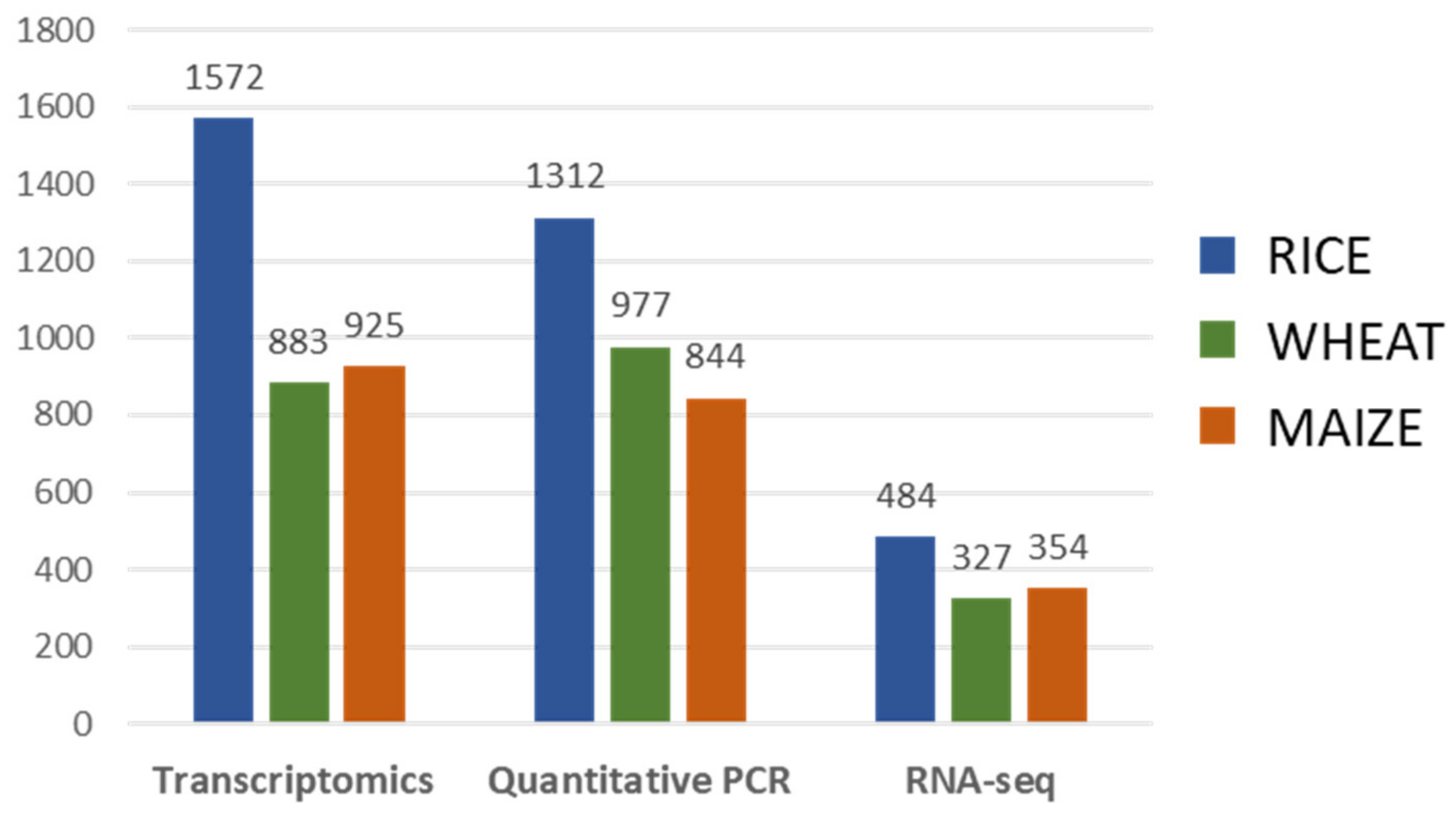
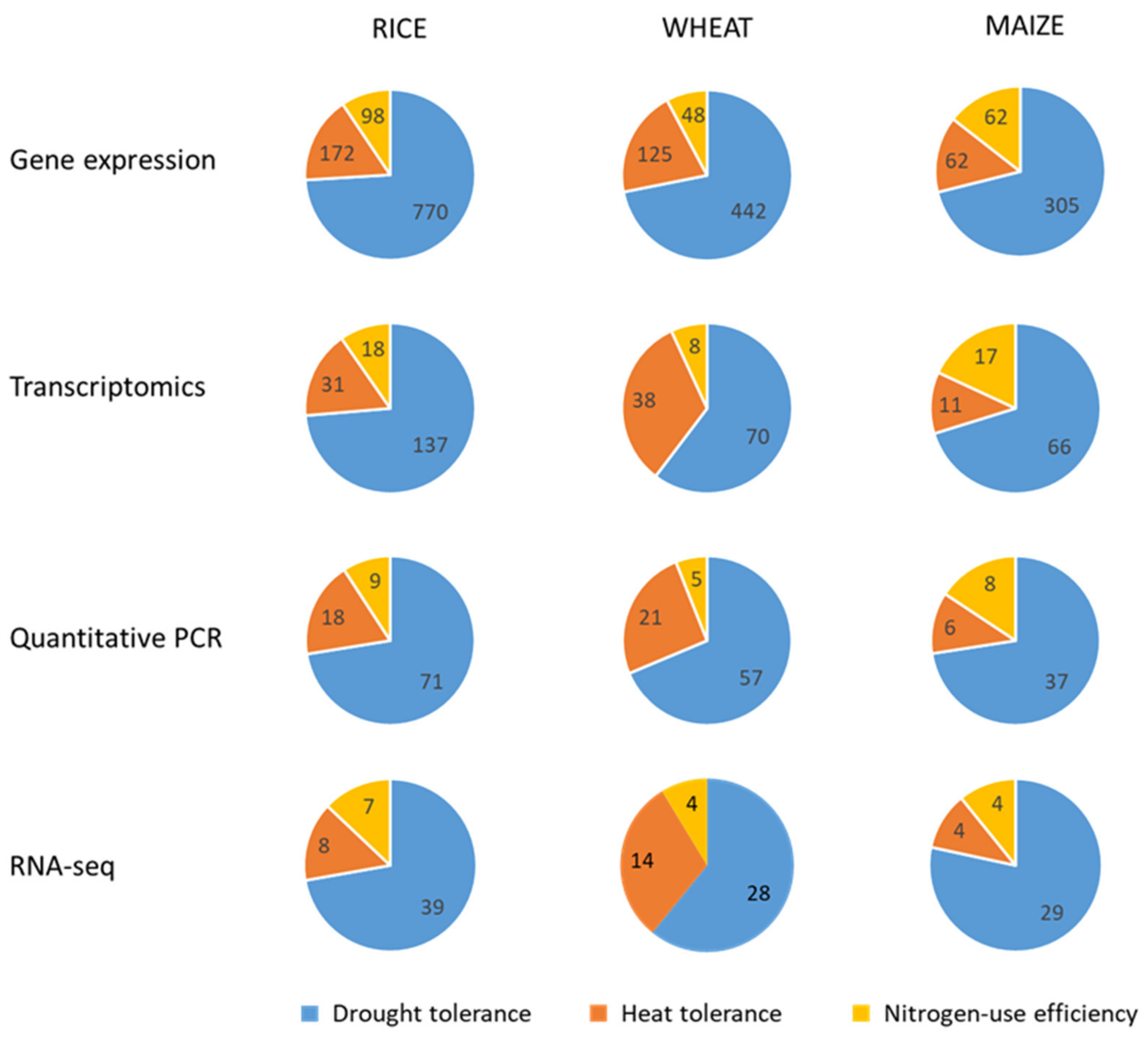
3. Gene Expression Analysis
3.1. Gene Expression Analysis Approaches
3.2. Gene Expression Analysis Oriented to Crop Improvement for Abiotic Limitations’ Response
3.2.1. Rice
3.2.2. Wheat
3.2.3. Maize
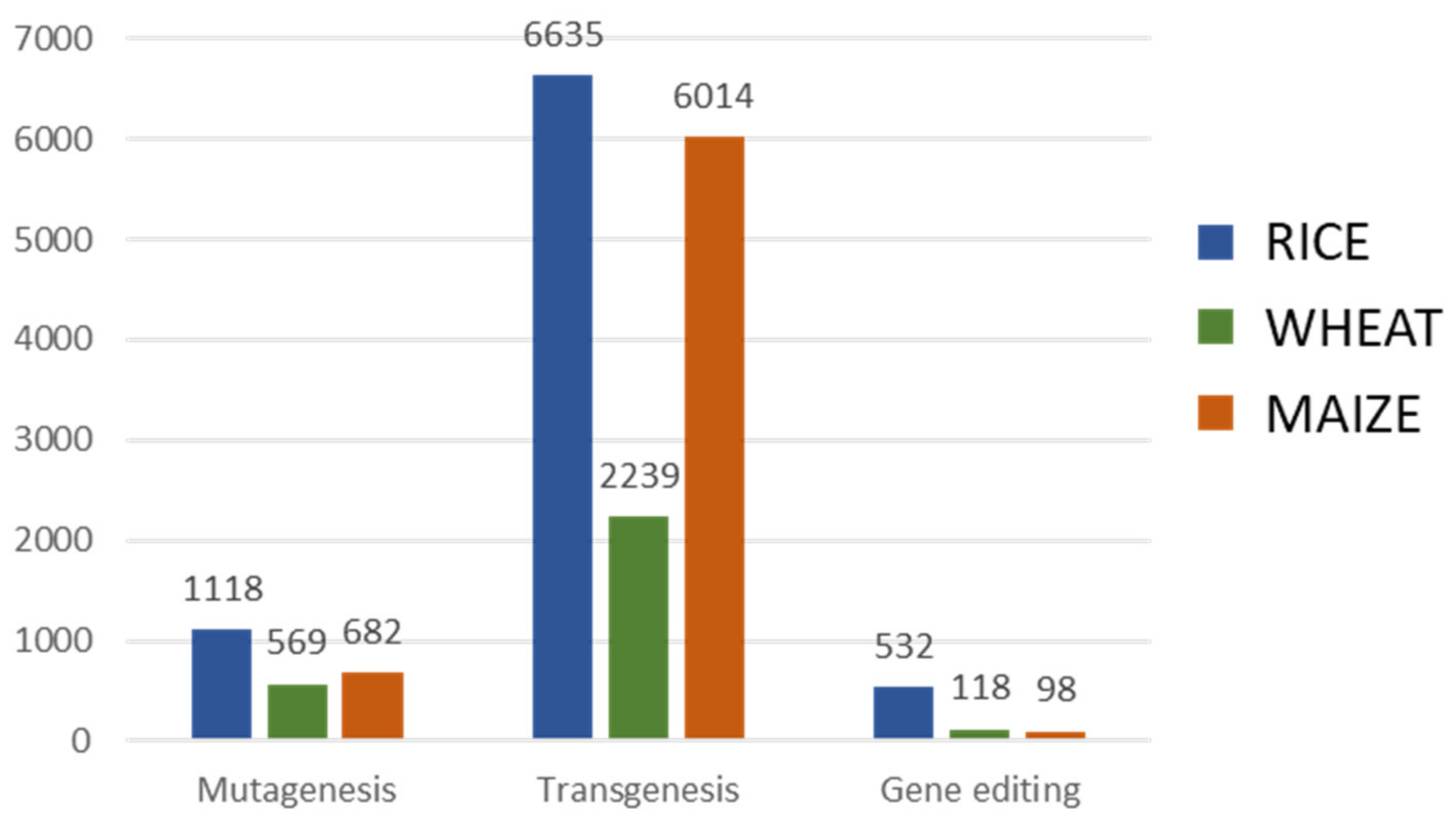
4. Genetic Modification
4.1. Mutagenesis
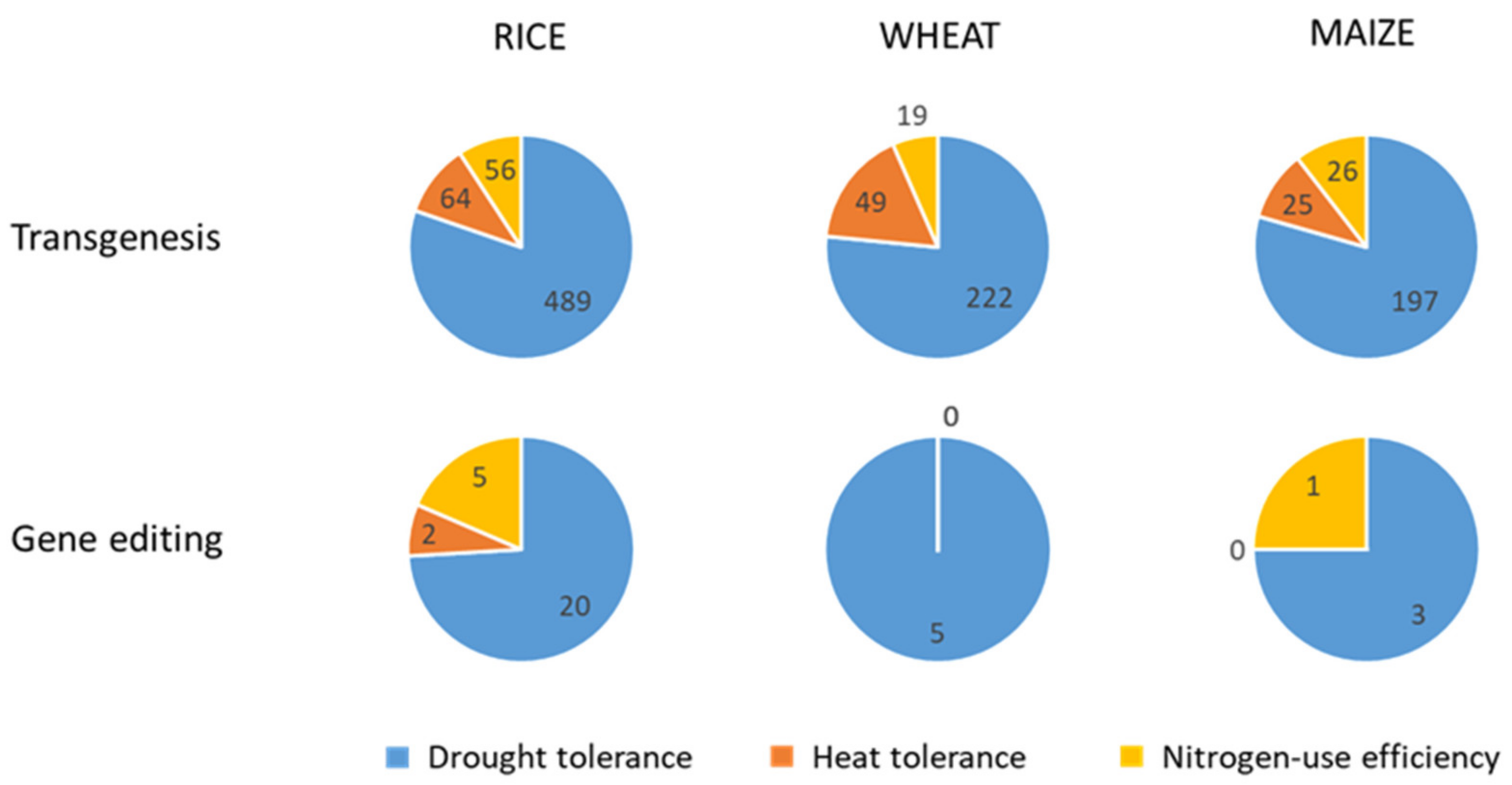
4.2. Transgenesis
4.3. Gene Editing
4.4. Genetic Modification Oriented to Crop Improvement for Abiotic Limitations’ Response
4.4.1. Rice
4.4.2. Wheat
4.4.3. Maize
5. Future Prospects
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gross, B.L.; Olsen, K.M. Genetic perspectives on crop domestication. Trends Plant Sci. 2010, 15, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langridge, P. Innovation in breeding and biotechnology. In Agriculture and Food Systems to 2050. Global Trends, Challenges and Opportunities; Serraj, R., Pingali, P.L., Eds.; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2019; pp. 245–284. [Google Scholar] [CrossRef]
- Acquaah, G. Principles of Plant Genetics and Breeding; John Wiley & Sons: Malden, MA, USA, 2009. [Google Scholar]
- Nejat, N.; Ramalingam, A.; Mantri, N. Advances in transcriptomics of plants. In Plant Genetics and Molecular Biology. Advances in Biochemical Engineering/Biotechnology; Varshney, R., Pandey, M., Chitikineni, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; Volume 164, pp. 161–185. [Google Scholar]
- Kumpatla, S.P.; Buyyarapu, R.; Abdurakhmonov, I.Y.; Mammadov, J.A. Genomics-assisted plant breeding in the 21st century: Technological advances and progress. In Plant Breeding; Abdurakhmonov, I., Ed.; Intechopen: London, UK, 2012; pp. 131–184. Available online: https://www.intechopen.com/books/plant-breeding/genomics-assisted-plant-breeding-in-the-21st-century-technological-advances-and-progresspp (accessed on 8 October 2020).
- Awika, J.M. Major Cereal Grains Production and Use around the World. In ACS Symposium Series; American Chemical Society (ACS): Washington, DC, USA, 2011; pp. 1–13. [Google Scholar] [CrossRef]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.-S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef]
- Jackson, S.A. Rice: The First Crop Genome. Rice 2016, 9, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Shimamoto, K. Genetic manipulation of rice: From protoplasts to transgenic plants. Jpn. J. Genet. 1992, 67, 273–290. [Google Scholar] [CrossRef] [Green Version]
- Helmy, M.; Tomita, M.; Ishihama, Y. OryzaPG-DB: Rice proteome database based on shotgun proteogenomics. BMC Plant Biol. 2011, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- McCouch, S.R.; Zhao, K.; Wright, M.; Tung, C.-W.; Ebana, K.; Thomson, M.; Reynolds, A.; Wang, D.; Declerck, G.; Ali, L.; et al. Development of genome-wide SNP assays for rice. Breed. Sci. 2010, 60, 524–535. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Tung, C.-W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.R.; Mezey, J.G.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef] [PubMed]
- Viana, V.E.; Pegoraro, C.; Busanello, C.; De Oliveira, A.C. Mutagenesis in Rice: The Basis for Breeding a New Super Plant. Front. Plant Sci. 2019, 10, 1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohapatra, T.; Robin, S.; Sarla, N.; Sheshasayee, M.; Singh, A.K.; Singh, K.; Mithra, S.V.A.; Sharma, R.P. EMS Induced Mutants of Upland Rice Variety Nagina22: Generation and Characterization. Proc. Indian Natl. Sci. Acad. 2014, 80, 163. [Google Scholar] [CrossRef]
- Li, J.-Y.; Wang, J.; Zeigler, R.S. The 3,000 rice genomes project: New opportunities and challenges for future rice research. Gigascience 2014, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, 7191. [Google Scholar] [CrossRef] [Green Version]
- Sears, E. Cytogenetic studies with polyploid species of wheat. I. Chromosomal aberrations in the progeny of a haploid of Triticum vulgare. Genetics 1939, 24, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Sears, E.; Miller, T. The history of Chinese Spring wheat. Cereal Res. Commun. 1985, 13, 261–263. [Google Scholar]
- De Caleya, R.F.; Hernandez-Lucas, C.; Carbonero, P.; Garcia-Olmedo, F. Gene Expression in Alloploids: Genetic Control of Lipopurothionins in Wheat. Genetics 1976, 83, 687–699. [Google Scholar]
- Gupta, P.K.; Vasistha, N.K. Wheat cytogenetics and cytogenomics: The present status. Nucleus 2018, 61, 195–212. [Google Scholar] [CrossRef]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G.; et al. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Garcia, D.F.; Zhou, Y.; Appels, R.; Li, A.; Mao, L. The Battle to Sequence the Bread Wheat Genome: A Tale of the Three Kingdoms. Genom. Proteom. Bioinform. 2020, 18, 221–229. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Helentjaris, T.; Weber, D.; Wright, S. Identification of the genomic locations of duplicate nucleotide sequences in maize by analysis of restriction fragment length polymorphisms. Genetics 1988, 118, 353–363. [Google Scholar] [CrossRef]
- Gale, M.D. Plant Comparative Genetics after 10 Years. Science 1998, 282, 656–659. [Google Scholar] [CrossRef] [Green Version]
- Fischer, R.; Byerlee, D.; Edmeades, G. Crop Yields and Global Food Security; Australian Centre for International Agricultural Research: Canberra, Australia, 2014. [Google Scholar]
- Jiang, G.-L. Molecular markers and marker-assisted breeding in plants. In Plant Breeding from Laboratories to Fields; Andersen, S.B., Ed.; IntechOpen: London, UK, 2013; pp. 45–83. [Google Scholar] [CrossRef] [Green Version]
- Mitchell-Olds, T. Complex-trait analysis in plants. Genome Biol. 2010, 11, 1–3. [Google Scholar] [CrossRef]
- Liller, C.B.; Walla, A.; Boer, M.P.; Hedley, P.; Macaulay, M.; Effgen, S.; Von Korff, M.; Van Esse, G.W.; Koornneef, M. Fine mapping of a major QTL for awn length in barley using a multiparent mapping population. Theor. Appl. Genet. 2016, 130, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S. Structure of Linkage Disequilibrium in Plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alqudah, A.M.; Sallam, A.; Baenziger, P.S.; Börner, A. GWAS: Fast-forwarding gene identification and characterization in temperate Cereals: Lessons from Barley—A review. J. Adv. Res. 2020, 22, 119–135. [Google Scholar] [CrossRef]
- Zhao, K.; Aranzana, M.J.; Kim, S.; Lister, C.; Shindo, C.; Tang, C.; Toomajian, C.; Zheng, H.; Dean, C.; Marjoram, P. An Arabidopsis example of association mapping in structured samples. PLoS Genet. 2007, 3, e4. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Mol. Plant 2017, 10, 359–374. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.-X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M. Breeding strategies: Optimum design of marker-assisted backcross programs. In Molecular Marker Systems in Plant Bredding and Crop Improvement; Lorz, H., Wenzel, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 55, pp. 319–334. [Google Scholar]
- Meuwissen, T. Genomic selection: Marker assisted selection on a genome wide scale. J. Anim. Breed. Genet. 2007, 124, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Voss-Fels, K.P.; Cooper, M.; Hayes, B.J. Accelerating crop genetic gains with genomic selection. Theor. Appl. Genet. 2019, 132, 669–686. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, Y.; Hu, Z.; Xu, C. Genomic selection methods for crop improvement: Current status and prospects. Crop. J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- Pham, J.-L. Identification of genetic markers for quantitative traits in rice (Oryza sativa L.). Comptes Rendus Acad. Sci. Ser. 3 Sci. Vie 1990, 310, 477–483. [Google Scholar]
- Miura, H.; Parker, B.B.; Snape, J.W. The location of major genes and associated quantitative trait loci on chromosome arm 5BL of wheat. Theor. Appl. Genet. 1992, 85, 197–204. [Google Scholar] [CrossRef]
- Edwards, M.D.; Stuber, C.W.; Wendel, J.F. Molecular-Marker-Facilitated Investigations of Quantitative-Trait Loci in Maize. I.; Numbers, Genomic Distribution and Types of Gene Action. Genetics 1987, 116, 113–125. [Google Scholar] [CrossRef]
- Champoux, M.C.; Wang, G.; Sarkarung, S.; Mackill, D.J.; O’Toole, J.C.; Huang, N.; McCouch, S.R. Locating genes associated with root morphology and drought avoidance in rice via linkage to molecular markers. Theor. Appl. Genet. 1995, 90, 969–981. [Google Scholar] [CrossRef]
- Quarrie, S.A.; Gullì, M.; Calestani, C.; Steed, A.; Marmiroli, N. Location of a gene regulating drought-induced abscisic acid production on the long arm of chromosome 5A of wheat. Theor. Appl. Genet. 1994, 89, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, C.; Lazić-Jančić, V.; Steed, A.; Pekić, S.; Quarrie, S. Identification of QTL for drought responses in maize and their use in testing causal relationships between traits. J. Exp. Bot. 1995, 46, 853–865. [Google Scholar] [CrossRef]
- Tripathy, J.N.; Zhang, J.; Robin, S.; Nguyen, T.T.; Nguyen, H.T. QTLs for cell-membrane stability mapped in rice (Oryza sativa L.) under drought stress. Theor. Appl. Genet. 2000, 100, 1197–1202. [Google Scholar] [CrossRef]
- Yang, J.; Sears, R.; Gill, B.; Paulsen, G. Quantitative and molecular characterization of heat tolerance in hexaploid wheat. Euphytica 2002, 126, 275–282. [Google Scholar] [CrossRef]
- Ottaviano, E.; Gorla, M.S.; Pè, E.; Frova, C. Molecular markers (RFLPs and HSPs) for the genetic dissection of thermotolerance in maize. Theor. Appl. Genet. 1991, 81, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, K.; Kobayashi, N.; Ono, K.; Yano, M.; Ohsugi, R. Are contents of Rubisco, soluble protein and nitrogen in flag leaves of rice controlled by the same genetics? J. Exp. Bot. 2001, 52, 1827–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, V.; Foulkes, M.J.; Worland, A.J.; Sylvester-Bradley, R.; Caligari, P.D.S.; Snape, J.W. Mapping quantitative trait loci for flag leaf senescence as a yield determinant in winter wheat under optimal and drought-stressed environments. Euphytica 2004, 135, 255–263. [Google Scholar] [CrossRef]
- Agrama, H.; Zakaria, A.; Said, F.; Tuinstra, M. Identification of quantitative trait loci for nitrogen use efficiency in maize. Mol. Breed. 1999, 5, 187–195. [Google Scholar] [CrossRef]
- Yang, X.; Jawdy, S.; Tschaplinski, T.J.; Tuskan, G.A. Genome-wide identification of lineage-specific genes in Arabidopsis, Oryza and Populus. Genomics 2009, 93, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Wang, L.; Liu, L.; Zhao, C.; Zheng, Y. Association mapping of agronomic traits on chromosome 2A of wheat. Genetica 2009, 137, 67–75. [Google Scholar] [CrossRef]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flintgarcia, S.A.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Nuruzzaman, M.; Manimekalai, R.; Sharoni, A.M.; Satoh, K.; Kondoh, H.; Ooka, H.; Kikuchi, S. Genome-wide analysis of NAC transcription factor family in rice. Gene 2010, 465, 30–44. [Google Scholar] [CrossRef]
- Edae, E.A.; Byrne, P.F.; Haley, S.D.; Lopes, M.S.; Reynolds, M.P. Genome-wide association mapping of yield and yield com-ponents of spring wheat under contrasting moisture regimes. Theor. Appl. Genet. 2014, 127, 791–807. [Google Scholar] [CrossRef]
- Li, L.; Hao, Z.; Li, X.; Xie, C.; Li, M.; Zhang, D.; Weng, J.; Su, Z.; Liang, X.; Zhang, S. An analysis of the polymorphisms in a gene for being involved in drought tolerance in maize. Genetica 2011, 139, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Dingkuhn, M.; Pasco, R.; Pasuquin, J.M.; Damo, J.; Soulié, J.-C.; Raboin, L.-M.; Dusserre, J.; Sow, A.; Manneh, B.; Shrestha, S.; et al. Crop-model assisted phenomics and genome-wide association study for climate adaptation of indica rice. 2. Thermal stress and spikelet sterility. J. Exp. Bot. 2017, 68, 4389–4406. [Google Scholar] [CrossRef] [PubMed]
- Elbasyoni, I.; Saadalla, M.; Baenziger, S.; Bockelman, H.; Morsy, S. Cell Membrane Stability and Association Mapping for Drought and Heat Tolerance in a Worldwide Wheat Collection. Sustainability 2017, 9, 1606. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, S.; Zhou, Z.; Wang, S.; Dong, C.; Mu, C.; Song, Y.; Ma, P.; Li, C.; Wang, Z.; et al. Linkage mapping and genome-wide association reveal candidate genes conferring thermotolerance of seed-set in maize. J. Exp. Bot. 2019, 70, 4849–4864. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Ye, J.; Yao, X.; Zhao, P.; Xuan, W.; Tian, Y.; Zhang, Y.; Xu, S.; An, H.; Chen, G.; et al. Genome-wide associated study identifies NAC42-activated nitrate transporter conferring high nitrogen use efficiency in rice. Nat. Commun. 2019, 10, 5279. [Google Scholar] [CrossRef] [Green Version]
- Cormier, F.; Le Gouis, J.; Dubreuil, P.; Lafarge, S.; Praud, S. A genome-wide identification of chromosomal regions determining nitrogen use efficiency components in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2014, 127, 2679–2693. [Google Scholar] [CrossRef] [PubMed]
- Morosini, J.S.; Mendonça, L.D.F.; Lyra, D.H.; Galli, G.; Vidotti, M.S.; Fritsche-Neto, R. Association mapping for traits related to nitrogen use efficiency in tropical maize lines under field conditions. Plant Soil 2017, 421, 453–463. [Google Scholar] [CrossRef]
- Xu, S.; Zhu, D.; Zhang, Q. Predicting hybrid performance in rice using genomic best linear unbiased prediction. Proc. Natl. Acad. Sci. USA 2014, 111, 12456–12461. [Google Scholar] [CrossRef] [Green Version]
- Heffner, E.L.; Jannink, J.L.; Sorrells, M.E. Genomic selection accuracy using multifamily prediction models in a wheat breeding program. Plant Genome 2011, 4, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, R.; Yu, J. Prospects for Genomewide Selection for Quantitative Traits in Maize. Crop. Sci. 2007, 47, 1082–1090. [Google Scholar] [CrossRef] [Green Version]
- Ben Hassen, M.; Bartholomé, J.; Valè, G.; Cao, T.-V.; Ahmadi, N. Genomic Prediction Accounting for Genotype by Environment Interaction Offers an Effective Framework for Breeding Simultaneously for Adaptation to an Abiotic Stress and Performance Under Normal Cropping Conditions in Rice. G3 Genes Genomes Genet. 2018, 8, 2319–2332. [Google Scholar] [CrossRef] [PubMed]
- Ly, D.; Huet, S.; Gauffreteau, A.; Rincent, R.; Touzy, G.; Mini, A.; Jannink, J.-L.; Cormier, F.; Paux, E.; Lafarge, S.; et al. Whole-genome prediction of reaction norms to environmental stress in bread wheat (Triticum aestivum L.) by genomic random regression. Field Crop. Res. 2018, 216, 32–41. [Google Scholar] [CrossRef]
- Ziyomo, C.; Bernardo, R. Drought Tolerance in Maize: Indirect Selection through Secondary Traits versus Genomewide Selection. Crop. Sci. 2013, 53, 1269–1275. [Google Scholar] [CrossRef] [Green Version]
- Van Inghelandt, D.; Frey, F.P.; Ries, D.; Stich, B. QTL mapping and genome-wide prediction of heat tolerance in multiple connected populations of temperate maize. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, C.; Jiang, Y.; Tian, Y.; Yu, J.; An, H.; Tang, W.; Sun, J.; Tang, J.; Chen, G. Association mapping and genetic dis-section of nitrogen use efficiency-related traits in rice (Oryza sativa L.). Funct. Integr. Genom. 2016, 16, 323–333. [Google Scholar] [CrossRef]
- Michel, S.; Löschenberger, F.; Ametz, C.; Pachler, B.; Sparry, E.; Bürstmayr, H. Simultaneous selection for grain yield and protein content in genomics-assisted wheat breeding. Theor. Appl. Genet. 2019, 132, 1745–1760. [Google Scholar] [CrossRef]
- Semagn, K.; Beyene, Y.; Babu, R.; Nair, S.; Gowda, M.; Das, B.; Tarekegne, A.; Mugo, S.; Mahuku, G.; Worku, M.; et al. Quantitative Trait Loci Mapping and Molecular Breeding for Developing Stress Resilient Maize for Sub-Saharan Africa. Crop. Sci. 2015, 55, 1449–1459. [Google Scholar] [CrossRef]
- McCough, S.R.; Doerge, R.W. QTL mapping in rice. Trends Genet. 1995, 11, 482–487. [Google Scholar] [CrossRef]
- Jagadish, S.V.K.; Cairns, J.; Lafitte, R.; Wheeler, T.R.; Price, A.H.; Craufurd, P.Q. Genetic Analysis of Heat Tolerance at Anthesis in Rice. Crop. Sci. 2010, 50, 1633–1641. [Google Scholar] [CrossRef]
- Wei, D.; Cui, K.; Ye, G.; Pan, J.; Xiang, J.; Huang, J.; Nie, L. QTL mapping for nitrogen-use efficiency and nitrogen-deficiency tolerance traits in rice. Plant Soil 2012, 359, 281–295. [Google Scholar] [CrossRef]
- Raju, B.R.; Mohankumar, M.V.; Sumanth, K.K.; Rajanna, M.P.; Udayakumar, M.; Prasad, T.G.; Sheshshayee, M.S. Discovery of QTLs for water mining and water use efficiency traits in rice under water-limited condition through association mapping. Mol. Breed. 2016, 36, 35. [Google Scholar] [CrossRef]
- Roja, V.; Patil, S.; Deborah, D.A.; Srividhya, A.; Ranjitkumar, N.; Kadambari, G.; Ramanarao, P.V.; Siddiq, E.A.; Vemireddy, L.R. Finding genomic regions and candidate genes governing water use efficiency in rice. Biol. Plant. 2016, 60, 757–766. [Google Scholar] [CrossRef]
- An, H.; Liu, K.; Wang, B.; Tian, Y.; Ge, Y.; Zhang, Y.; Tang, W.; Chen, G.; Yu, J.; Wu, W.; et al. Genome-wide association study identifies QTLs conferring salt tolerance in rice. Plant Breed. 2020, 139, 73–82. [Google Scholar] [CrossRef]
- Ghomi, K.; Rabiei, B.; Sabouri, H.; Sabouri, A. Mapping QTLs for Traits Related to Salinity Tolerance at Seedling Stage of Rice (Oryza sativa L.): An Agrigenomics Study of an Iranian Rice Population. OMICS J. Integr. Biol. 2013, 17, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, P.; Dvorak, J.; Mackell, D.; Deal, K.; Gregorio, G. RFLP and SSLP mapping of salinity tolerance genes in chromosome 1 of rice (Oryza sativa L.) using recombinant inbred lines. Philipp. Agric. Sci. 2002, 85, 68–76. [Google Scholar]
- Fukuda, A.; Kondo, K.; Ikka, T.; Takai, T.; Tanabata, T.; Yamamoto, T. A novel QTL associated with rice canopy temperature difference affects stomatal conductance and leaf photosynthesis. Breed. Sci. 2018, 68, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, T.; Ohsumi, A.; Arai, Y.; Iwasawa, N.; Yano, M.; Yamamoto, T.; Yoshinaga, S.; Kondo, M. QTL Analysis of Leaf Photosynthesis in Rice. Jpn. Agric. Res. Q. JARQ 2013, 47, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Vinod, K.K.; Heuer, S. Approaches towards nitrogen- and phosphorus-efficient rice. AoB Plants 2012, 2012, pls028. [Google Scholar] [CrossRef] [Green Version]
- Hartley, T.N.; Thomas, A.S.; Maathuis, F.J.M. A role for the OsHKT 2;1 sodium transporter in potassium use efficiency in rice. J. Exp. Bot. 2020, 71, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Pariasca-Tanaka, J.; Baertschi, C.; Wissuwa, M. Identification of Loci Through Genome-Wide Association Studies to Improve Tolerance to Sulfur Deficiency in Rice. Front. Plant Sci. 2020, 10, 1668. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Burow, M.D.; Le, B.T.; Le, T.D.; Paterson, A.H. Molecular mapping of genes conferring aluminum tolerance in rice (Oryza sativa L.). Theor. Appl. Genet. 2001, 102, 1002–1010. [Google Scholar] [CrossRef]
- Singh, R.; Singh, Y.; Xalaxo, S.; Verulkar, S.; Yadav, N.; Singh, S.; Singh, N.; Prasad, K.; Kondayya, K.; Rao, P.R. From QTL to variety-harnessing the benefits of QTLs for drought, flood and salt tolerance in mega rice varieties of India through a multi-institutional network. Plant Sci. 2016, 242, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Chen, K.; Cui, Y.; Wu, Z.; Zheng, T.; Zhu, Y.; Ali, J.; Wang, B.; Xu, J.; Zhang, W.; et al. Genetic Dissection and Simultaneous Improvement of Drought and Low Nitrogen Tolerances by Designed QTL Pyramiding in Rice. Front. Plant Sci. 2018, 9, 306. [Google Scholar] [CrossRef] [Green Version]
- Dharmappa, P.M.; Doddaraju, P.; Malagondanahalli, M.V.; Rangappa, R.B.; Mallikarjuna, N.M.; Rajendrareddy, S.H.; Ramanjinappa, R.; Mavinahalli, R.P.; Prasad, T.G.; Udayakumar, M.; et al. Introgression of Root and Water Use Efficiency Traits Enhances Water Productivity: An Evidence for Physiological Breeding in Rice (Oryza sativa L.). Rice 2019, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Aversano, R.; Ercolano, M.R.; Caruso, I.; Fasano, C.; Rosellini, D.; Carputo, D. Molecular Tools for Exploring Polyploid Genomes in Plants. Int. J. Mol. Sci. 2012, 13, 10316–10335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthukumar, C.; Subathra, T.; Aiswarya, J.; Gayathri, V.; Babu, R.C. Comparative genome-wide association studies for plant production traits under drought in diverse rice (Oryza sativa L.) lines using SNP and SSR markers. Curr. Sci. 2015, 109, 139–147. [Google Scholar]
- Bhandari, A.; Bartholomé, J.; Cao-Hamadoun, T.-V.; Kumari, N.; Frouin, J.; Kumar, A.; Ahmadi, N. Selection of trait-specific markers and multi-environment models improve genomic predictive ability in rice. PLoS ONE 2019, 14, e0208871. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.A.; Sorrells, M.E.; Tanksley, S.D. RFLP Analysis of Genomic Regions Associated with Resistance to Preharvest Sprouting in Wheat. Crop. Sci. 1993, 33, 453–459. [Google Scholar] [CrossRef]
- Gupta, P.K.; Balyan, H.S.; Sharma, S.; Kumar, R. Genetics of yield, abiotic stress tolerance and biofortification in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2020, 133, 1569–1602. [Google Scholar] [CrossRef]
- Cormier, F.; Foulkes, J.; Hirel, B.; Gouache, D.; Moënne-Loccoz, Y.; Le Gouis, J. Breeding for increased nitrogen-use efficiency: A review for wheat (T. aestivum L.). Plant Breed. 2016, 135, 255–278. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Gao, M.; Zheng, H.; Yuan, Y.; Zhou, X.; Guo, Y.; Zhang, G.; Zhao, Y.; Kong, F.; An, Y.; et al. QTL mapping for nitrogen use efficiency and agronomic traits at the seedling and maturity stages in wheat. Mol. Breed. 2019, 39, 71. [Google Scholar] [CrossRef]
- Mérida-García, R.; Bentley, A.R.; Gálvez, S.; Dorado, G.; Solís, I.; Ammar, K.; Hernandez, P. Mapping Agronomic and Quality Traits in Elite Durum Wheat Lines under Differing Water Regimes. Agronomy 2020, 10, 144. [Google Scholar] [CrossRef] [Green Version]
- Pascual, L.; Ruiz, M.; López-Fernández, M.; Pérez-Peña, H.; Benavente, E.; Vázquez, J.F.; Sansaloni, C.; Giraldo, P. Genomic analysis of Spanish wheat landraces reveals their variability and potential for breeding. BMC Genom. 2020, 21, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Dong, Z.; Zhao, L.; Ren, Y.; Zhang, N.; Chen, F. The Wheat 660K SNP array demonstrates great potential for marker-assisted selection in polyploid wheat. Plant Biotechnol. J. 2020, 18, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M. Molecular marker-assisted selection of HMW glutenin alleles related to wheat bread quality by PCR-generated DNA markers. Theor. Appl. Genet. 2000, 101, 892–896. [Google Scholar] [CrossRef]
- Vida, G.; Gál, M.; Uhrin, A.; Veisz, O.; Syed, N.H.; Flavell, A.J.; Wang, Z.; Bedö, Z. Molecular markers for the identification of resistance genes and marker-assisted selection in breeding wheat for leaf rust resistance. Euphytica 2009, 170, 67–76. [Google Scholar] [CrossRef]
- Mohammadi, R. Breeding for increased drought tolerance in wheat: A review. Crop. Pasture Sci. 2018, 69, 223–241. [Google Scholar] [CrossRef]
- Juliana, P.; Poland, J.; Huerta-Espino, J.; Shrestha, S.; Crossa, J.; Crespo-Herrera, L.; Toledo, F.H.; Govindan, V.; Mondal, S.; Kumar, U.; et al. Improving grain yield, stress resilience and quality of bread wheat using large-scale genomics. Nat. Genet. 2019, 51, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Guzman, C.; Peña, R.J.; Singh, R.; Autrique, E.; Dreisigacker, S.; Crossa, J.; Rutkoski, J.; Poland, J.; Battenfield, S. Wheat quality improvement at CIMMYT and the use of genomic selection on it. Appl. Transl. Genom. 2016, 11, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Zaïm, M.; Kabbaj, H.; Kehel, Z.; Gorjanc, G.; Filali-Maltouf, A.; Belkadi, B.; Nachit, M.M.; Bassi, F.M. Combining QTL Analysis and Genomic Predictions for Four Durum Wheat Populations Under Drought Conditions. Front. Genet. 2020, 11, 316. [Google Scholar] [CrossRef]
- Hao, Z.; Li, X.; Liu, X.; Xie, C.; Li, M.; Zhang, D.; Zhang, S. Meta-analysis of constitutive and adaptive QTL for drought tol-erance in maize. Euphytica 2010, 174, 165–177. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, B.; Yu, F.; Li, L.; Wang, M.; Xue, Y.; Zhang, Z.; Yan, J.; Yue, B.; Zheng, Y.; et al. Identification of Major QTL for Waterlogging Tolerance Using Genome-Wide Association and Linkage Mapping of Maize Seedlings. Plant Mol. Biol. Rep. 2012, 31, 594–606. [Google Scholar] [CrossRef]
- Pestsova, E.; Lichtblau, D.; Wever, C.; Presterl, T.; Bolduan, T.; Ouzunova, M.; Westhoff, P. QTL mapping of seedling root traits associated with nitrogen and water use efficiency in maize. Euphytica 2015, 209, 585–602. [Google Scholar] [CrossRef]
- Luo, M.; Zhao, Y.; Zhang, R.; Xing, J.; Duan, M.; Li, J.; Wang, N.; Wang, W.; Zhang, S.; Chen, Z.; et al. Mapping of a major QTL for salt tolerance of mature field-grown maize plants based on SNP markers. BMC Plant Biol. 2017, 17, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zehr, B.E.; Dudley, J.W.; Chojecki, J.; Maroof, M.A.S.; Mowers, R.P. Use of RFLP markers to search for alleles in a maize population for improvement of an elite hybrid. Theor. Appl. Genet. 1992, 83, 903–911. [Google Scholar] [CrossRef]
- Silvela, L.; Ayuso, M.; Gil-Delgado, L.; Salaices, L. Genetic and environmental contributions to bread-wheat flour quality using the SDS sedimentation test as an index. Theor. Appl. Genet. 1993, 86, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.G.; Eun, M.Y.; McCouch, S.R.; Chae, Y.A. The semidwarf gene, sd-1, of rice (Oryza sativa L.). II. Molecular mapping and marker-assisted selection. Theor. Appl. Genet. 1994, 89, 54–59. [Google Scholar] [CrossRef]
- Scandalios, J.G.; Felder, M.R. Developmental expression of alcohol dehydrogenases in maize. Dev. Biol. 1971, 25, 641–654. [Google Scholar] [CrossRef]
- Luig, N.; Rajaram, S. The effect of temperature and genetic background on host gene expression and interaction to Puccinia graminis tritici. Phytopathology 1972, 62, 1171–1174. [Google Scholar] [CrossRef]
- Sano, Y. Differential regulation of waxy gene expression in rice endosperm. Theor. Appl. Genet. 1984, 68, 467–473. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; Mafra, V.S.; Machado, M.A. Transcriptomics. In Omics in Plant Breeding; Borem, A., Fritsche-Neto, R., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2014; pp. 33–57. [Google Scholar]
- Morozova, O.; Hirst, M.; Marra, M.A. Applications of New Sequencing Technologies for Transcriptome Analysis. Annu. Rev. Genom. Hum. Genet. 2009, 10, 135–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, J.H.; Tan, S.S.; Martin, B.K.; Wittwer, C.T. Detection of rare mRNAs via quantitative RT-PCR. Trends Genet. 1992, 8, 263–264. [Google Scholar] [CrossRef]
- Parker, R.M.; Barnes, N.M. mRNA: Detection by in Situ and northern hybridization. Methods Mol. Biol. 1999, 106, 247–283. [Google Scholar] [PubMed]
- Bustin, S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000, 25, 169–193. [Google Scholar] [CrossRef] [Green Version]
- Deepak, S.; Kottapalli, K.; Rakwal, R.; Oros, G.; Rangappa, K.; Iwahashi, H.; Masuo, Y.; Agrawal, G. Real-time PCR: Revolutionizing detection and expression analysis of genes. Curr. Genom. 2007, 8, 234–251. [Google Scholar] [CrossRef]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative Monitoring of Gene Expression Patterns with a Complementary DNA Microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [Green Version]
- Zik, M.; Irish, V.F. Global Identification of Target Genes Regulated by APETALA3 and PISTILLATA Floral Homeotic Gene Action. Plant Cell 2002, 15, 207–222. [Google Scholar] [CrossRef] [Green Version]
- Harmer, S.L.; HogenEsch, J.B.; Straume, M.; Chang, H.-S.; Han, B.; Zhu, T.; Wang, X.; Kreps, J.A.; Kay, S.A. Orchestrated Transcription of Key Pathways in Arabidopsis by the Circadian Clock. Science 2000, 290, 2110–2113. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, Q.; Zhang, Y.; Zhang, Y.; Xing, J.; Yang, B.; Mi, G.; Li, Z.; Zhang, M. The Role of Gibberellins in Regulation of Nitrogen Uptake and Physiological Traits in Maize Responding to Nitrogen Availability. Int. J. Mol. Sci. 2020, 21, 1824. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, H.; Shao, H.; Tang, X. Recent Advances in Utilizing Transcription Factors to Improve Plant Abiotic Stress Tolerance by Transgenic Technology. Front. Plant Sci. 2016, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Mundy, J.; Chua, N.H. Abscisic acid and water-stress induce the expression of a novel rice gene. EMBO J. 1988, 7, 2279–2286. [Google Scholar] [CrossRef]
- Curry, J.; Morris, C.F.; Walker-Simmons, M.K. Sequence analysis of a cDNA encoding a Group 3 LEA mRNA inducible by ABA or dehydration stress in wheat. Plant Mol. Biol. 1991, 16, 1073–1076. [Google Scholar] [CrossRef]
- Bochicchio, A.; Vazzana, C.; Velasco, R.; Singh, M.; Bartels, D. Exogenous ABA induces desiccation tolerance and leads to the synthesis of specific gene transcription in immature embryos of maize. Maydica (Italy) 1991, 36, 11–16. [Google Scholar]
- Borkird, C.; Claes, B.; Caplan, A.; Simoens, C.; Van Montagu, M. Differential Expression of Water-Stress Associated Genes in Tissues of Rice Plants. J. Plant Physiol. 1991, 138, 591–595. [Google Scholar] [CrossRef]
- Vierling, R.A.; Nguyen, H.T. Heat-Shock Protein Gene Expression in Diploid Wheat Genotypes Differing in Thermal Tolerance. Crop. Sci. 1992, 32, 370–377. [Google Scholar] [CrossRef]
- Frova, C.; Gorla, M.S. Quantitative expression of maize HSPs: Genetic dissection and association with thermotolerance. Theor. Appl. Genet. 1993, 86, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Wang, S.; Zhang, J.; Feng, Q.; Zhang, L.; Fan, D.; Li, X.; Yuan, D.; Han, B.; Zhang, Q. Expression Profiles of 10,422 Genes at Early Stage of Low Nitrogen Stress in Rice Assayed using a cDNA Microarray. Plant Mol. Biol. 2006, 60, 617–631. [Google Scholar] [CrossRef]
- Bernard, S.M.; Møller, A.L.B.; Dionisio, G.; Kichey, T.; Jahn, T.P.; Dubois, F.; Baudo, M.; Lopes, M.S.; Tercé-Laforgue, T.; Foyer, C.H.; et al. Gene expression, cellular localisation and function of glutamine synthetase isozymes in wheat (Triticum aestivum L.). Plant Mol. Biol. 2008, 67, 89–105. [Google Scholar] [CrossRef]
- Martin, A.; Lee, J.; Kichey, T.; Gerentes, D.; Zivy, M.; Tatout, C.; Dubois, F.; Balliau, T.; Valot, B.; Davanture, M.; et al. Two Cytosolic Glutamine Synthetase Isoforms of Maize Are Specifically Involved in the Control of Grain Production. Plant Cell 2006, 18, 3252–3274. [Google Scholar] [CrossRef] [Green Version]
- Kohli, A.; Xiong, J.; Greco, R.; Christou, P.; Pereira, A. Tagged Transcriptome Display (TTD) in indica rice using Ac transposition. Mol. Genet. Genom. 2001, 266, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kashkush, K.; Feldman, M.; Levy, A.A. Gene loss, silencing and activation in a newly synthesized wheat allotetraploid. Genetics 2002, 160, 1651–1659. [Google Scholar]
- Wang, H.; Miyazaki, S.; Kawai, K.; Deyholos, M.; Galbraith, D.W.; Bohnert, H.J. Temporal progression of gene expression responses to salt shock in maize roots. Plant Mol. Biol. 2003, 52, 873–891. [Google Scholar] [CrossRef]
- Wu, C.-Q.; Hu, H.-H.; Zeng, Y.; Liang, D.-C.; Xie, K.-B.; Zhang, J.-W.; Chu, Z.-H.; Xiong, L.-Z. Identification of Novel Stress-responsive Transcription Factor Genes in Rice by cDNA Array Analysis. J. Integr. Plant Biol. 2006, 48, 1216–1224. [Google Scholar] [CrossRef]
- Ergen, N.Z.; Thimmapuram, J.; Bohnert, H.J.; Budak, H. Transcriptome pathways unique to dehydration tolerant relatives of modern wheat. Funct. Integr. Genom. 2009, 9, 377–396. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Ren, G.; Yue, G.; Li, Z.; Qu, X.; Hou, G.; Zhu, Y.; Zhang, J. Effects of water-deficit stress on the transcriptomes of developing immature ear and tassel in maize. Plant Cell Rep. 2007, 26, 2137–2147. [Google Scholar] [CrossRef]
- Antoine, W.; Stewart, J.M.; Reyes, B.G.D.L. The rice homolog of the sodium/lithium tolerance gene functions as molecular chaperon in vitro. Physiol. Plant. 2005, 125, 299–310. [Google Scholar] [CrossRef]
- Hays, D.; Mason, E.; Do, J.H.; Menz, M.; Reynolds, M. Expression quantitative trait loci mapping heat tolerance during reproductive development in wheat (Triticum aestivum). In Wheat Production in Stressed Environments; Buck, H.T., Nisi, J.E., Salomon, N., Eds.; Springer: Dordrecht, The Netherlands, 2007; Volume 12, p. 373. [Google Scholar]
- Dutra, S.; Von Pinho, E.; Santos, H.; Lima, A.; Von Pinho, R.; Carvalho, M. Genes related to high temperature tolerance during maize seed germination. Genet. Mol. Res. 2015, 14, 18047–18058. [Google Scholar] [CrossRef]
- Gregersen, P.L.; Holm, P.B.; Krupinska, K. Leaf senescence and nutrient remobilisation in barley and wheat. Plant Biol. 2008, 10, 37–49. [Google Scholar] [CrossRef]
- Clay, S.A.; Clay, D.E.; Horvath, D.P.; Pullis, J.; Carlson, C.G.; Hansen, S.; Reicks, G. Corn Response to Competition: Growth Alteration vs. Yield Limiting Factors. Agron. J. 2009, 101, 1522–1529. [Google Scholar] [CrossRef] [Green Version]
- Wasaki, J.; Yonetani, R.; Shinano, T.; Kai, M.; Osaki, M. Expression of the OsPI1 gene, cloned from rice roots using cDNA microarray, rapidly responds to phosphorus status. New Phytol. 2003, 158, 239–248. [Google Scholar] [CrossRef]
- Terzi, V.; Ferrari, B.; Finocchiaro, F.; Di Fonzo, N.; Stanca, A.M.; Lamacchia, C.; Napier, J.; Shewry, P.R.; Faccioli, P. TaqMan PCR for detection of genetically modified durum wheat. J. Cereal Sci. 2003, 37, 157–163. [Google Scholar] [CrossRef]
- Vaïtilingom, M.; Pijnenburg, H.; Gendre, F.; Brignon, P. Real-Time Quantitative PCR Detection of Genetically Modified Maximizer Maize and Roundup Ready Soybean in Some Representative Foods. J. Agric. Food Chem. 1999, 47, 5261–5266. [Google Scholar] [CrossRef]
- Kumar, K.; Rao, K.P.; Sharma, P.; Sinha, A.K. Differential regulation of rice mitogen activated protein kinase kinase (MKK) by abiotic stress. Plant Physiol. Biochem. 2008, 46, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Gallé, Á.; Csiszár, J.; Secenji, M.; Guóth, A.; Cseuz, L.; Tari, I.; Györgyey, J.; Erdei, L. Glutathione transferase activity and expression patterns during grain filling in flag leaves of wheat genotypes differing in drought tolerance: Response to water deficit. J. Plant Physiol. 2009, 166, 1878–1891. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.; Kakimoto, M.; Sakuma, Y.; Maruyama, K.; Osakabe, Y.; Tran, L.-S.P.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Regulation and functional analysis of ZmDREB2A in response to drought and heat stresses in Zea mays L. Plant J. 2007, 50, 54–69. [Google Scholar] [CrossRef]
- Khurana, P.; Chauhan, H. Characterization and expression of high temperature stress responsive genes in bread wheat (Triticum aestivum L.). Czech J. Genet. Plant Breed. 2011, 47, S94–S97. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.H.; Zhang, Y.L.; Ye, L.T.; Fan, X.R.; Xu, G.H.; Shen, Q.R. Responses of Rice Cultivars with Different Nitrogen Use Efficiency to Partial Nitrate Nutrition. Ann. Bot. 2007, 99, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Nigro, D.; Gu, Y.Q.; Huo, N.; Marcotuli, I.; Blanco, A.; Gadaleta, A.; Anderson, O.D. Structural Analysis of the Wheat Genes Encoding NADH-Dependent Glutamine-2-oxoglutarate Amidotransferases and Correlation with Grain Protein Content. PLoS ONE 2013, 8, e73751. [Google Scholar] [CrossRef]
- Chen, R.; Tian, M.; Wu, X.; Huang, Y. Differential global gene expression changes in response to low nitrogen stress in two maize inbred lines with contrasting low nitrogen tolerance. Genes Genom. 2011, 33, 491–497. [Google Scholar] [CrossRef]
- Zhang, G.; Guo, G.; Hu, X.; Zhang, Y.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X.; et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [Green Version]
- Pont, C.; Murat, F.; Confolent, C.; Balzergue, S.; Salse, J. RNA-seq in grain unveils fate of neo- and paleopolyploidization events in bread wheat (Triticum aestivum L.). Genome Biol. 2011, 12, 119. [Google Scholar] [CrossRef] [Green Version]
- Davidson, R.M.; Hansey, C.N.; Gowda, M.; Childs, K.L.; Lin, H.; Vaillancourt, B.; Sekhon, R.S.; De Leon, N.; Kaeppler, S.M.; Jiang, N.; et al. Utility of RNA Sequencing for Analysis of Maize Reproductive Transcriptomes. Plant Genome 2011, 4, 191–203. [Google Scholar] [CrossRef]
- Silveira, R.; Abreu, F.; Mamidi, S.; McClean, P.; Vianello, R.; Lanna, A.; Carneiro, N.; Brondani, C. Expression of drought tolerance genes in tropical upland rice cultivars (Oryza sativa). Genet. Mol. Res. 2015, 14, 8181–8200. [Google Scholar] [CrossRef]
- Okay, S.; Derelli, E.; Unver, T. Transcriptome-wide identification of bread wheat WRKY transcription factors in response to drought stress. Mol. Genet. Genom. 2014, 289, 765–781. [Google Scholar] [CrossRef]
- Kakumanu, A.; Ambavaram, M.M.; Klumas, C.; Krishnan, A.; Batlang, U.; Myers, E.; Grene, R.; Pereira, A. Effects of Drought on Gene Expression in Maize Reproductive and Leaf Meristem Tissue Revealed by RNA-Seq. Plant Physiol. 2012, 160, 846–867. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.-L.; Zhou, H.-W.; Peng, Q.; Zhong, P.-A.; Zhang, H.-Y.; He, C.; Huang, Y.-J. Transcriptome changes in rice (Oryza sativa L.) in response to high night temperature stress at the early milky stage. BMC Genom. 2015, 16, 18. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.R.; Goswami, S.; Sharma, S.K.; Kala, Y.K.; Rai, G.K.; Mishra, D.C.; Grover, M.; Singh, G.P.; Pathak, H.; Rai, A.; et al. Harnessing Next Generation Sequencing in Climate Change: RNA-Seq Analysis of Heat Stress-Responsive Genes in Wheat (Triticum aestivum L.). OMICS J. Integr. Biol. 2015, 19, 632–647. [Google Scholar] [CrossRef] [Green Version]
- Frey, F.P.; Urbany, C.; Hüttel, B.; Reinhardt, R.; Stich, B. Genome-wide expression profiling and phenotypic evaluation of European maize inbreds at seedling stage in response to heat stress. BMC Genom. 2015, 16, 123. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.K.; Sevanthi, V.A.M.; Chaudhary, S.; Tyagi, P.; Venkadesan, S.; Rani, M.; Mandal, P.K. Transcriptome analysis of two rice varieties contrasting for nitrogen use efficiency under chronic N starvation reveals differences in chloroplast and starch metabolism-related genes. Genes 2018, 9, 206. [Google Scholar] [CrossRef] [Green Version]
- Camilios-Neto, D.; Bonato, P.; Wassem, R.; Tadra-Sfeir, M.Z.; Brusamarello-Santos, L.C.C.; Valdameri, G.; Donatti, L.; Faoro, H.; Weiss, V.A.; Chubatsu, L.S.; et al. Dual RNA-seq transcriptional analysis of wheat roots colonized by Azospirillum brasilense reveals up-regulation of nutrient acquisition and cell cycle genes. BMC Genom. 2014, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Trevisan, S.; Manoli, A.; Ravazzolo, L.; Botton, A.; Pivato, M.; Masi, A.; Quaggiotti, S. Nitrate sensing by the maize root apex transition zone: A merged transcriptomic and proteomic survey. J. Exp. Bot. 2015, 66, 3699–3715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleck, K.; Levine, A.; Eulgem, T.; Morgan, A.; Schmid, J.; Lawton, K.A.; Dangl, J.L.; Dietrich, R.A. The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nat. Genet. 2000, 26, 403–410. [Google Scholar] [CrossRef]
- Swift, J.; Adame, M.; Tranchina, D.; Henry, A.; Coruzzi, G.M. Water impacts nutrient dose responses genome-wide to affect crop production. Nat. Commun. 2019, 10, 1374. [Google Scholar] [CrossRef] [PubMed]
- Wasaki, J.; Yonetani, R.; Kuroda, S.; Shinano, T.; Yazaki, J.; Fujii, F.; Shimbo, K.; Yamamoto, K.; Sakata, K.; Sasaki, T. Tran-scriptomic analysis of metabolic changes by phosphorus stress in rice plant roots. Plant Cell Environ. 2003, 26, 1515–1523. [Google Scholar] [CrossRef]
- Pillai, M.A.; Yanagihara, S.; Akiyama, T. Molecular cloning and characterization of salt responsive genes in rice (Oryza sativa). J. Plant Physiol. 2001, 158, 1189–1194. [Google Scholar] [CrossRef]
- Baulcombe, D.C.; Buffard, D. Gibberellic-acid-regulated expression of α-amylase and six other genes in wheat aleurone layers. Planta 1983, 157, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Winfield, M.O.; Lu, C.; Wilson, I.D.; Coghill, J.A.; Edwards, K.J. Plant responses to cold: Transcriptome analysis of wheat. Plant Biotechnol. J. 2010, 8, 749–771. [Google Scholar] [CrossRef] [Green Version]
- Henry, R.J.; Furtado, A.; Rangan, P. Wheat seed transcriptome reveals genes controlling key traits for human preference and crop adaptation. Curr. Opin. Plant Biol. 2018, 45, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Xie, Y.; Fan, S.; Wang, Z.; Wang, F.; Zhang, B.; Li, H.; Song, J.; Kong, L. Comparative analysis of root transcriptome profiles between drought-tolerant and susceptible wheat genotypes in response to water stress. Plant Sci. 2018, 272, 276–293. [Google Scholar] [CrossRef]
- Amirbakhtiar, N.; Ismaili, A.; Ghaffari, M.R.; Firouzabadi, F.N.; Shobbar, Z.-S. Transcriptome response of roots to salt stress in a salinity-tolerant bread wheat cultivar. PLoS ONE 2019, 14, e0213305. [Google Scholar] [CrossRef]
- Curci, P.L.; Cigliano, R.A.; Zuluaga, D.L.; Janni, M.; Sanseverino, W.; Sonnante, G. Transcriptomic response of durum wheat to nitrogen starvation. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Link, G.; Coen, N.M.; Bogorad, L. Differential expression of the gene for the large subunit of ribulose bisphosphate carboxylase in maize leaf cell types. Cell 1978, 15, 725–731. [Google Scholar] [CrossRef]
- Bedbrook, J.R.; Link, G.; Coen, D.M.; Bogorad, L. Maize plastid gene expressed during photoregulated development. Proc. Natl. Acad. Sci. USA 1978, 75, 3060–3064. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, Y.; Zhang, Y.; Li, C.; Gong, S.; Yan, S.; Li, G.; Hu, G.; Ren, H.; Yang, J.; et al. Comparative transcriptome analysis of salt-sensitive and salt-tolerant maize reveals potential mechanisms to enhance salt resistance. Genes Genom. 2019, 41, 781–801. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.S.; Wu, J.; Ziegler, T.E.; Yang, X.; Zayed, A.; Rajani, M.; Zhou, D.; Basra, A.S.; Schachtman, D.P.; Peng, M.; et al. Gene Expression Biomarkers Provide Sensitive Indicators of in Planta Nitrogen Status in Maize. Plant Physiol. 2011, 157, 1841–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamboni, A.; Astolfi, S.; Zuchi, S.; Pii, Y.; Guardini, K.; Tononi, P.; Varanini, Z. Nitrate induction triggers different transcrip-tional changes in a high and a low nitrogen use efficiency maize inbred line. J. Integr. Plant Biol. 2014, 56, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Marino, R.; Ponnaiah, M.; Krajewski, P.; Frova, C.; Gianfranceschi, L.; Pè, M.E.; Sari-Gorla, M. Addressing drought tolerance in maize by transcriptional profiling and mapping. Mol. Genet. Genom. 2008, 281, 163–179. [Google Scholar] [CrossRef]
- Guo, J.; Li, C.; Zhang, X.; Li, Y.; Zhang, D.; Shi, Y.; Song, Y.; Li, Y.; Yang, D.; Wang, T. Transcriptome and GWAS analyses reveal candidate gene for seminal root length of maize seedlings under drought stress. Plant Sci. 2020, 292, 110380. [Google Scholar] [CrossRef]
- Ren, J.; Xie, T.; Wang, Y.; Li, H.; Liu, T.; Zhang, S.; Yin, L.; Wang, S.; Deng, X.; Ke, Q. Coordinated regulation of carbon and nitrogen assimilation confers drought tolerance in maize (Zea mays L.). Environ. Exp. Bot. 2020, 176, 104086. [Google Scholar] [CrossRef]
- Humbert, S.; Subedi, S.; Cohn, J.; Zeng, B.; Bi, Y.-M.; Chen, X.; Zhu, T.; McNicholas, P.D.; Rothstein, S.J. Genome-wide ex-pression profiling of maize in response to individual and combined water and nitrogen stresses. BMC Genomics 2013, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Koornneeff, M.; Dellaert, L.; Van Der Veen, J. EMS- and relation-induced mutation frequencies at individual loci in Arabidopsis thaliana (L.) Heynh. Mutat. Res. Mol. Mech. Mutagen. 1982, 93, 109–123. [Google Scholar] [CrossRef]
- Pathirana, R. Plant mutation breeding in agriculture. CAB Rev. Perspect. Agric. Veter. Sci. Nutr. Nat. Resour. 2011, 6, 107–126. [Google Scholar] [CrossRef]
- McCallum, C.M.; Comai, L.; Greene, E.A.; Henikoff, S. Targeting induced locallesions in genomes (TILLING) for plant functional genomics. Plant Physiol. 2000, 123, 439–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taheri, S.; Abdullah, T.L.; Jain, S.M.; Sahebi, M.; Azizi, P. TILLING, high-resolution melting (HRM), and next-generation sequencing (NGS) techniques in plant mutation breeding. Mol. Breed. 2017, 37, 40. [Google Scholar] [CrossRef]
- Holme, I.B.; Wendt, T.; Holm, P.B. Intragenesis and cisgenesis as alternatives to transgenic crop development. Plant Biotechnol. J. 2013, 11, 395–407. [Google Scholar] [CrossRef]
- Cardi, T. Cisgenesis and genome editing: Combining concepts and efforts for a smarter use of genetic resources in crop breeding. Plant Breed. 2016, 135, 139–147. [Google Scholar] [CrossRef]
- Shewry, P.R.; Jones, H.D.; Halford, N.G. Plant Biotechnology: Transgenic Crops. In Food Biotechnology; Stahl, U., Donalies, U.E.B., Nevoigt, E., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 111, pp. 149–186. [Google Scholar]
- International Service for Acquisition of Agri-biotech Applications. Brief 55: Global Status of Commercialized Biotech/GM Crops: 2019; ISAAA: Ithaca, NY, USA, 2019. [Google Scholar]
- Kozaki, A.; Sakamoto, A.; Tanaka, K.; Takeba, G. The promoter of the gene for glutamine synthetase from rice shows or-gan-specific and substrate-induced expression in transgenic tobacco plants. Plant Cell Physiol. 1991, 32, 353–358. [Google Scholar] [CrossRef]
- López-Calcagno, P.E.; Fisk, S.; Brown, K.L.; Bull, S.E.; South, P.F.; Raines, C.A. Overexpressing the H-protein of the glycine cleavage system increases biomass yield in glasshouse and field-grown transgenic tobacco plants. Plant Biotechnol. J. 2019, 17, 141–151. [Google Scholar] [CrossRef]
- Bosher, J.M.; Labouesse, M. RNA interference: Genetic wand and genetic watchdog. Nat. Cell Biol. 2000, 2, E31–E36. [Google Scholar] [CrossRef]
- Abe, K.; Ichikawa, H. Gene Overexpression Resources in Cereals for Functional Genomics and Discovery of Useful Genes. Front. Plant Sci. 2016, 7, 1359. [Google Scholar] [CrossRef] [Green Version]
- Kamburova, V.S.; Nikitina, E.V.; Shermatov, S.E.; Buriev, Z.T.; Kumpatla, S.P.; Emani, C.; Abdurakhmonov, I.Y. Genome Editing in Plants: An Overview of Tools and Applications. Int. J. Agron. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.K.; Alper, H.S. The genome editing toolbox: A spectrum of approaches for targeted modification. Curr. Opin. Biotechnol. 2014, 30, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Noman, A.; Aqeel, M.; He, S. CRISPR-Cas9: Tool for Qualitative and Quantitative Plant Genome Editing. Front. Plant Sci. 2016, 7, 1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, W.J.; Golic, K.G. Ends-out, or replacement, gene targeting in Drosophila. Proc. Natl. Acad. Sci. USA 2003, 100, 2556–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156. [Google Scholar] [CrossRef] [Green Version]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [Green Version]
- Puchta, H.; Fauser, F. Gene targeting in plants: 25 years later. Int. J. Dev. Biol. 2013, 57, 629–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratho, S.; Jachuck, P. A method of inducing awnless condition in rice by chemical mutagenesis. Curr. Sci. 1971, 40, 274–276. [Google Scholar]
- Rao, H.S.; Sears, E. Chemical mutagenesis in Triticum aestivum. Mutat. Res. Mol. Mech. Mutagen. 1964, 1, 387–399. [Google Scholar] [CrossRef]
- Bellini, G.; Bianchi, A.; Ottaviano, E. The use of interchanges involving B-type chromosomes in studying artificial mutagenesis in maize. Mol. Genet. Genom. 1961, 92, 85–99. [Google Scholar] [CrossRef]
- Jiang, S.-Y.; Bachmann, D.; La, H.; Ma, Z.; Venkatesh, P.N.; Ramamoorthy, R.; Ramachandran, S. Ds insertion mutagenesis as an efficient tool to produce diverse variations for rice breeding. Plant Mol. Biol. 2007, 65, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.J.; Hassan, S.; Tariq, M.; Khan, T. Haploidy breeding and mutagenesis for drought tolerance in wheat. In Mutations, In Vitro and Molecular Techniques for Environmentally Sustainable Crop Improvement; Springer International Publishing: Berlin/Heidelberg, Germany, 2002; pp. 75–82. [Google Scholar]
- Gao, Z.; Liu, H.; Wang, H.; Li, N.; Wang, D.; Song, Y.; Miao, Y.; Song, C. Generation of the genetic mutant population for the screening and characterization of the mutants in response to drought in maize. Chin. Sci. Bull. 2014, 59, 766–775. [Google Scholar] [CrossRef]
- Agarwal, M.; Sahi, C.; Katiyar-Agarwal, S.; Agarwal, S.; Young, T.; Gallie, D.R.; Sharma, V.M.; Ganesan, K.; Grover, A. Molecular characterization of rice hsp101: Complementation of yeast hsp104 mutation by disaggregation of protein granules and differential expression in indica and japonica rice types. Plant Mol. Biol. 2003, 51, 543–553. [Google Scholar] [CrossRef]
- Xue, G.-P.; Sadat, S.; Drenth, J.; McIntyre, C.L. The heat shock factor family from Triticum aestivumin response to heat and other major abiotic stresses and their role in regulation of heat shock protein genes. J. Exp. Bot. 2014, 65, 539–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Du, H.; Wang, Y.; Wang, H.; Yang, S.; Li, C.; Chen, N.; Yang, H.; Zhang, Y.; Zhu, Y.; et al. The calcium-dependent protein kinase ZmCDPK7 functions in heat-stress tolerance in maize (Zea mays L.). J. Integr. Plant Biol. 2020. [Google Scholar] [CrossRef]
- Brauer, E.K.; Rochon, A.; Bi, Y.M.; Bozzo, G.G.; Rothstein, S.J.; Shelp, B.J. Reappraisal of nitrogen use efficiency in rice over-expressing glutamine synthetase1. Physiol. Plant. 2011, 141, 361–372. [Google Scholar] [CrossRef]
- Toriyama, K.; Arimoto, Y.; Uchimiya, H.; Hinata, K. Transgenic Rice Plants After Direct Gene Transfer into Protoplasts. Nat. Biotechnol. 1988, 6, 1072–1074. [Google Scholar] [CrossRef]
- Hess, D.; Dressler, K.; Nimmrichter, R. Transformation experiments by pipetting Agrobacterium into the spikelets of wheat (Triticum aestivum L.). Plant Sci. 1990, 72, 233–244. [Google Scholar] [CrossRef]
- Klein, T.M.; Fromm, M.; Weissinger, A.; Tomes, D.; Schaaf, S.; Sletten, M.; Sanford, J.C. Transfer of foreign genes into intact maize cells with high-velocity microprojectiles. Proc. Natl. Acad. Sci. USA 1988, 85, 4305–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.; Shen, Q.; Ho, T.-H.D.; Wu, R. Dehydration-Stress-Regulated Transgene Expression in Stably Transformed Rice Plants. Plant Physiol. 1998, 117, 913–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivamani, E.; Bahieldin, A.; Wraith, J.M.; Alniemi, T.S.; Dyer, W.E.; Ho, T.-H.D.; Qu, R. Improved biomass productivity and water use efficiency under water deficit conditions in transgenic wheat constitutively expressing the barley HVA1 gene. Plant Sci. 2000, 155, 1–9. [Google Scholar] [CrossRef]
- Jeanneau, M.; Gerentes, D.; Foueillassar, X.; Zivy, M.; Vidal, J.; Toppan, A.; Perez, P. Improvement of drought tolerance in maize: Towards the functional validation of the Zm-Asr1 gene and increase of water use efficiency by over-expressing C4–PEPC. Biochimie 2002, 84, 1127–1135. [Google Scholar] [CrossRef]
- Kishitani, S.; Takanami, T.; Suzuki, M.; Oikawa, M.; Yokoi, S.; Ishitani, M.; Alvarez-Nakase, A.M.; Takabe, T. Compatibility of glycinebetaine in rice plants: Evaluation using transgenic rice plants with a gene for peroxisomal betaine aldehyde dehydrogenase from barley. Plant Cell Environ. 2000, 23, 107–114. [Google Scholar] [CrossRef]
- Fu, J.; Momcilović, I.; Clemente, T.E.; Nersesian, N.; Trick, H.N.; Ristic, Z. Heterologous expression of a plastid EF-Tu reduces protein thermal aggregation and enhances CO2 fixation in wheat (Triticum aestivum) following heat stress. Plant Mol. Biol. 2008, 68, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Makino, A.; Shimada, T.; Takumi, S.; Kaneko, K.; Matsuoka, M.; Shimamoto, K.; Nakano, H.; Miyao-Tokutomi, M.; Mae, T.; Yamamoto, N. Does Decrease in Ribulose-1,5-Bisphosphate Carboxylase by Antisense RbcS Lead to a Higher N-Use Efficiency of Photosynthesis under Conditions of Saturating CO2 and Light in Rice Plants? Plant Physiol. 1997, 114, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Habash, D.Z.; Massiah, A.J.; Rong, H.L.; Wallsgrove, R.M.; Leigh, R.A. The role of cytosolic glutamine synthetase in wheat. Ann. Appl. Biol. 2001, 138, 83–89. [Google Scholar] [CrossRef]
- Liu, G.-W.; Sun, A.-L.; Li, D.-Q.; Athman, A.; Gilliham, M.; Liu, L.-H. Molecular identification and functional analysis of a maize (Zea mays) DUR3 homolog that transports urea with high affinity. Planta 2014, 241, 861–874. [Google Scholar] [CrossRef]
- Li, T.; Liu, B.; Spalding, M.H.; Weeks, D.P.; Yang, B. High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 2012, 30, 390–392. [Google Scholar] [CrossRef]
- Upadhyay, S.K.; Kumar, J.; Alok, A.; Tuli, R. RNA-guided genome editing for target gene mutations in wheat. G3 Genes Genomes Genet. 2013, 3, 2233–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Zhang, K.; Chen, K.; Gao, C. Targeted Mutagenesis in Zea mays Using TALENs and the CRISPR/Cas System. J. Genet. Genom. 2014, 41, 63–68. [Google Scholar] [CrossRef]
- Lou, D.; Wang, H.; Liang, G.; Yu, D. OsSAPK2 Confers Abscisic Acid Sensitivity and Tolerance to Drought Stress in Rice. Front. Plant Sci. 2017, 8, 993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chilcoat, D.; Liu, Z.-B.; Sander, J. Use of CRISPR/Cas9 for Crop Improvement in Maize and Soybean. Prog. Mol. Biol. Transl. Sci. 2017, 149, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhong, Z.; Wang, X.; Han, X.; Yu, D.; Wang, C.; Song, W.; Zheng, X.; Chen, C.; Zhang, Y. Knockout of the OsNAC006 Transcription Factor Causes Drought and Heat Sensitivity in Rice. Int. J. Mol. Sci. 2020, 21, 2288. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, X.; Sun, Y.; Zhang, J.; Du, W.; Guo, X.; Li, S.; Zhao, Y.; Xia, L. Efficient allelic replacement in rice by gene editing: A case study of the NRT1.1B gene. J. Integr. Plant Biol. 2018, 60, 536–540. [Google Scholar] [CrossRef] [Green Version]
- Todaka, D.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Recent advances in the dissection of drought-stress regulatory networks and strategies for development of drought-tolerant transgenic rice plants. Front. Plant Sci. 2015, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Duan, X.; Wang, B.; Hong, B.; Ho, T.; Wu, R. Expression of a Late Embryogenesis Abundant Protein Gene, HVA1, from Barley Confers Tolerance to Water Deficit and Salt Stress in Transgenic Rice. Plant Physiol. 1996, 110, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Targolli, J.; Huang, X.; Wu, R. Wheat LEA genes, PMA80 and PMA1959, enhance dehydration tolerance of trans-genic rice (Oryza sativa L.). Mol. Breed. 2002, 10, 71–82. [Google Scholar] [CrossRef]
- Yin, X.; Anand, A.; Quick, P.; Bandyopadhyay, A. Editing a stomatal developmental gene in rice with CRISPR/Cpf1. In Methods in Molecular Biology; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; Volume 1917, pp. 257–268. [Google Scholar]
- Gaudin, A.C.M.; Henry, A.; Sparks, A.H.; Slamet-Loedin, I.H. Taking transgenic rice drought screening to the field. J. Exp. Bot. 2013, 64, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamppa, G.; Nagy, F.; Chua, N.-H. Light-regulated and organ-specific expression of a wheat Cab gene in transgenic tobacco. Nat. Cell Biol. 1985, 316, 750–752. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, W.R.; Russell, S.H.; Quatrano, R.S. Abscisic acid-responsive sequences from the em gene of wheat. Plant Cell 1989, 1, 969–976. [Google Scholar] [PubMed] [Green Version]
- Shewry, P. Transgenic wheat: Where do we stand after the first 12 years? Ann. Appl. Biol. 2005, 147, 1–14. [Google Scholar] [CrossRef]
- Khan, S.; Anwar, S.; Yu, S.; Sun, M.; Yang, Z.; Gao, Z.-Q. Development of Drought-Tolerant Transgenic Wheat: Achievements and Limitations. Int. J. Mol. Sci. 2019, 20, 3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaponenko, A.K.; Shulga, O.A.; Mishutkina, Y.B.; Tsarkova, E.A.; Timoshenko, A.A.; Spechenkova, N.A. Perspectives of Use of Transcription Factors for Improving Resistance of Wheat Productive Varieties to Abiotic Stresses by Transgenic Technologies. Russ. J. Genet. 2018, 54, 27–35. [Google Scholar] [CrossRef]
- Hu, M.; Zhao, X.; Liu, Q.; Hong, X.; Zhang, W.; Zhang, Y.; Sun, L.; Li, H.; Tong, Y. Transgenic expression of plastidic glutamine synthetase increases nitrogen uptake and yield in wheat. Plant Biotechnol. J. 2018, 16, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Rochester, D.E.; Winer, J.A.; Shah, D.M. The structure and expression of maize genes encoding the major heat shock protein, hsp70. EMBO J. 1986, 5, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Streatfield, S.J.; Magallanes-Lundback, M.E.; Beifuss, K.K.; Brooks, C.A.; Harkey, R.L.; Love, R.T.; Bray, J.; Howard, J.A.; Jilka, J.M.; Hood, E.E. Analysis of the maize polyubiquitin-1 promoter heat shock elements and generation of promoter variants with modified expression characteristics. Transgenic Res. 2004, 13, 299–312. [Google Scholar] [CrossRef]
- Di, H.; Tian, Y.; Zu, H.; Meng, X.; Zeng, X.; Wang, Z. Enhanced salinity tolerance in transgenic maize plants expressing a BADH gene from Atriplex micrantha. Euphytica 2015, 206, 775–783. [Google Scholar] [CrossRef]
- Puskaric, V. Maize Inbred Line PH1MD Useful for Producing F1. Hybrid. Patent US6127610-A, 24 October 2000. [Google Scholar]
- Wu, J.; Lawit, S.J.; Weers, B.; Sun, J.; Mongar, N.; Van Hemert, J.; Melo, R.; Meng, X.; Rupe, M.; Clapp, J. Overexpression of zmm28 increases maize grain yield in the field. Proc. Natl. Acad. Sci. USA 2019, 116, 23850–23858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castiglioni, P.; Warner, D.; Bensen, R.J.; Anstrom, D.C.; Harrison, J.; Stoecker, M.; Abad, M.; Kumar, G.; Salvador, S.; D’Ordine, R.; et al. Bacterial RNA Chaperones Confer Abiotic Stress Tolerance in Plants and Improved Grain Yield in Maize under Water-Limited Conditions. Plant Physiol. 2008, 147, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Gao, H.; Wang, H.; Lafitte, H.R.; Archibald, R.L.; Yang, M.; Hakimi, S.M.; Mo, H.; Habben, J.E. ARGOS8 variants generated by CRISPR-Cas9 improve maize grain yield under field drought stress conditions. Plant Biotechnol. J. 2016, 15, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss-Fels, K.P.; Stahl, A.; Hickey, L.T. Q&A: Modern crop breeding for future food security. BMC Biol. 2019, 17, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Blum, A. Genomics for drought resistance–getting down to earth. Funct. Plant Biol. 2014, 41, 1191–1198. [Google Scholar] [CrossRef]
- Araus, J.L.; Cairns, J.E. Field high-throughput phenotyping: The new crop breeding frontier. Trends Plant Sci. 2014, 19, 52–61. [Google Scholar] [CrossRef]
- Mayer, M.; Hölker, A.C.; González-Segovia, E.; Bauer, E.; Presterl, T.; Ouzunova, M.; Melchinger, A.E.; Schön, C.-C. Discovery of beneficial haplotypes for complex traits in maize landraces. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Sansaloni, C.; Franco, J.; Santos, B.; Percival-Alwyn, L.; Singh, S.; Petroli, C.; Campos, J.; Dreher, K.; Payne, T.; Marshall, D.; et al. Diversity analysis of 80,000 wheat accessions reveals consequences and opportunities of selection footprints. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Curatti, L.; Rubio, L.M. Challenges to develop nitrogen-fixing cereals by direct nif-gene transfer. Plant Sci. 2014, 225, 130–137. [Google Scholar] [CrossRef]
- Rogers, C.; Oldroyd, G.E.D. Synthetic biology approaches to engineering the nitrogen symbiosis in cereals. J. Exp. Bot. 2014, 65, 1939–1946. [Google Scholar] [CrossRef] [Green Version]
- National Academies of Sciences, Engineering and Medicine. Future genetically engineered crops. In Genetically Engineered Crops: Experiences and Prospects; National Academies Press: Washington, DC, USA, 2016. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approach | Topic | Rice | Wheat | Maize |
|---|---|---|---|---|
| QTL mapping | (Any) | 1990 [42] | 1992 [43] | 1987 [44] |
| Drought tolerance | 1995 [45] | 1994 [46] | 1995 [47] | |
| Heat tolerance | 2000 [48] | 2002 [49] | 1991 [50] | |
| NUE | 2001 [51] | 2004 [52] | 1999 [53] | |
| GWAS | (Any) | 2009 [54] | 2009 [55] | 2011 [56] |
| Drought tolerance | 2010 [57] | 2014 [58] | 2011 [59] | |
| Heat tolerance | 2017 [60] | 2017 [61] | 2019 [62] | |
| NUE | 2019 [63] | 2014 [64] | 2017 [65] | |
| Genome selection | (Any) | 2014 [66] | 2011 [67] | 2007 [68] |
| Drought tolerance | 2018 [69] | 2018 [70] | 2013 [71] | |
| Heat tolerance | - | 2018 [70] | 2019 [72] | |
| NUE | 2016 [73] | 2019 [74] | 2015 [75] |
| Approach | Topic | Rice | Wheat | Maize |
|---|---|---|---|---|
| Gene expression | (Any) | 1984 [118] | 1972 [117] | 1971 [116] |
| Drought tolerance | 1988 [131] | 1991 [132] | 1991 [133] | |
| Heat tolerance | 1991 [134] | 1992 [135] | 1993 [136] | |
| NUE | 2006 [137] | 2008 [138] | 2006 [139] | |
| Transcriptomics | (Any) | 2001 [140] | 2002 [141] | 2003 [142] |
| Drought tolerance | 2006 [143] | 2009 [144] | 2007 [145] | |
| Heat tolerance | 2005 [146] | 2007 [147] | 2015 [148] | |
| NUE | 2006 [137] | 2008 [149] | 2009 [150] | |
| Quantitative PCR | (Any) | 2003 [151] | 2003 [152] | 1999 [153] |
| Drought tolerance | 2008 [154] | 2009 [155] | 2007 [156] | |
| Heat tolerance | 2008 [154] | 2011 [157] | 2007 [156] | |
| NUE | 2007 [158] | 2013 [159] | 2011 [160] | |
| RNA-seq | (Any) | 2010 [161] | 2011 [162] | 2011 [163] |
| Drought tolerance | 2015 [164] | 2014 [165] | 2012 [166] | |
| Heat tolerance | 2015 [167] | 2015 [168] | 2015 [169] | |
| NUE | 2018 [170] | 2014 [171] | 2015 [172] |
| Approach | Topic | Rice | Wheat | Maize |
|---|---|---|---|---|
| Mutagenesis | (Any) | 1971 [212] | 1964 [213] | 1961 [214] |
| Drought tolerance, WUE | 2007 [215] | 2001 [216] | 2014 [217] | |
| Heat tolerance | 2003 [218] | 2014 [219] | 2020 [220] | |
| NUE | 2011 [221] | - | 2006 [139] | |
| Transgenesis | (Any) | 1988 [222] | 1990 [223] | 1988 [224] |
| Drought tolerance, WUE | 1998 [225] | 2000 [226] | 2002 [227] | |
| Heat tolerance | 2000 [228] | 2008 [229] | 2007 [156] | |
| NUE | 1997 [230] | 2001 [231] | 2015 [232] | |
| Gene editing | (Any) | 2012 [233] | 2013 [234] | 2014 [235] |
| Drought tolerance, WUE | 2017 [236] | - | 2017 [237] | |
| Heat tolerance | 2020 [238] | - | - | |
| NUE | 2018 [239] | - | 2020 [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benavente, E.; Giménez, E. Modern Approaches for the Genetic Improvement of Rice, Wheat and Maize for Abiotic Constraints-Related Traits: A Comparative Overview. Agronomy 2021, 11, 376. https://doi.org/10.3390/agronomy11020376
Benavente E, Giménez E. Modern Approaches for the Genetic Improvement of Rice, Wheat and Maize for Abiotic Constraints-Related Traits: A Comparative Overview. Agronomy. 2021; 11(2):376. https://doi.org/10.3390/agronomy11020376
Chicago/Turabian StyleBenavente, Elena, and Estela Giménez. 2021. "Modern Approaches for the Genetic Improvement of Rice, Wheat and Maize for Abiotic Constraints-Related Traits: A Comparative Overview" Agronomy 11, no. 2: 376. https://doi.org/10.3390/agronomy11020376